β-blockers Reverse Agonist-Induced β2-AR Downregulation Regardless of Their Signaling Profile

 , and
, and 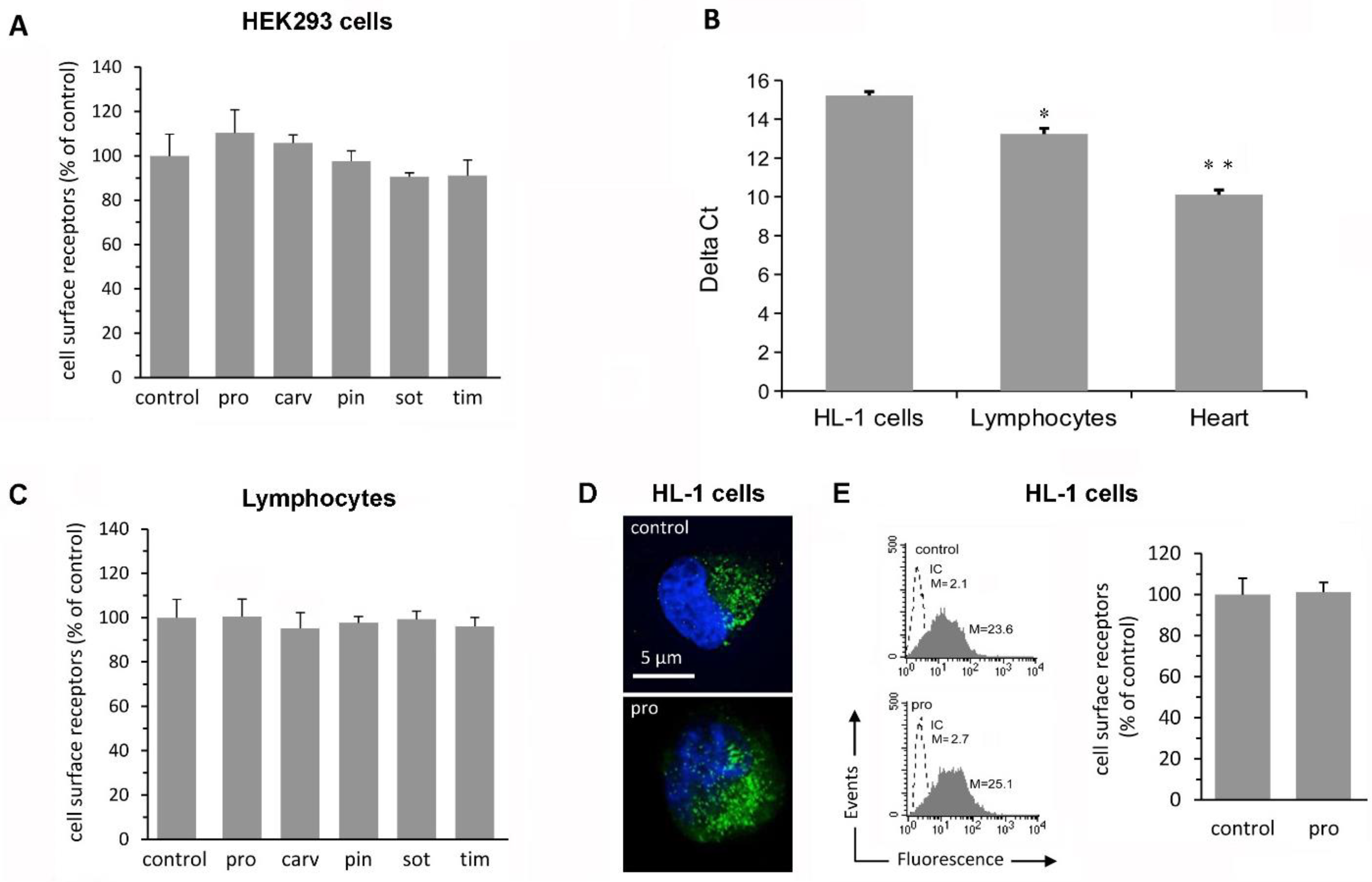{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. β-Blockers Do Not Affect Receptor Density in Simple Cell Systems
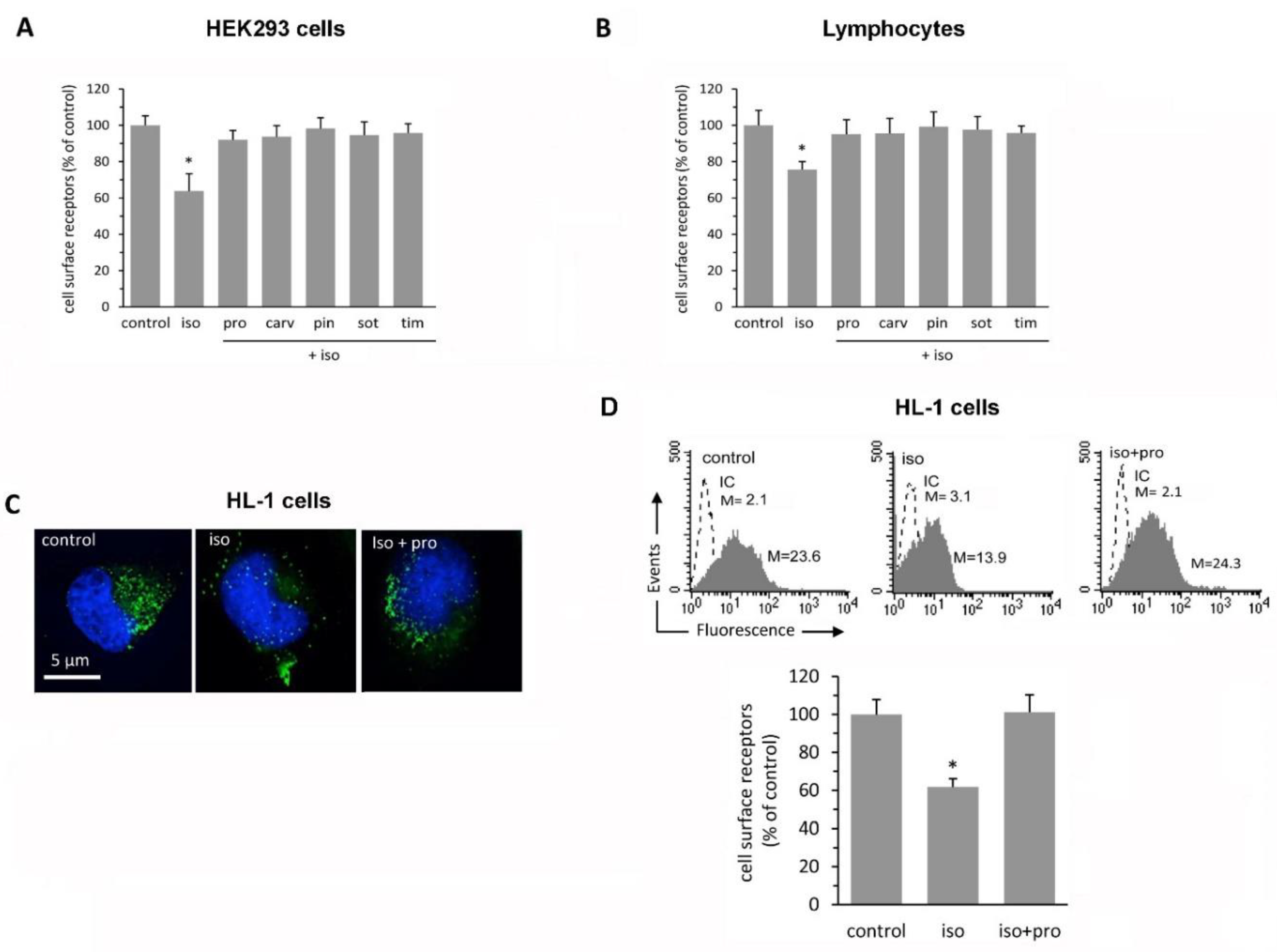
2.2. β-Blockers Restore β2-AR Density after Agonist-Induced Receptor Downregulation
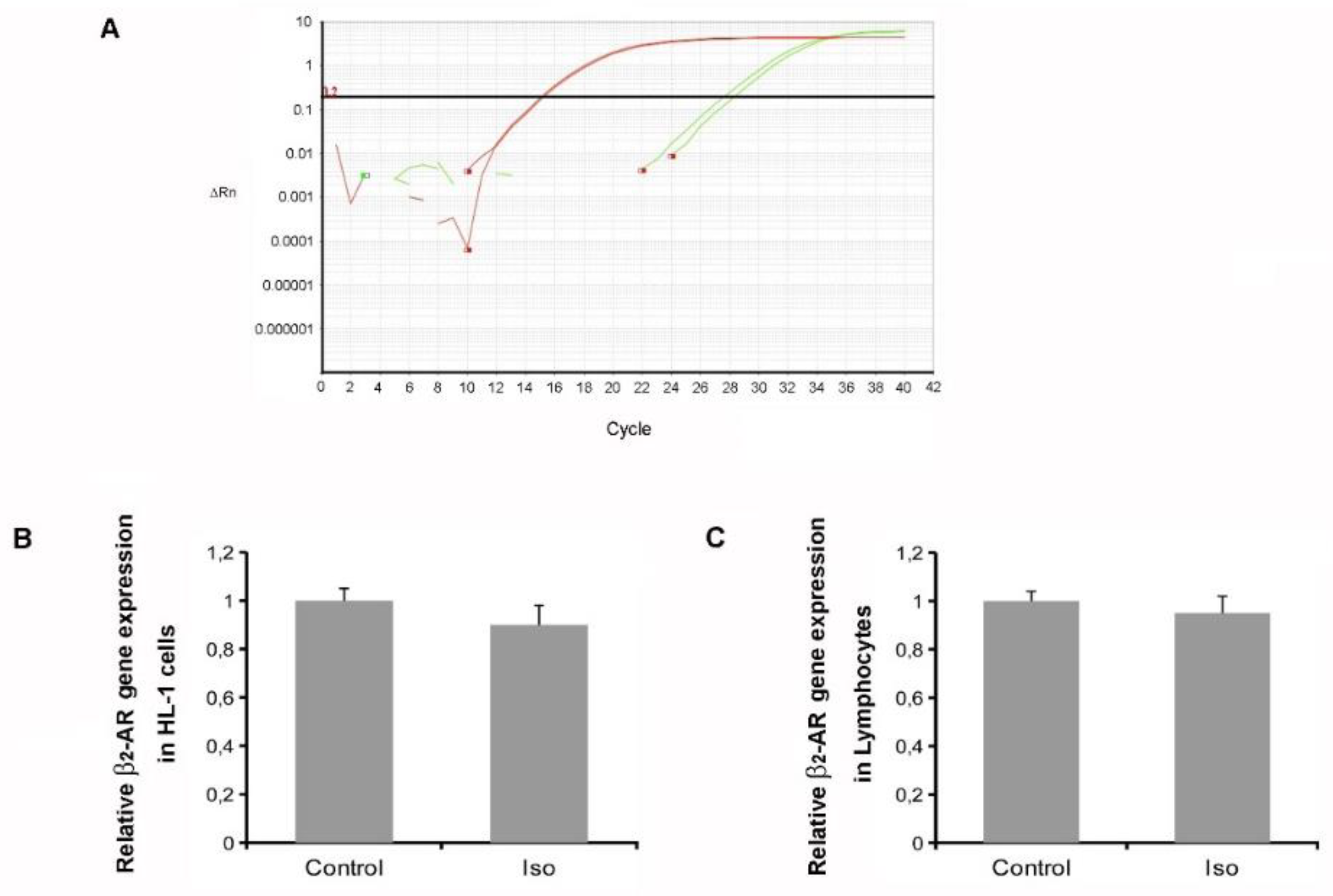
2.3. Isoproterenol Did Not Affect β2-AR Gene Expression
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Isolation of Human Lymphocytes
4.4. Fluorescence Microscopy
4.5. Flow Cytometry
4.6. Receptor Downregulation and Recycling Assays
4.7. RNA Isolation and Quantification
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aarons, R.D.; Molinoff, P.B. Changes in the density of β-adrenergic receptors in rat lymphocytes, heart and lung after chronic treatment with propranolol. J. Pharmacol. Exp. Ther. 1982, 221, 439–443. [Google Scholar] [PubMed]
- van den Meiracker, A.H.; Man in’t Veld, A.J.; Boomsma, F.; Fischberg, D.J.; Molinoff, P.B.; Schalekamp, M.A. Hemodynamic and β-adrenergic receptor adaptations during long-term β-adrenoceptor blockade. Studies with acebutolol, atenolol, pindolol, and propranolol in hypertensive patients. Circulation 1989, 80, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Hedberg, A.; Gerber, J.G.; Nies, A.S.; Wolfe, B.B.; Molinoff, P.B. Effects of pindolol and propranolol on β-adrenergic receptors on human lymphocytes. Pharmacol. Exp. Ther. 1986, 239, 117–123. [Google Scholar]
- Michel, M.C.; Pingsmann, A.; Beckeringh, J.J.; Zerkowski, H.R.; Doetsch, N.; Brodde, O.E. Selective regulation of β-1- and β-2-adrenoceptors in the human heart by chronic β-adrenoceptor antagonist treatment. Br. J. Pharmacol. 1988, 94, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, E.M.; Abraham, W.T.; Olsen, S.; Hattler, B.; White, M.; Mealy, P.; Larrabee, P.; Bristow, M.R. Comparative hemodynamic, left ventricular functional, and antiadrenergic effects of chronic treatment with metoprolol versus carvedilol in the failing heart. Circulation 1996, 94, 2817–2825. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.F. β1-adrenergic receptor regulation revisited. Circ. Res. 2018, 123, 1199–1201. [Google Scholar] [CrossRef]
- Nooh, M.M.; Mancarella, S.; Bahouth, S.W. Novel paradigms governing β1-adrenergic receptor trafficking in primary adult rat cardiac myocytes. Mol. Pharmacol. 2018, 94, 862–875. [Google Scholar] [CrossRef] [Green Version]
- Zuckerman, D.M.; Hicks, S.W.; Charron, G.; Hang, H.C.; Machamer, C.E. Differential regulation of two palmitoylation sites in the cytoplasmic tail of the β1-adrenergic receptor. J. Biol. Chem. 2001, 286, 19014–19023. [Google Scholar] [CrossRef] [Green Version]
- AlOkda, A.M.; Nasr, M.M.; Amin, S.N. Between an ugly truth and a perfect lie: Wiping off fearful memories using beta-adrenergic receptors antagonists. J. Cell. Physiol. 2019, 234, 5722–5727. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Sorriento, D.; Coscioni, E.; Iaccarino, G.; Santulli, G. Adrenergic Receptors. In Endocrinology of the Heart in Health and Disease; Academic Press: Amsterdam, The Netherlands, 2017; pp. 285–315. [Google Scholar] [CrossRef]
- Lorton, D.; Bellinger, D.L. Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisler, J.W.; DeWire, S.M.; Whalen, E.J.; Violin, J.D.; Drake, M.T.; Ahn, S.; Shenoy, S.K.; Lefkowitz, R.J. A unique mechanism of beta-blocker action: Carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Certo, M.G.; Batassa, E.M.; Casella, I.; Serafino, A.; Floridi, A.; Passananti, C.; Molinari, P.; Mattei, E. Delayed internalization and lack of recycling in a β2-adrenergic receptor fused to the G protein alpha-subunit. BMC Cell. Biol. 2008, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, L.T.; Snyderman, R.; Lefkowitz, R.J. Identification of β-adrenergic receptors in human lymphocytes by (-)[3H] alprenolol binding. J. Clin. Invest. 1976, 2, 149–155. [Google Scholar] [CrossRef]
- Warner, A.L.; Bellah, K.L.; Raya, T.E.; Roeske, W.R.; Goldman, S. Effects of β-adrenergic blockade on papillary muscle function and the beta-adrenergic receptor system in noninfarcted myocardium in compensated ischemic left ventricular dysfunction. Circulation 1992, 86, 1584–1595. [Google Scholar] [CrossRef] [Green Version]
- Glaubiger, G.; Lefkowitz, R.J. Elevated β-adrenergic receptor number after chronic propranolol treatment. Biochem. Biophys. Res. Commun. 1977, 78, 720–725. [Google Scholar] [CrossRef]
- Filipeanu, C.M.; Zhou, F.; Lam, M.L.; Kerut, K.E.; Claycomb, W.C.; Wu, G. Enhancement of the recycling and activation of β-adrenergic receptor by Rab4 GTPase in cardiac myocytes. J. Biol. Chem. 2006, 281, 11097–11103. [Google Scholar] [CrossRef] [Green Version]
- Dangel, V.; Giray, J.; Ratge, D.; Wisser, H. Regulation of β-adrenoceptor density and mRNA levels in the rat heart cell-line H9c2. Biochem. J. 1996, 317, 925–931. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, E.E.; Molinoff, P.B. Down regulation of β-adrenergic receptors in S49 lymphoma cells induced by atypical agonists. J. Pharmacol. Exp. Ther. 1986, 239, 654–660. [Google Scholar]
- Asano, K.; Zisman, L.S.; Yoshikawa, T.; Headley, V.; Bristow, M.R.; Port, J.D. Bucindolol, a nonselective β1- and β2-adrenergic receptor antagonist, decreases β-adrenergic receptor density in cultured embryonic chick cardiac myocyte membranes. J. Cardiovasc. Pharmacol. 2001, 37, 678–691. [Google Scholar] [CrossRef]
- Flesch, M.; Ettelbrück, S.; Rosenkranz, S.; Maack, C.; Cremers, B.; Schlüter, K.D.; Zolk, O.; Böhm, M. Differential effects of carvedilol and metoprolol on isoprenaline-induced changes in β-adrenoceptor density and systolic function in rat cardiac myocytes. Cardiovasc. Res. 2001, 49, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Hughes, R.J.; Howard, M.J.; Allen, J.M.; Insel, P.A. Decreased β2-adrenergic receptor mRNA expression in receptor-deficient S49 lymphoma cells. Mol. Pharmacol. 1991, 40, 974–979. [Google Scholar]
- Kindermann, M.; Maack, C.; Schaller, S.; Finkler, N.; Schmidt, K.I.; Läer, S.; Wuttke, H.; Schäfers, H.J.; Böhm, M. Carvedilol but not metoprolol reduces β-adrenergic responsiveness after complete elimination from plasma in vivo. Circulation 2004, 109, 3182–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110γ. Mol. Cell. 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzo, R.; Cipolletta, E.; Ciccarelli, M.; Campanile, A.; Santulli, G.; Palumbo, G.; Vasta, A.; Formisano, S.; Trimarco, B.; Iaccarino, G. Enhanced GRK2 expression and desensitization of β-AR vasodilatation in hypertensive patients. Clin. Transl. Sci. 2008, 1, 215–220. [Google Scholar] [CrossRef]
- Ambrosio, C.; Molinari, P.; Fanelli, F.; Chuman, Y.; Sbraccia, M.; Ugur, O.; Costa, T. Different structural requirements for the constitutive and the agonist-induced activities of the beta2-adrenergic receptor. J. Biol. Chem. 2005, 280, 23464–23474. [Google Scholar] [CrossRef] [Green Version]
- Makita, N.; Kabasawa, Y.; Otani, Y.; Firman; Sato, J.; Hashimoto, M.; Nakaya, M.; Nishihara, H.; Nangaku, M.; Kurose, H.; et al. Attenuated desensitization of β-adrenergic receptor by water-soluble N-nitrosamines that induce S-nitrosylation without NO release. Circ. Res. 2013, 112, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Kumari, P.; Srivastava, A.; Banerjee, R.; Ghosh, E.; Gupta, P.; Ranjan, R.; Chen, X.; Gupta, B.; Gupta, C.; Jaiman, D.; et al. Functional competence of a partially engaged GPCR-β-arrestin complex. Nat. Commun. 2016, 7, 13416. [Google Scholar] [CrossRef] [Green Version]
- Tyurin-Kuzmin, P.A.; Fadeeva, J.I.; Kanareikina, M.A.; Kalinina, N.I.; Sysoeva, V.Y.; Dyikanov, D.T.; Stambolsky, D.V.; Tkachuk, V.A. Activation of β-adrenergic receptors is required for elevated α1A-adrenoreceptors expression and signaling in mesenchymal stromal cells. Sci. Rep. 2016, 6, 32835. [Google Scholar] [CrossRef]
- Tyurin-Kuzmin, P.A.; Dyikanov, D.T.; Fadeeva, J.I.; Sysoeva, V.Y.; Kalinina, N.I. Flow cytometry analysis of adrenoceptors expression in human adipose-derived mesenchymal stem/stromal cells. Sci. Data 2018, 5, 180196. [Google Scholar] [CrossRef]
- Saygin, D.; Wanner, N.; Rose, J.A.; Naga Prasad, S.V.; Tang, W.H.W.; Erzurum, S.; Asosingh, K. Relative quantification of β-adrenergic receptor in peripheral blood cells using flow cytometry. Cytometry A 2018, 93, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, J.G.; Hall, I.P.; Hill, S.J. Agonist and inverse agonist actions of beta-blockers at the human β2-adrenoceptor provide evidence for agonist-directed signaling. Mol. Pharmacol. 2003, 64, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Casella, I.; Ambrosio, C.; Grò, M.C.; Molinari, P.; Costa, T. Divergent agonist selectivity in activating β1- and β2-adrenoceptors for G-protein and arrestin coupling. Biochem. J. 2011, 438, 191–202. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maccari, S.; Vezzi, V.; Barbagallo, F.; Stati, T.; Ascione, B.; Grò, M.C.; Catalano, L.; Marano, G.; Matarrese, P.; Ambrosio, C.; et al. β-blockers Reverse Agonist-Induced β2-AR Downregulation Regardless of Their Signaling Profile. Int. J. Mol. Sci. 2020, 21, 512. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020512
Maccari S, Vezzi V, Barbagallo F, Stati T, Ascione B, Grò MC, Catalano L, Marano G, Matarrese P, Ambrosio C, et al. β-blockers Reverse Agonist-Induced β2-AR Downregulation Regardless of Their Signaling Profile. International Journal of Molecular Sciences. 2020; 21(2):512. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020512
Chicago/Turabian StyleMaccari, Sonia, Vanessa Vezzi, Federica Barbagallo, Tonino Stati, Barbara Ascione, Maria Cristina Grò, Liviana Catalano, Giuseppe Marano, Paola Matarrese, Caterina Ambrosio, and et al. 2020. "β-blockers Reverse Agonist-Induced β2-AR Downregulation Regardless of Their Signaling Profile" International Journal of Molecular Sciences 21, no. 2: 512. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21020512