Periodontal Disease and Senescent Cells: New Players for an Old Oral Health Problem?


Abstract
:1. Introduction
2. Cellular Senescence
2.1. Stress-Induced Premature Senescence
2.2. Senescence-Associated Secretory Phenotype (SASP)
2.3. Paracrine Senescence
2.4. Senescent Cells: Friends, Foes, or Both?
3. Cellular Senescence as a DNA Damage Response
3.1. Immune-Mediated Senescent Cells Clearance
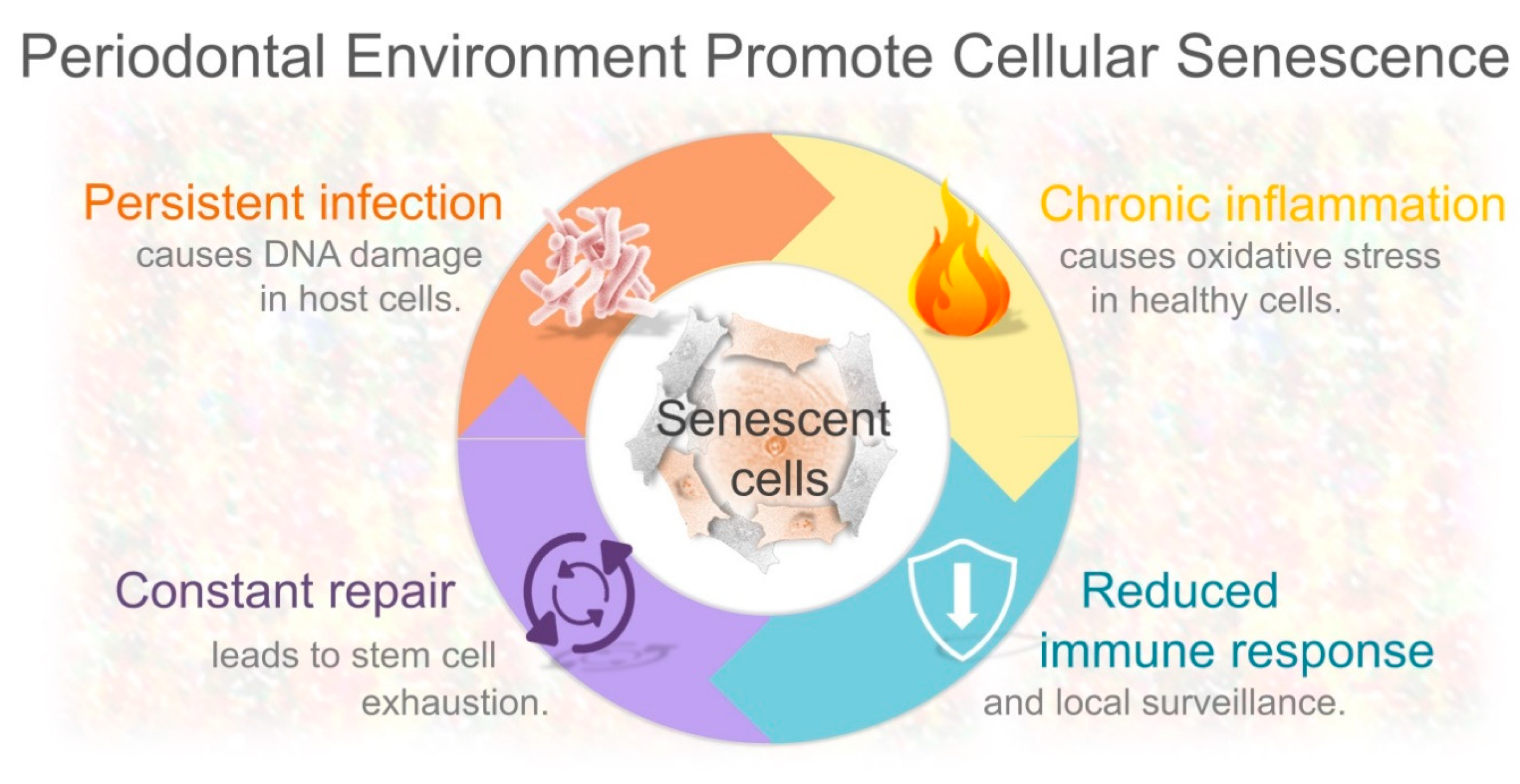
4. Periodontal Inflammation Creates a Permissive Environment for Cellular Senescence
4.1. DNA Damage-Induced Senescence as a Result of Persistent Gram-Negative Bacterial Infection
4.2. Chronic Inflammation Itself Induces DNA Damage-Driven Cellular Senescence
4.3. Constant Renewal of Damaged Periodontal Tissues and Replicative Senescence
4.4. Could Bacteria-Mediated Local Immunosuppression Promote Senescent Cell Accumulation?
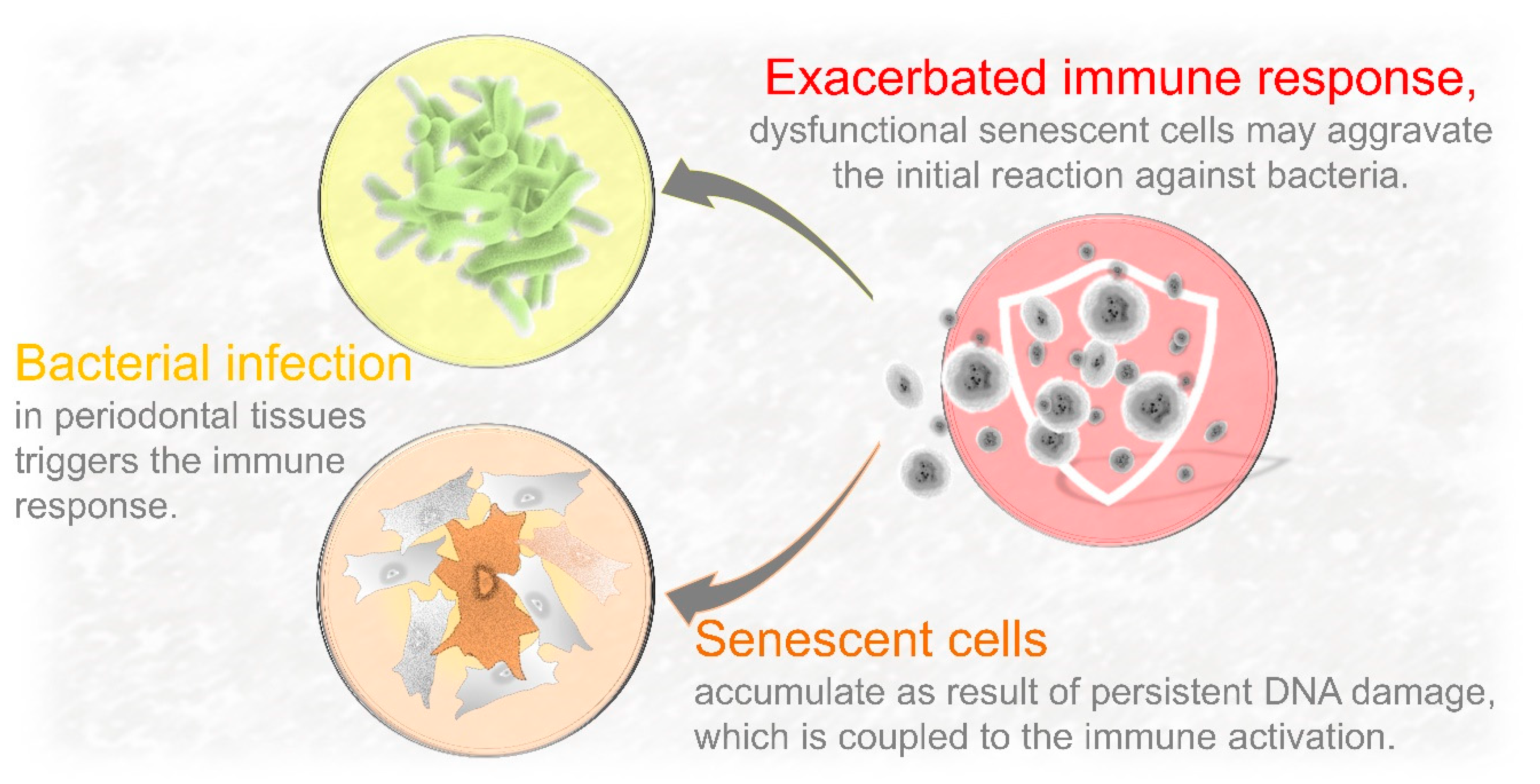
5. Can DNA Damage-Driven Senescence Contribute to Increase Local Infiltration of Immune Cells?
Bacterial Recognition and Nuclear DNA Damage Converge on NF-κB Activation
6. Clinical Implications and Therapeutic Opportunities
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| H2O2 | Hydrogen peroxide |
| ROS | Reactive oxygen species |
| DDR | DNA damage response |
| SASP | Senescence-associated secretory phenotype |
| IL | Interleukin |
| CDT | Cytolethal distending toxin |
| LPS | Lipopolysaccharide |
| MMP | Matrix metallopeptidase |
| HMGB1 | High Mobility Group Box 1 |
References
- Darveau, R.P. Periodontitis: A polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010, 8, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Haffajee, A.D. Microbial mechanisms in the pathogenesis of destructive periodontal diseases: A critical assessment. J. Periodontal Res. 1991, 26, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Paster, B.J.; Boches, S.K.; Galvin, J.L.; Ericson, R.E.; Lau, C.N.; Levanos, V.A.; Sahasrabudhe, A.; Dewhirst, F.E. Bacterial Diversity in Human Subgingival Plaque. J. Bacteriol. 2001, 183, 3770–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezow, A.B.; Darveau, R.P. Microbial shift and periodontitis. Periodontology 2000 2011, 55, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Bartold, P.M.; Van Dyke, T.E. Host modulation: Controlling the inflammation to control the infection. Periodontology 2000 2017, 75, 317–329. [Google Scholar] [CrossRef]
- Bartold, P.M.; Van Dyke, T.E. Periodontitis: A host-mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontology 2000 2013, 62, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Page, R.C.; Schroeder, H.E. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab. Investig. 1976, 34, 235. [Google Scholar]
- Hajishengallis, G.; Korostoff, J.M. Revisiting the Page & Schroeder model: The good, the bad and the unknowns in the periodontal host response 40 years later. Periodontology 2000 2017, 75, 116–151. [Google Scholar] [CrossRef]
- Miyasaki, K.T.; Wilson, M.E.; Brunetti, A.J.; Genco, R.J. Oxidative and nonoxidative killing of Actinobacillus actinomycetemcomitans by human neutrophils. Infect. Immun. 1986, 53, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, A.; Yamamoto, K.-I. DNA damage responses to oxidative stress. DNA Repair 2004, 3, 1109–1115. [Google Scholar] [CrossRef]
- Chapple, I.L.C.; Matthews, J.B. The role of reactive oxygen and antioxidant species in periodontal tissue destruction. Periodontology 2000 2007, 43, 160–232. [Google Scholar] [CrossRef]
- Cheng, R.; Choudhury, D.; Liu, C.; Billet, S.; Hu, T.; Bhowmick, N.A. Gingival fibroblasts resist apoptosis in response to oxidative stress in a model of periodontal diseases. Cell Death Discov. 2015, 1, 15046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquino-Martinez, R.; Rowsey, J.L.; Fraser, D.G.; Eckhardt, B.A.; Khosla, S.; Farr, J.N.; Monroe, D.G. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone 2020, 132, 115220. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, O.C.B.; Frisan, T. Bacterial Genotoxin-Induced DNA Damage and Modulation of the Host Immune Microenvironment. Toxins (Basel) 2020, 12, 63. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Campisi, J.; Di Fagagna, F.D. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Vaziri, H.; Benchimol, S. From telomere loss to p53 induction and activation of a DNA-damage pathway at senescence: The telomere loss/DNA damage model of cell aging. Exp. Gerontol. 1996, 31, 295–301. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasco, M.A. Telomere length, stem cells and aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette Smoke Induces Cellular Senescence. Am. J. Respir. Cell Mol. Biol. 2006, 35, 681–688. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Chen, Q.; Ames, B.N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4130–4134. [Google Scholar] [CrossRef] [Green Version]
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling. J. Cell. Mol. Med. 2010, 14, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Toussaint, O.; Medrano, E.E.; Von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimozi, A.; Mavrogonatou, E.; Sklirou, A.; Kletsas, D. Oxidative stress inhibits the proliferation, induces premature senescence and promotes a catabolic phenotype in human nucleus pulposus intervertebral disc cells. Eur. Cells Mater. 2015, 30, 89–103. [Google Scholar] [CrossRef]
- Duan, J.; Duan, J.; Zhang, Z.; Tong, T. Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int. J. Biochem. Cell Biol. 2005, 37, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-M.; Zhao, Y.-M.; Luo, X.; Feng, Y.; Ren, Y.; Shang, H.; He, Z.-Y.; Luo, X.-M.; Chen, S.-D.; Wang, X.-Y. Repeated Lipopolysaccharide Stimulation Induces Cellular Senescence in BV2 Cells. Neuroimmunomodulation 2012, 19, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Feng, G.; Xing, J.; Shen, B.; Tan, W.; Huang, D.; Lu, X.; Tao, T.; Zhang, J.; Li, L.; et al. Repeated lipopolysaccharide stimulation promotes cellular senescence in human dental pulp stem cells (DPSCs). Cell Tissue Res. 2014, 356, 369–380. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, X. Effect of lipopolysaccharides on adipogenic potential and premature senescence of adipocyte progenitors. Am. J. Physiol.-Endocrinol. Metab. 2015, 309, E334–E344. [Google Scholar] [CrossRef] [Green Version]
- Correia-Melo, C.; Hewitt, G.; Passos, J.F. Telomeres, oxidative stress and inflammatory factors: Partners in cellular senescence? Longev. Health 2014, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [Green Version]
- Levi, N.; Papismadov, N.; Solomonov, I.; Sagi, I.; Krizhanovsky, V. The ECM path of senescence in aging: Components and modifiers. FEBS J. 2020. [Google Scholar] [CrossRef]
- Le Maitre, C.L.; Freemont, A.J.; Hoyland, J.A. Accelerated cellular senescence in degenerate intervertebral discs: A possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res. Ther. 2007, 9, R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philipot, D.; Guérit, D.; Platano, D.; Chuchana, P.; Olivotto, E.; Espinoza, F.; Dorandeu, A.; Pers, Y.-M.; Piette, J.; Borzi, R.M.; et al. p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 2014, 16, R58. [Google Scholar] [CrossRef] [Green Version]
- Victorelli, S.; Passos, J.F. Reactive oxygen species detection in senescent cells. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019. [Google Scholar]
- Hoare, M.; Narita, M. Transmitting senescence to the cell neighbourhood. Nat. Cell Biol. 2013, 15, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; Von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Bird, T.G.; Müller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.-Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, eaan1230. [Google Scholar] [CrossRef] [Green Version]
- Kojima, H.; Inoue, T.; Kunimoto, H.; Nakajima, K. IL-6-STAT3 signaling and premature senescence. JAK-STAT 2013, 2, e25763. [Google Scholar] [CrossRef]
- Severino, V.; Alessio, N.; Farina, A.; Sandomenico, A.; Cipollaro, M.; Peluso, G.; Galderisi, U.; Chambery, A. Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis. 2013, 4, e911. [Google Scholar] [CrossRef]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; Von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Ovadya, Y.; Krizhanovsky, V. Senescent cells: SASPected drivers of age-related pathologies. Biogerontology 2014, 15, 627–642. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef] [Green Version]
- Sone, H.; Kagawa, Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 2005, 48, 58–67. [Google Scholar] [CrossRef]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Van Gool, F.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060.e10. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Tavazzi, G.; Pellegrini, C.; Maurelli, M.; Belliato, M.; Sciutti, F.; Bottazzi, A.; Sepe, P.A.; Resasco, T.; Camporotondo, R.; Bruno, R.; et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur. J. Heart Fail. 2020, 22, 911–915. [Google Scholar] [CrossRef] [Green Version]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [Green Version]
- Hampel, B.; Wagner, M.; Teis, D.; Zwerschke, W.; Huber, L.A.; Jansen-Dürr, P. Apoptosis resistance of senescent human fibroblasts is correlated with the absence of nuclear IGFBP-3. Aging Cell 2005, 4, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Kale, A.; Sharma, A.; Stolzing, A.; Desprez, P.-Y.; Campisi, J. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 2020, 17, 16. [Google Scholar] [CrossRef]
- Song, P.; An, J.; Zou, M.-H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.C.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Haber, J.E. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Kurosaka, K.; Takahashi, M.; Watanabe, N.; Kobayashi, Y. Silent Cleanup of Very Early Apoptotic Cells by Macrophages. J. Immunol. 2003, 171, 4672–4679. [Google Scholar] [CrossRef] [Green Version]
- Fielder, E.; Von Zglinicki, T.; Jurk, D. The DNA Damage Response in Neurons: Die by Apoptosis or Survive in a Senescence-Like State? J. Alzheimer’s Dis. 2017, 60, S107–S131. [Google Scholar] [CrossRef]
- Chen, Q.M.; Liu, J.; Merrett, J.B. Apoptosis or senescence-like growth arrest: Influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem. J. 2000, 347, 543. [Google Scholar] [CrossRef]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002, 1, 639–649. [Google Scholar]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoenicke, L.; Zender, L. Immune surveillance of senescent cells-biological significance in cancer-and non-cancer pathologies. Carcinogenesis 2012, 33, 1123–1126. [Google Scholar] [PubMed] [Green Version]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.-P.; Rodier, F.; Campisi, J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Idan, C.; Peleg, R.; Elena, V.; Martin, T.; Cicerone, T.; Mareike, W.; Lydia, B.; Marina, F.; Gerhard, M.; Elisa, F.M.; et al. IL-1α is a DNA damage sensor linking genotoxic stress signaling to sterile inflammation and innate immunity. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STI NG pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar]
- Wang, Y.; Andrukhov, O.; Rausch-Fan, X. Oxidative Stress and Antioxidant System in Periodontitis. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef]
- Kim, I.D.; Ha, B.J. Paeoniflorin protects RAW 264.7 macrophages from LPS-induced cytotoxicity and genotoxicity. Toxicol. In Vitro 2009, 23, 1014–1019. [Google Scholar] [CrossRef]
- Glukhov, I.L.; Sirota, N.P.; Kuznetsova, E.A. Dna damage in human mononuclear cells induced by bacterial endotoxin. Bull. Exp. Biol. Med. 2008, 146, 301–303. [Google Scholar] [CrossRef]
- Bezine, E.; Vignard, J.; Mirey, G. The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective. Cells 2014, 3, 592. [Google Scholar] [CrossRef] [Green Version]
- Belibasakis, G.N.; Mattsson, A.; Wang, Y.; Chen, C.; Johansson, A. Cell cycle arrest of human gingival fibroblasts and periodontal ligament cells by Actinobacillus actinomycetemcomitans: Involvement of the cytolethal distending toxin. APMIS 2004, 112, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Frisan, T. Bacterial genotoxins: The long journey to the nucleus of mammalian cells. Biochim. Biophys. Acta Biomembr. 2016, 1858, 567–575. [Google Scholar] [CrossRef]
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dörsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair 2014, 18, 31–43. [Google Scholar] [CrossRef] [PubMed]
- DiRienzo, J.M. Breaking the Gingival Epithelial Barrier: Role of the Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin in Oral Infectious Disease. Cells 2014, 3, 476–499. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; Guidi, R.; Frisan, T. Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumor progression? FEBS J. 2011, 278, 4577–4588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, F.; Frisan, T. Bacterial Genotoxins: Merging the DNA Damage Response into Infection Biology. Biomolecules 2015, 5, 1762–1782. [Google Scholar] [CrossRef] [Green Version]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Van Houten, B.; Santa-Gonzalez, G.A.; Camargo, M. DNA repair after oxidative stress: Current challenges. Curr. Opin. Toxicol. 2018, 7, 9–16. [Google Scholar] [CrossRef]
- Davies, K. The Broad Spectrum of Responses to Oxidants in Proliferating Cells: A New Paradigm for Oxidative Stress. IUBMB Life 1999, 48, 41–47. [Google Scholar] [CrossRef]
- Kiyoshima, T.; Enoki, N.; Kobayashi, I.; Sakai, T.; Nagata, K.; Wada, H.; Fujiwara, H.; Ookuma, Y.; Sakai, H. Oxidative stress caused by a low concentration of hydrogen peroxide induces senescence-like changes in mouse gingival fibroblasts. Int. J. Mol. Med. 2012, 30, 1007–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.-Y.; Lee, S.-Y.; Son, Y.-O.; Shi, X.; Park, S.-S.; Lee, J.-C. Continuous presence of H2O2 induces mitochondrial-mediated, MAPK- and caspase-independent growth inhibition and cytotoxicity in human gingival fibroblasts. Toxicol. In Vitro 2012, 26, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Sun, M.; Xie, Y.-F.; Shu, R. mTOR Inhibition Rejuvenates the Aging Gingival Fibroblasts through Alleviating Oxidative Stress. Oxidative Med. Cell. Longev. 2017, 2017, 6292630. [Google Scholar] [CrossRef] [PubMed]
- D’Aiuto, F.; Nibali, L.; Parkar, M.; Patel, K.; Suvan, J.; Donos, N. Oxidative stress, systemic inflammation, and severe periodontitis. J. Dent. Res. 2010, 89, 1241–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, I.; Machado Moreira, E.A.; Filho, D.W.; De Oliveira, T.B.; Da Silva, M.B.S.; Fröde, T.S. Proinflammatory and oxidative stress markers in patients with periodontal disease. Mediat. Inflamm. 2007. [Google Scholar] [CrossRef]
- Parsonnet, J. Bacterial infection as a cause of cancer. Environ. Health Perspect. 1995, 103, 263–268. [Google Scholar] [PubMed] [Green Version]
- Moffatt-Jauregui, C.E.; Robinson, B.; De Moya, A.V.; Brockman, R.D.; Roman, A.V.; Cash, M.N.; Culp, D.J.; Lamont, R.J. Establishment and characterization of a telomerase immortalized human gingival epithelial cell line. J. Periodontal Res. 2013, 48, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.; Choi, Y.S.; Choi, Y. Bacterial invasion and persistence: Critical events in the pathogenesis of periodontitis? J. Periodontal Res. 2014, 50, 570–585. [Google Scholar] [CrossRef]
- Bulut, S.; Uslu, H.; Ozdemir, B.H.; Bulut, O.E. Analysis of proliferative activity in oral gingival epithelium in immunosuppressive medication induced gingival overgrowth. Head Face Med. 2006, 2, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kuboniwa, M.; Hasegawa, Y.; Mao, S.; Shizukuishi, S.; Amano, A.; Lamont, R.J.; Yilmaz, Ö. P. gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes Infect. 2008, 10, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Ashida, H.; Ogawa, M.; Yoshikawa, Y.; Mimuro, H.; Sasakawa, C. Bacterial Interactions with the Host Epithelium. Cell Host Microbe 2010, 8, 20–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damek-Poprawa, M.; Haris, M.; Volgina, A.; Korostoff, J.; DiRienzo, J.M. Cytolethal Distending Toxin Damages the Oral Epithelium of Gingival Explants. J. Dent. Res. 2011, 90, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Ohara, M.; Miyauchi, M.; Tsuruda, K.; Takata, T.; Sugai, M. Topical application of Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces cell cycle arrest in the rat gingival epithelium in vivo. J. Periodontal Res. 2011, 46, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Ara, T.; Kurata, K.; Hirai, K.; Uchihashi, T.; Uematsu, T.; Imamura, Y.; Furusawa, K.; Kurihara, S.; Wang, P.-L. Human gingival fibroblasts are critical in sustaining inflammation in periodontal disease. J. Periodontal Res. 2009, 44, 21–27. [Google Scholar] [CrossRef]
- Páez, J.; Hernández, R.; Espinoza, J.; Rojas, L.; Martínez, C.E.; Tobar, N.; Martínez, J.; Smith, P.C. Uncoupled inflammatory, proliferative, and cytoskeletal responses in senescent human gingival fibroblasts. J. Periodontal Res. 2020, 55, 432–440. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.-Y.; Campisi, J. Tumor Suppressor and Aging Biomarker p16INK4a Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [Green Version]
- Belibasakis, G.N.; Johansson, A.; Wang, Y.; Chen, C.; Lagergard, T.; Kalfas, S.; Lerner, U.H. Cytokine responses of human gingival fibroblasts to Actinobacillus actinomycetemcomitans cytolethal distending toxin. Cytokine 2005, 30, 56–63. [Google Scholar] [CrossRef]
- Steffens, J.P.; Masi, S.; D’Aiuto, F.; Spolidorio, L.C. Telomere length and its relationship with chronic diseases—New perspectives for periodontal research. Arch. Oral Biol. 2013, 58, 111–117. [Google Scholar] [CrossRef]
- Masi, S.; Gkranias, N.; Li, K.; Salpea, K.D.; Parkar, M.; Orlandi, M.; Suvan, J.E.; Eng, H.L.; Taddei, S.; Patel, K.; et al. Association Between Short Leukocyte Telomere Length, Endotoxemia, and Severe Periodontitis in People With Diabetes: A Cross-Sectional Survey. Diabetes Care 2014, 37, 1140–1147. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Nishida, H.; Takeda, H.; Shin, K. Telomere Length in Leukocytes and Cultured Gingival Fibroblasts from Patients with Aggressive Periodontitis. J. Periodontol. 2004, 75, 84–90. [Google Scholar] [CrossRef]
- Sanders, A.E.; Divaris, K.; Naorungroj, S.; Heiss, G.; Risques, R.A. Telomere length attrition and chronic periodontitis: An ARIC Study nested case-control study. J. Clin. Periodontol. 2015, 42, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Olsen, I.; Taubman, M.A.; Singhrao, S.K. Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis, and Alzheimer’s disease. J. Oral Microbiol. 2016, 8, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darveau, R.P.; Belton, C.M.; Reife, R.A.; Lamont, R.J. Local Chemokine Paralysis, a Novel Pathogenic Mechanism for Porphyromonas gingivalis. Infect. Immun. 1998, 66, 1660–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, E.; Khalaf, H.; Bengtsson, T. Porphyromonas gingivalis downregulates the immune response of fibroblasts. BMC Microbiol. 2013, 13, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenker, B.J.; McKay, T.; Datar, S.; Miller, M.; Chowhan, R.; Demuth, D. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J. Immunol. 1999, 162, 4773–4780. [Google Scholar]
- Rabie, G.; Lally, E.T.; Shenker, B.J. Immunosuppressive properties of Actinobacillus actinomycetemcomitans leukotoxin. Infect. Immun. 1988, 56, 122–127. [Google Scholar] [CrossRef] [Green Version]
- Sekot, G.; Posch, G.; Messner, P.; Matejka, M.; Rausch-Fan, X.; Andrukhov, O.; Schäffer, C. Potential of the Tannerella forsythia S-layer to Delay the Immune Response. J. Dent. Res. 2010, 90, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Graves, D.T. Cytokines That Promote Periodontal Tissue Destruction. J. Periodontol. 2008, 79, 1585–1591. [Google Scholar] [CrossRef] [Green Version]
- Garlet, G.P. Critical reviews in oral biology & medicine: Destructive and protective roles of cytokines in periodontitis: A re-appraisal from host defense and tissue destruction viewpoints. J. Dent. Res. 2010, 89, 1349–1363. [Google Scholar]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The Sterile Inflammatory Response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzinikolaou, G.; Karakasilioti, I.; Garinis, G.A. DNA damage and innate immunity: Links and trade-offs. Trends Immunol. 2014, 35, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [PubMed] [Green Version]
- Tripathi, P.; Aggarwal, A. NF-kB transcription factor: A key player in the generation of immune response. Curr. Sci. 2006, 90, 519. [Google Scholar]
- Chow, J.C.; Young, D.W.; Golenbock, D.T.; Christ, W.J.; Gusovsky, F. Toll-like Receptor-4 Mediates Lipopolysaccharide-induced Signal Transduction. J. Biol. Chem. 1999, 274, 10689–10692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Mani, A.M.; Wu, Z.-H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef] [PubMed]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef]
- Gölz, L.; Memmert, S.; Rath-Deschner, B.; Jäger, A.; Appel, T.; Baumgarten, G.; Götz, W.; Frede, S. Hypoxia and P. gingivalis Synergistically Induce HIF-1 and NF-κB Activation in PDL Cells and Periodontal Diseases. Mediat. Inflamm. 2015, 2015, 438085. [Google Scholar] [CrossRef] [Green Version]
- Nareika, A.; Im, Y.-B.; Game, B.A.; Slate, E.H.; Sanders, J.J.; London, S.D.; Lopes-Virella, M.F.; Huang, Y. High glucose enhances lipopolysaccharide-stimulated CD14 expression in U937 mononuclear cells by increasing nuclear factor κB and AP-1 activities. J. Endocrinol. 2008, 196, 45–55. [Google Scholar] [CrossRef]
- Busik, J.V.; Mohr, S.; Grant, M.B. Hyperglycemia-Induced Reactive Oxygen Species Toxicity to Endothelial Cells Is Dependent on Paracrine Mediators. Diabetes 2008, 57, 1952–1965. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. NF-κB inhibition delays DNA damage–induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. Targeting normal and cancer senescent cells as a strategy of senotherapy. Ageing Res. Rev. 2019, 55, 100941. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; Van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Robbins, P.D. Senotherapeutics for healthy ageing. Nat. Rev. Drug Discov. 2018, 17, 377. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.-C.; Kim, J.-R. Senotherapeutics: Emerging strategy for healthy aging and age-related disease. BMB Rep. 2019, 52, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef] [PubMed]
- An, J.Y.; Kerns, K.A.; Ouellette, A.; Robinson, L.; Morris, H.D.; Kaczorowski, C.; Park, S.-I.; Mekvanich, T.; Kang, A.; McLean, J.S.; et al. Rapamycin rejuvenates oral health in aging mice. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhu, M.; Che, X.; Wang, H.; Liang, X.-J.; Wu, C.; Xue, X.; Yang, J. Lipopolysaccharide induces neuroinflammation in microglia by activating the MTOR pathway and downregulating Vps34 to inhibit autophagosome formation. J. Neuroinflamm. 2020, 17, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Wang, S.; Ma, D.; Tang, L.; Duan, Y.; Jin, Y. Loss of Proliferation and Differentiation Capacity of Aged Human Periodontal Ligament Stem Cells and Rejuvenation by Exposure to the Young Extrinsic Environment. Tissue Eng. Part A 2009, 15, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Hu, B.; Feng, G.; Xiang, M.; Deng, Y.; Tan, M.; Li, J.; Song, J. Metformin prevents against oxidative stress-induced senescence in human periodontal ligament cells. Biogerontology 2019, 21, 13–27. [Google Scholar] [CrossRef]
- Fan, C.; Ji, Q.; Zhang, C.; Xu, S.; Sun, H.; Li, Z. TGF-β induces periodontal ligament stem cell senescence through increase of ROS production. Mol. Med. Rep. 2019, 20, 3123–3130. [Google Scholar] [CrossRef]
- Honda, Y.; Huang, A.; Tanaka, T.; Han, X.; Gao, B.; Liu, H.; Wang, X.; Zhao, J.; Hashimoto, Y.; Yamamoto, K.; et al. Augmentation of Bone Regeneration by Depletion of Stress-Induced Senescent Cells Using Catechin and Senolytics. Int. J. Mol. Sci. 2020, 21, 4213. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transient Presence and Beneficial Effects | Accumulation and Detrimental Effects |
|---|---|
| Tumor suppression mechanism [63] | Source inflammatory cytokines (IL-1, TNF-β, IL-6) [16,39] |
| Embryonic development [52,54] | Source proteolytic enzymes (MMPs) [16,40] |
| Tissue remodeling and regeneration [53] | Source or ROS [51] |
| Wound healing [56,64] | Stem cell exhaustion and tissue decline [22,23] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aquino-Martinez, R.; Khosla, S.; Farr, J.N.; Monroe, D.G. Periodontal Disease and Senescent Cells: New Players for an Old Oral Health Problem? Int. J. Mol. Sci. 2020, 21, 7441. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207441
Aquino-Martinez R, Khosla S, Farr JN, Monroe DG. Periodontal Disease and Senescent Cells: New Players for an Old Oral Health Problem? International Journal of Molecular Sciences. 2020; 21(20):7441. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207441
Chicago/Turabian StyleAquino-Martinez, Ruben, Sundeep Khosla, Joshua N. Farr, and David G. Monroe. 2020. "Periodontal Disease and Senescent Cells: New Players for an Old Oral Health Problem?" International Journal of Molecular Sciences 21, no. 20: 7441. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207441