Influence of the TGF-β Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions


Abstract
:1. Introduction
2. Osteoblast/Osteoclast Balance in Bone Remodeling and Repair
2.1. Bone Forming Cells
2.1.1. Osteogenic Differentiation
2.1.2. Osteoblast and Osteocyte Functions
2.2. Bone Resorbing Cells
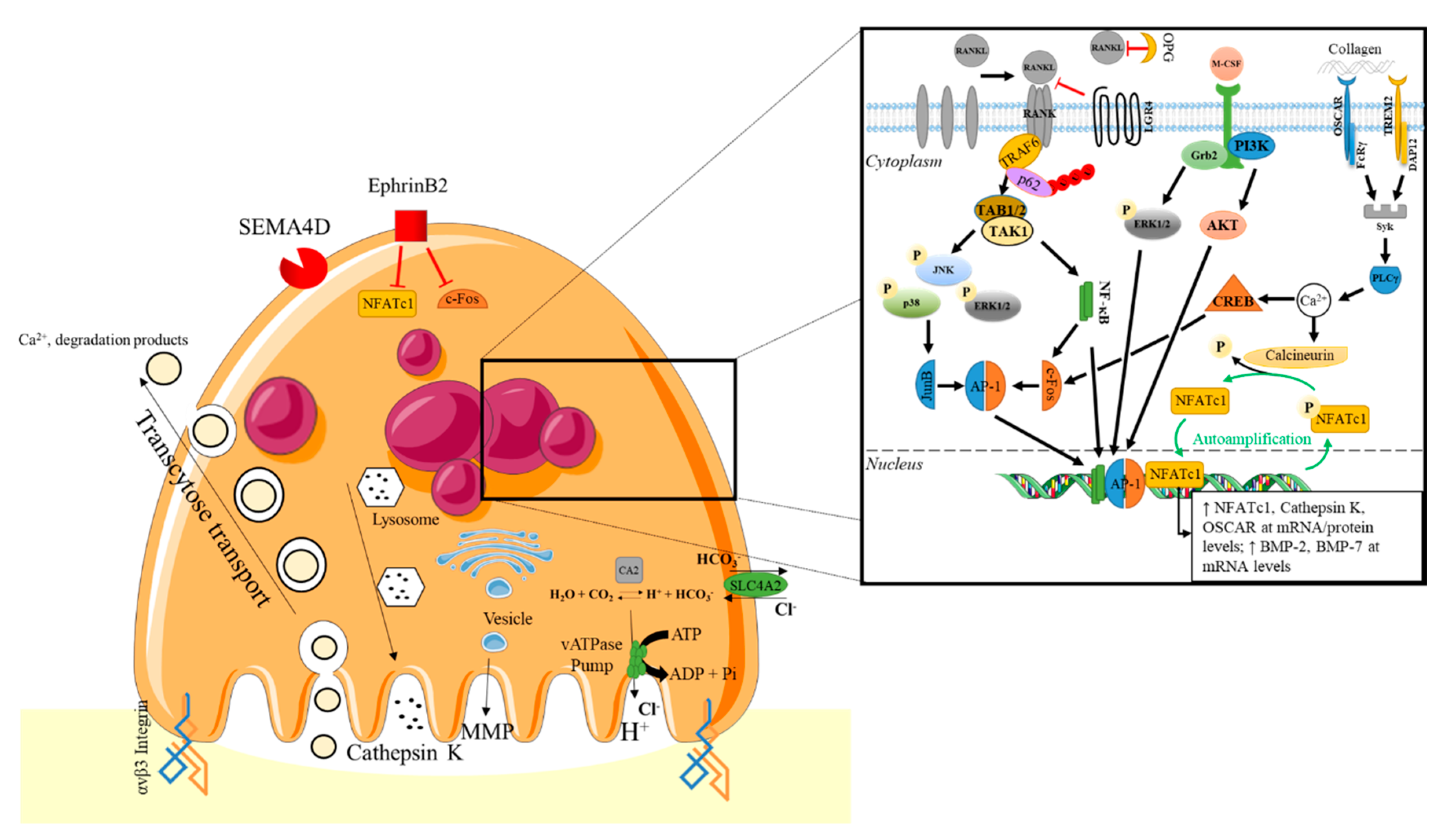
2.2.1. Osteoclastogenesis

2.2.2. Mature Osteoclast Functions
2.3. Osteoblasts/Osteoclasts Balance
2.3.1. Bone Remodeling
2.3.2. Bone Fracture Healing
3. The TGF-β Superfamily
3.1. Members of the TGF-β Superfamily
3.1.1. TGF-β /Nodal/Activin Family
- TGF-β
- Activin/Nodal
3.1.2. BMP/GDF Family
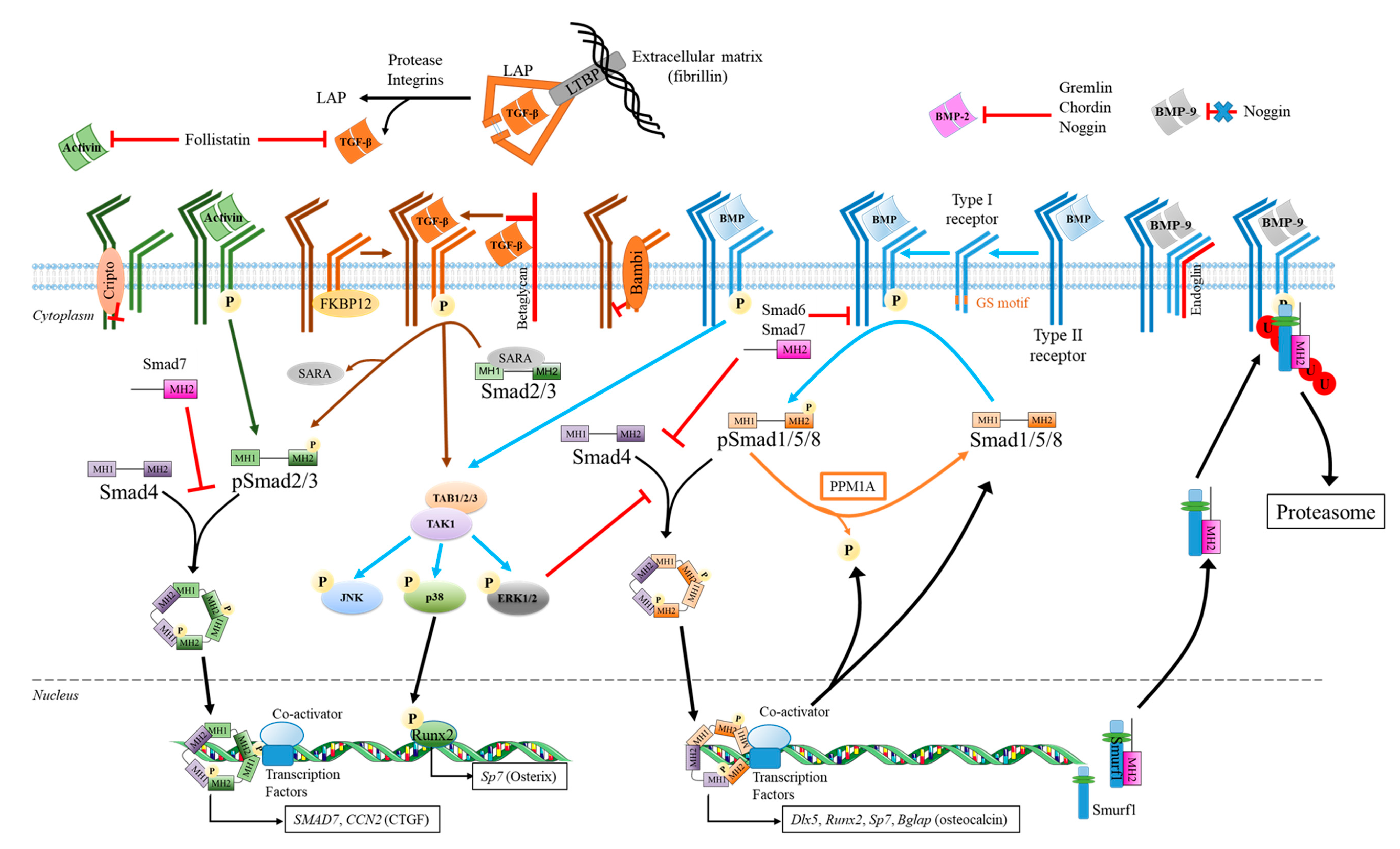
3.2. TGF-β Superfamily Signaling Pathways and Their Regulation
3.2.1. The Canonical Pathways Used by Members of TGF-β Superfamily

- Smad 2/3 Pathway
- Smad1/5/8 Pathway
- Regulation Mechanisms of the Canonical Smad Pathways
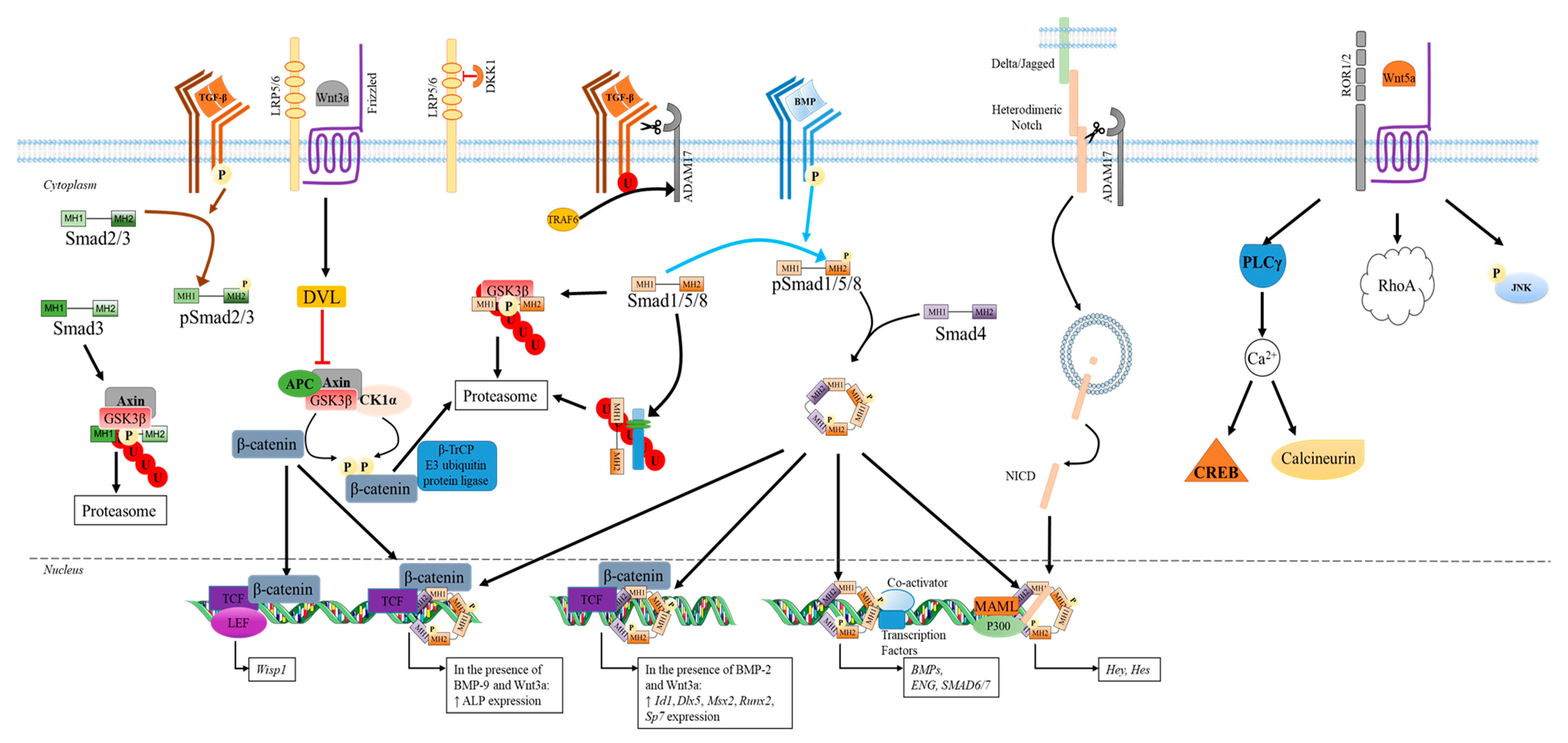
3.2.2. Non-Canonical Pathways Used by Members of TGF-β Superfamily

3.2.3. Other Regulators of the TGF-β Superfamily
- Wnt Signaling Pathways
- Canonical Wnt pathways
- Non-canonical Wnt signaling pathways
- Notch Signaling Pathways
4. Effect of TGF-β Superfamily on Bone Homeostasis and Disease
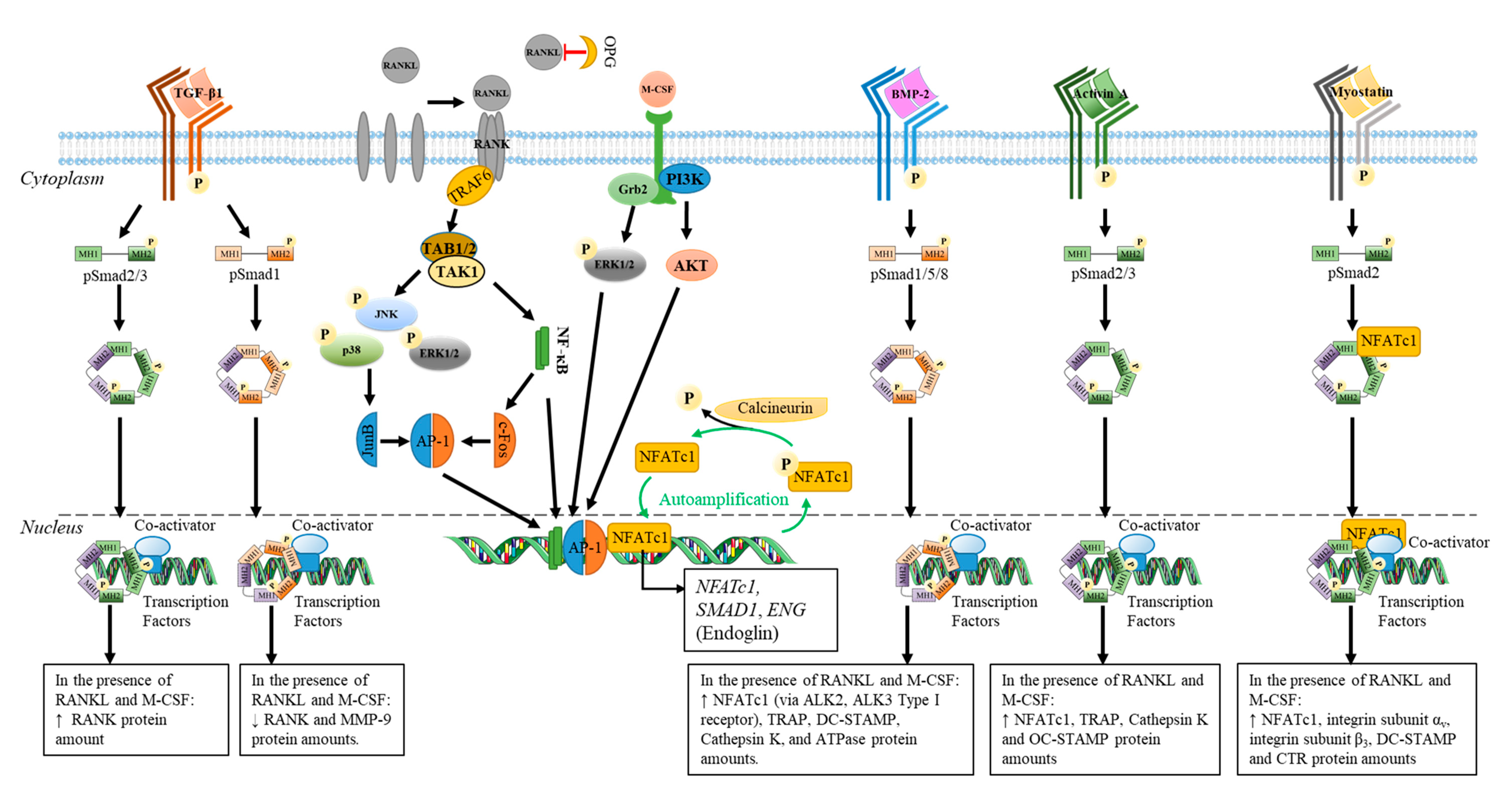
4.1. The Role Played by Members of TGF-β on Osteoblast and Osteoclast Differentiation
4.1.1. Osteogenic Differentiation
4.1.2. Osteoclastogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Members of TGF-β Superfamily | Experimental Conditions | Impact on Gene and Protein Expression | Impact on Osteoclast Function | Refs |
|---|---|---|---|---|
| TGF-β/Nodal/Activin family | ||||
| TGF-β1 | Cells: Murine RAW264.7; Treatment: M-CSF (20 ng/mL), RANK-L (50 ng/mL) and TGF-β1 (0.1 to 20 ng/mL); Time: 2-7 days | TGF-β1 dose dependently ↑ TNFRSF11A (RANK) at 48 h TGF-β1 5 ng/mL ↑ RANK protein amount after 3 days TGF-β1 dose dependently ↑ both CTR and VTR mRNA levels at day 7 | TGF-β1 dose dependently ↑ number of TRAP+ multinucleated cells (plateau at 1 ng/mL) | [327] |
| Cells: murine primary osteoblasts co-cultured with spleen cells; Treatment: 1,25(OH)2D3 (10 nM) plus Dex (100 nM) with or without rhTGF-β1 (0.3 to 10 ng/mL), M-CSF ((25 ng/mL), RANKL (50–200 ng/mL); Time: 7 days | N.A. | TGF-β1 dose-dependently ↓ osteoclast formation (TRAP+ cells) in the presence of 1,25(OH)2D3 plus dexamethasone. TGF-β1 dose-dependently ↑ RANKL-induced osteoclastogenesis (TRAP+ cells) of M-CSF stimulated spleen cells cultured alone. RANKL/TGF-β effect is inhibited by OPG (100 ng/mL). | [328] | |
| Cells: Marrow-derived osteoclasts precursors co-cultured with ST2 stromal cells; Treatment: TGF-β1 (2 × 10−5 to 2 ng/mL) | N.A. | Biphasic effect of TGF-β1 on osteoclast differentiation: ↑ number of TRAP+ multinucleated cells at 1 × 10−4 ng/mL Complete inhibition at 2 ng/mL | [329] | |
| Cells: marrow and spleen cells (osteoclast precursors); Treatment: ascorbic acid (7 × 10−3 M) and TGF-β1 (2 × 10−5 or 1 ng/mL), M-CSF (25 ng/mL) and RANKL (30 ng/mL). | N.A. | Only TGF-β1 at 1 ng/mL ↑ number of TRAP+ multinucleated cells (spleen cells). TGF-β1 dose dependently ↑ number of TRAP+ multinucleated cells (marrow cells) | ||
| Cells: human mononuclear leukocytes from umbilical cord blood differentiated in osteoclasts; Treatment: rhTGF-1 (0.1–1 ng/mL) | TGF-β1 ↑ pERK1/2, phosphorylated p38 and pSmad 2 TGF-1 ↑ amount of pro-apoptotic proteins (Bax/Bim). TGF-β1 ↑ expression of Bim through Smad 2. | TGF-β1 dose-dependently ↑ apoptosis of human osteoclasts through caspase 9 | [330] | |
| Cells: monocytes from normal human peripheral blood; Treatment: 20 ng/mL M-CSF for 2 days and then RANKL (40 ng/mL) for an additional 6 days with or without TGF-β1 (10 ng/mL); Time: 8 days | In the presence of M-CSF/RANKL: TGF-β1 ↑ Endoglin expression (mRNA and protein) compared to M-CSF/RANKL control. TGF-β1 ↓ levels of mRNA encoding NFAT-c1, TRAP and Cathepsin K. TGF-β1 ↓ levels of mRNA encoding RANK and MMP-9 through Smad1 activation | In the presence of M-CSF/RANKL: TGF-β1 ↓ number of TRAP+ multinucleated cells in a Smad1 dependent manner TGF-β1 inhibits osteoclastogenesis only when added within 48 h TGF-β1 ↑osteoclastogenesis through a Smad3 dependent manner | [325] | |
| TGF-β2 | Cells: marrow and spleen cells; Treatment: ascorbic acid and TGF-β2 (2 × 10−5 to 2 ng/mL) with or without M-CSF (25 ng/mL), RANKL (30 ng/mL). | N.A. | TGF-β2 biphasic effect on osteoclast differentiation: A maximal number of TRAP+ multinucleated cells at 2 × 10-4 ng/mL No TRAP+ cells at 2 ng/mL | [329] |
| Activin A | Cells: murine bone marrow cells (BMC); Treatment: rhM-CSF (20 ng/mL) and rhRANKL (40 ng/mL) with or without rh activinA (50 ng/mL); Time: 4 days Cells: Murine monocyte/macrophage cell line RAW264.7; Treatment: rhRANKL (40 ng/mL) with or without rh activinA (50 ng/mL); Time: 2, 3, 4 or 7 days | ActivinA ↑ RANKL-induced NFATc1 expression in both BMC and RAW264.7 via Smad2 phosphorylation ActivinA ↑ RANKL-induced osteoclastogenic gene (TRAP, OC-STAMP and Cathepsin K) expression in RAW264.7 at 3 days | ActivinA ↑ differentiation of both BMC and RAW264.7 in osteoclasts (as shown by TRAP+ cells at 4 and 7 days, respectively) in the presence of M-CSF and RANKL | [184] |
| BMP/GDF family | ||||
| BMP-2 | Cells: murine primary osteoclast; Treatment: 10 ng/mL of M-CSF for 3 days before adding 30 ng/mL of RANKL with or without BMP-2 (30 ng/mL) for 5 days | BMP-2 ↑ RANKL-induced genes encoding osteoclast markers (NFATc1, TRAP, DC-STAMP, cathepsin K and ATP6v0d2) at day 3 BMP-2 plus RANKL had no effect on RANKL or OPG expression at day 3 | BMP-2 from day 3 to day 4 ↑ RANKL-induced osteoclast formation as shown by an increase in TRAP+ multinuclear cells Suppression of BMPRII expression by specific shRNA inhibits osteoclastogenesis | [331] |
| BMP-2 | Cells: bone marrow mononuclear cells incubated Treatment: 20 ng/mL of M-CSF for 4 days, followed by another 5 days with 20 ng/mL M-CSF and 50 ng/mL of RANKL with or without BMP-2 or BMP-7 at 100 ng/mL. | BMP-2 ↑the amount of pSmad1/5/9 through ALK2 and ALK3 BMP-2 via Smad activation ↑ NFATc1 protein levels and its nuclear translocation in osteoclasts | BMP-2 alone had no effect on osteoclast differentiation BMP-2 ↑ RANKL-induced osteoclastogenesis as shown by TRAP+ cells (with three or more nuclei) at day 5 BMP-2 plus RANKL ↑ the area of demineralized pits on OsteoAssay surface plates | [59] |
| BMP-7 | BMP-7 ↑ the amount of pSmad1/5/9 through ALK2 BMP-7 via Smad activation ↑ NFATc1 protein levels and its nuclear translocation in osteoclasts | BMP-7 alone had no effect on osteoclast differentiation BMP-7 ↑ RANKL-induced osteoclast differentiation at day 5 BMP-7 plus RANKL ↑ demineralization activity | ||
| BMP-9 | Cells: human mononuclear leukocytes from umbilical cord blood are differentiated in osteoclasts; Treatment: Opti-MEM media supplemented with 2% FBS, 25 ng/mL M-CSF and 100 ng/mL of RANKL with or without BMP-9 (50 or 150 ng/mL) | BMP-9 acts via BMPR-II receptor to activate ERK1/2 pathways ↓ of BMPR-II by siRNA prevents bone resorption | In the presence of M-CSF/RANKL: No effect of BMP-9 on osteoclast formation (no change in % of multinucleated cells expressing RANK or CTR) BMP-9 ↑ bone resorption (30–40%) BMP-9 (50 ng/mL) protects osteoclasts from apoptosis by ↓ the % of cleaved caspase 9 and its activity | [171] |
| Myostatin | Cells: Bone marrow–derived macrophages Treatment: 50 ng/mL M-CSF for 72h. Then cells are incubated for 4–6 days with M-CSF (50 ng/mL) and RANKL (50 ng/mL) with or without myostatin (30 ng/mL) | Myostatin ↑ RANKL-induced expression of NFATc1; integrin αv, integrin β3, DC-STAMP and CTR Myostatin activates Smad2 to enhance RANKL-induced osteoclastogenesis NFATC1 and pSmad2 can interact together favoring their nuclear translocation | No effect of Myostatin alone on osteoclast formation, apoptosis, and proliferation Myostatin + M-CSF/RANKL ↑ osteoclastogenesis (3.8-fold more osteoclasts after 4 days compared with M-CSF/RANKL control) ALK4/ALK5/ALK7 inhibitor ↓ number of osteoclasts | [332] |
4.2. Temporal Expression of the Members of TGF-β Superfamily during the Bone Fracture Healing Process
4.3. TGF-β Family Members and Bone Diseases
4.3.1. TGF-β Signaling and Osteoporosis
4.3.2. TGF-β Signaling and Osteogenesis Imperfecta
4.3.3. TGF-β Signaling in Bone Malignancies
- Bone metastases
- Multiple Myeloma
- Targeting Activin A in Myeloma
- TGF-β Family in Monogenic Developmental Bone Diseases
5. The Use of Members of the TGF-β Superfamily in Clinical Application and Their Potential Adverse Effect
| rhBMP | Clinical Application | Methodology | Dose | Conclusion and Adverse Effect | Refs |
|---|---|---|---|---|---|
| BMP-2 | Anterolateral interbody fusion | 3105 patients (anterolateral interbody fusion: 2000–2012) from 14 trials (PubMed database and FDA approval document) | 2.1–18 mg | Safe under FDA-approved recommendations (i.e., one-level anterolateral interbody fusion surgery with an LT-cage); Low complications (subsidence, cancer, infection); Equal efficiency (fusion rate, pain disability, patient satisfaction, risk of re-operations) between BMP-2, allogenic or autologous bone graft; Safety and effectiveness of BMP-2 in off-label use: not established. | [388] |
| BMP-2 | Spinal fusion surgery/degenerative disc disease (control: iliac crest bone graft (ICBG)) | 1408 patients (spinal fusion: 1997–2012) from 12 trials (mostly sponsored by Medtronic) | Infuse® (1.5 mg/mL) Amplify® (2.0 mg/mL) | ↑ early postsurgical pain compared with ICBG; Evidence of ↑ cancer incidence is inconclusive; ↑ fusion rates at 24 months. | [403] |
| BMP-2 | Spinal fusion (control: bone graft) | 1984 patients (spinal fusion: 1996–2012) from 13 trials (sponsored by Medtronic and Norton Healthcare) | 0.6 to 16.8 mg (11 trials); 15.0 to 63.0 mg (5 trials of posterolateral lumbar fusion studies) | ↑ complication in anterior cervical fusion: wound complication and dysphagia.; No proven clinical advantage over bone graft in spinal. fusion: May be associated with important harms (retrograde ejaculation and urogenital problems); ↑ cancer risk at 24 months. | [394] |
| BMP-2 | Spinal fusion | 55,862 patients (spinal fusion: 2004–2007) from the Scoliosis Research Society database (BMP used in 21% of all spinal fusions) | N.A. | ↑↑ incidence of complications and wound infections in anterior cervical fusions; Not associated with ↑ complications in thoracolumbar and posterior cervical fusions. | [393] |
| BMP-2 | Spinal fusion | 780 patients (1995–2010) from 13 trials (sponsored by industry). | 0.6–40 mg | ↑↑ complications and adverse events in spinal fusion; Possible study design bias in the original trials: risk of adverse events around 10 to 50 fold that of the original estimates reported in publications sponsored by industry; Higher doses of BMP-2: associated ↑ risk of new malignancy. | [395] |
| BMP-2 | Lumbar and lumbosacral fusion | 129 patients (2000–2008) from the New York Harbor Health Care System Manhattan Veterans Administration operating room record | 12 and 24 mg | Higher doses of rhBMP2 in lumbar and lumbosacral fusion: may ↑ risk of renal insufficiency. | [404] |
| BMP-7 | Single-level lumbar fusion (control: ICBG) | 539 patients (2002–2016) from 5 trials (PubMed, EMBASE, Scopus, and the Cochrane Collaboration Library databases) | 3.5 mg of (rh)BMP-7 (Osigraft or Putty) per side | Shorter operation times; No additional beneficial effect (clinical success, revision rates and duration of hospitalization) between BMP-7 and ICBG; ↓ lumbar fusion rate (in instrumented posterolateral fusion). | [389] |
| BMP-2 and/or BMP-7 | Lumbar fusion | 2185 patients (2000–2016) from 21 trials | 12–48 mg | ↑ lumbar fusion success rate (BMP-2) and ↓ risk of re-operation; No difference in complication rate between BMPs and ICBG. | [390] |
| BMP-2 and/or BMP-7 | Treatment of fractures, non-union and osteonecrosis | 3324 patients (1601 fracture, 1654 non-unions and 69 osteonecrosis: from 2000 to 2016) from 43 trials (PubMed database) | Inductos® (0.75, 1.5 or 2.0 mg/mL); Infuse®(1.5 mg/mL); OP-1 Stryker (3.3 and 3.5 mg/mL); Osigraft (3.5 mg/mL) | Controversial clinical evidence (fractures, non-union, and osteonecrosis); Preliminary knowledge and few low quality reports; Positive findings in many studies, but mixed efficacy and adverse events in overall literature; Unclear conclusions (heterogeneity of studies: different BMPs, doses and delivery method for each bone pathology). | [402] |
| BMP-2 and/or BMP-7 | Tibial fracture and nonunion | 1113 patients (tibial fracture and nonunion: 1997 to 2011) from 8 trials (MEDLINE, EMABSE, BIOSIS and Cochrane central data bases) | 3.5, 6 or 12 mg | ↑ effectiveness of bone union and ↓ risk of re-operation (tibial fractures); Equal efficiency (bone union, infection, or re-operations rate) between BMPs and autologous bone graft to treat tibial fractures non-union. | [391] |
| BMP-2 and BMP-7 | Spinal fusion | 941 patients from 7 trials from Pubmed, Cochrane, National Guideline Clearinghouse databases, FDA safety summaries (2012) | 4–40 mg | ↑ cancer risk dependent on the dose of BMP used. | [405] |
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1,25-(OH)2D3 | 1α,25-dihydroxyvitamin D3 |
| ActRIIA | Type II activin receptor |
| ActRIIB | Type IIB activin receptor |
| ALK | Activin receptor-like kinases receptor |
| ALP | Alkaline phosphatase |
| AMH/MIS | Anti-Müllerian hormone/Müllerian inhibiting substance |
| AMHRII | Anti-Mullerian hormone receptor type II |
| AP-1 | Activator protein 1 |
| APC | Adenomatous polyposis coli |
| BAMBI | Bone morphogenetic protein and activin membrane-bound inhibitor |
| BMMs | Bone marrow–derived macrophage |
| BMP | Bone morphogenetic proteins |
| BMPRII | Type II BMP receptor |
| BMU | Basic multicellular unit |
| BSP | Bone sialoprotein |
| CA2 | Carbonic anhydrase enzymes |
| CAMKII | Calmodulin-dependent kinase II |
| CDK8 | Cyclin-dependent kinase 8 |
| CEBPα | CCAAT-enhancer binding protein α |
| CHO | Chinese hamster ovary |
| CKI | Casein kinase I |
| CRD | Extracellular N-terminal cysteine-rich domain |
| CREB | Cyclic AMP response element-binding protein |
| CSF-1 | Colony stimulating factor 1 |
| CTR | Calcitonin receptor |
| DAG | Diacylglycerol |
| DAP12 | DNAX associated protein 12kD size |
| Dkk | Dickkopf |
| DLL | Delta-like |
| dpp | Drosophila decapentaplegic |
| DVL | Disheveled |
| ENG | Endoglin |
| ERK | Extracellular signal-regulated kinase |
| FGF | Fibroblast growth factor |
| FKBP12 | FK506 binding protein of 12 kDa |
| FOP | Fibrodysplasia ossifying |
| FSH | Follicle-stimulating hormone |
| Fz | Frizzled |
| GDF | Growth differentiation factors |
| Grb2 | Growth factor receptor bound protein 2 |
| GSK3 β | Glycogen synthase kinase-3 β |
| GS motif | Gly/Ser rich motif |
| HAT | Histone acetyltransferases |
| HES | Hairy enhancer of split |
| HEY | HES-related with YRPW motif |
| HO | Heterotopic ossification |
| ID1 | DNA binding protein 1 |
| IGF | Insulin like growth factor |
| IL | Interleukins |
| IP3 | Inositol-1,4,5-trisphosphate |
| I-Smad | Inhibitory Smad |
| JAG | Jagged |
| JNK | c-Jun amino (N)-terminal kinases |
| LAP | Latency associated peptide |
| LEF | Lymphoid enhancer-binding factor |
| LGR4 | Leucine rich repeat containing G-coupled receptor 4 |
| LRP5/6 | Low-density-lipoprotein-related protein 5/6 |
| LTBP | Latent TGF-β binding protein |
| MAML | Adaptor protein Mastermind-like |
| MAPK | Mitogen-activated protein kinase |
| M-CSF | Macrophage- colony stimulating factor |
| MH1 | Mad homology 1 domain |
| miRNAs | microRNAs |
| MITF | Micropthalmia-associated transcription factor |
| MMP-9 | Matrix metalloproteases |
| MSCs | Mesenchymal stem cells |
| NFATc1 | Nuclear factor of activated T cells |
| NF-κB | Nuclear factor of κB |
| NICD | Notch intracellular domain |
| ODF | Osteoclast differentiation factor |
| OPG | Osteoprotegerin |
| OPGL | Osteoprotegerin ligand |
| OSCAR | Osteoclast-associated receptor |
| PI3K | Phosphoinositide 3-kinase |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| PPARγ | Peroxisome proliferation-activated receptor γ |
| PPM1A | Protein phosphatase magnesium-dependent 1A |
| RANKL | Receptor activator of nuclear factor kappa beta ligand |
| rhBMP2/7 | rhBMP-2/BMP-7 heterodimer |
| R-Smad | Receptor-regulated Smad proteins |
| Runx2 | Runt-related transcription factor 2 |
| SARA | Smad anchor for receptor activation protein |
| sFRPs | Fz-related proteins |
| SGFs | Sarcoma growth factors |
| SIBLING | Small integrin-binding ligand N-linked glycoprotein |
| Smad | Small mothers against decapentaplegic |
| Smurf | Smad ubiquitin regulatory factor |
| TAB1 | TAK1-binding protein 1 |
| TACE | Tumor Necrosis Factor α-converting enzyme |
| TAK1 | Transforming growth factor β-activated kinase 1 |
| TβRII | TGF-βRII |
| TCF | T cell factor |
| TNF | Tumor necrosis factor |
| TGF-β | Transforming growth factor β |
| TRAFs | Tumor necrosis factor receptor-associated factors |
| TRANCE | Tumor necrosis factor-related activation-induced cytokine |
| TRAP | Tartrate-resistant acid phosphatase |
| TREM2 | Triggering receptor expressed on myeloid cells-2 |
| TSP1 | Thrombospondin 1 |
| v-ATPase | Vacuolar protons -transporting adenosine triphosphatase pump |
| VEGF | Vascular endothelial growth factor |
References
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [Green Version]
- Marieb, E.N.; Hoen, K. Human Anatomy and Physiology, 5th ed.; Pearson Education Limited: London, UK, 2015. [Google Scholar]
- Datta, H.K.; Ng, W.F.; Walker, J.A.; Tuck, S.P.; Varanasi, S.S. The cell biology of bone metabolism. J. Clin. Pathol. 2008, 61, 577–587. [Google Scholar] [CrossRef]
- Sharma, S.; Mahajan, A.; Mittal, A.; Gohil, R.; Sachdeva, S.; Khan, S.; Dhillon, M. Epigenetic and transcriptional regulation of osteoclastogenesis in the pathogenesis of skeletal diseases: A systematic review. Bone 2020, 138, 115507. [Google Scholar] [CrossRef]
- Alejandro, P.; Constantinescu, F. A Review of Osteoporosis in the Older Adult: An Update. Rheum. Dis. Clin. N. Am. 2018, 44, 437–451. [Google Scholar] [CrossRef]
- Calori, G.M.; Tagliabue, L.; Mazza, E.; de Bellis, U.; Pierannunzii, L.; Marelli, B.M.; Colombo, M.; Albisetti, W. Tibial pilon fractures: Which method of treatment? Injury 2010, 41, 1183–1190. [Google Scholar] [CrossRef]
- Ho-Shui-Ling, A.; Bolander, J.; Rustom, L.E.; Johnson, A.W.; Luyten, F.P.; Picart, C. Bone regeneration strategies: Engineered scaffolds, bioactive molecules and stem cells current stage and future perspectives. Biomaterials 2018, 180, 143–162. [Google Scholar] [CrossRef]
- Maruyama, M.; Rhee, C.; Utsunomiya, T.; Zhang, N.; Ueno, M.; Yao, Z.; Goodman, S.B. Modulation of the Inflammatory Response and Bone Healing. Front. Endocrinol. 2020, 11, 386. [Google Scholar] [CrossRef]
- Walters, G.; Pountos, I.; Giannoudis, P.V. The cytokines and micro-environment of fracture haematoma: Current evidence. J. Tissue Eng. Regen. Med. 2018, 12, e1662–e1677. [Google Scholar] [CrossRef]
- Fischer, V.; Haffner-Luntzer, M.; Amling, M.; Ignatius, A. Calcium and vitamin D in bone fracture healing and post-traumatic bone turnover. Eur. Cells Mater. 2018, 35, 365–385. [Google Scholar] [CrossRef]
- Anderson, P.H. Vitamin D Activity and Metabolism in Bone. Curr. Osteoporos. Rep. 2017, 15, 443–449. [Google Scholar] [CrossRef]
- Linkhart, T.A.; Mohan, S.; Baylink, D.J. Growth factors for bone growth and repair: IGF, TGFβ and BMP. Bone 1996, 19, S1–S12. [Google Scholar] [CrossRef]
- Huntley, R.; Jensen, E.; Gopalakrishnan, R.; Mansky, K.C. Bone morphogenetic proteins: Their role in regulating osteoclast differentiation. Bone Rep. 2019, 10, 100207. [Google Scholar] [CrossRef]
- Feng, W.; Guo, J.; Li, M. RANKL-independent modulation of osteoclastogenesis. J. Oral. Biosci. 2019, 61, 16–21. [Google Scholar] [CrossRef]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFβ Signaling in Growth Control, Cancer, and Heritable Disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J.; Chen, Y.G. Controlling TGF-β signaling. Genes Dev. 2000, 14, 627–644. [Google Scholar] [CrossRef]
- Han, L.; Wang, B.; Wang, R.; Gong, S.; Chen, G.; Xu, W. The shift in the balance between osteoblastogenesis and adipogenesis of mesenchymal stem cells mediated by glucocorticoid receptor. Stem Cell Res. Ther. 2019, 10, 377. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Li, Q.; Luo, S.; Liu, Z.; Luo, D.; Zhang, B.; Zhang, D.; Rao, P.; Xiao, J. PPARgamma and Wnt Signaling in Adipogenic and Osteogenic Differentiation of Mesenchymal Stem Cells. Curr. Stem Cell Res. Ther. 2016, 11, 216–225. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Qian, F.; Ma, Q.; Zhang, P.; Chen, T.; Chen, C.; Zhang, Y.; Deng, P.; Zhou, Z.; Yu, Z. 50 Hz electromagnetic field exposure promotes proliferation and cytokine production of bone marrow mesenchymal stem cells. Int. J. Clin. Exp. Med. 2015, 8, 7394–7404. [Google Scholar]
- Moore, E.R.; Chen, J.C.; Jacobs, C.R. Prx1-Expressing Progenitor Primary Cilia Mediate Bone Formation in response to Mechanical Loading in Mice. Stem Cells Int. 2019, 2019, 3094154. [Google Scholar] [CrossRef]
- Root, S.H.; Wee, N.K.Y.; Novak, S.; Rosen, C.J.; Baron, R.; Matthews, B.G.; Kalajzic, I. Perivascular osteoprogenitors are associated with transcortical channels of long bones. Stem Cells 2020, 38, 769–781. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A Transcriptional Activator of Osteoblast Differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted Disruption of Cbfa1 Results in a Complete Lack of Bone Formation owing to Maturational Arrest of Osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Signaling networks in RUNX2-dependent bone development. J. Cell. Biochem. 2011, 112, 750–755. [Google Scholar] [CrossRef]
- Kawane, T.; Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, H.; Yoshida, C.A.; Matsuura-Kawata, V.; Sakane, C.; Matsuo, Y.; Nagai, K.; et al. Runx2 is required for the proliferation of osteoblast progenitors and induces proliferation by regulating Fgfr2 and Fgfr3. Sci. Rep. 2018, 8, 13551. [Google Scholar] [CrossRef]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The Novel Zinc Finger-Containing Transcription Factor Osterix Is Required for Osteoblast Differentiation and Bone Formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Greenblatt, M.B.; Shim, J.H.; Zou, W.; Sitara, D.; Schweitzer, M.; Hu, D.; Lotinun, S.; Sano, Y.; Baron, R.; Park, J.M.; et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Investig. 2010, 120, 2457–2473. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Yao, B.; Shi, K.; Lu, J.; Jin, Y.; Qi, B.; Li, H.; Pan, S.; Chen, L.; Ma, C. Phosphorylation of Serine422 increases the stability and transactivation activities of human Osterix. FEBS Lett. 2015, 589, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.H.; Li, F.G.; Chen, X.Y.; Li, J.T.; Wu, Y.H.; Huang, L.H.; Wang, Z.; Li, P.; Wang, T.; Lahn, B.T.; et al. PPARgamma suppression inhibits adipogenesis but does not promote osteogenesis of human mesenchymal stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 377–384. [Google Scholar] [CrossRef]
- Lin, L.; Dai, S.D.; Fan, G.Y. Glucocorticoid-induced differentiation of primary cultured bone marrow mesenchymal cells into adipocytes is antagonized by exogenous Runx2. Apmis 2010, 118, 595–605. [Google Scholar] [CrossRef]
- Stechschulte, L.A.; Lecka-Czernik, B. Reciprocal regulation of PPARgamma and RUNX2 activities in marrow mesenchymal stem cells: Fine balance between p38 MAPK and Protein Phosphatase 5. Curr. Mol. Biol. Rep. 2017, 3, 107–113. [Google Scholar] [CrossRef]
- Bonewald, L.F. Osteocytes as dynamic multifunctional cells. Ann. N. Y. Acad. Sci. 2007, 1116, 281–290. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Jikko, A.; Harris, S.E.; Chen, D.; Mendrick, D.L.; Damsky, C.H. Collagen integrin receptors regulate early osteoblast differentiation induced by BMP-2. J. Bone Miner. Res. 1999, 14, 1075–1083. [Google Scholar] [CrossRef]
- Nagae, M.; Re, S.; Mihara, E.; Nogi, T.; Sugita, Y.; Takagi, J. Crystal structure of alpha5beta1 integrin ectodomain: Atomic details of the fibronectin receptor. J. Cell Biol. 2012, 197, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Srinivasan, A.; Nikolajeff, F.; Kumar, S. Biomineralization process in hard tissues: The interaction complexity within protein and inorganic counterparts. Acta Biomater. 2020. [Google Scholar] [CrossRef]
- Tresguerres, F.G.F.; Torres, J.; Lopez-Quiles, J.; Hernandez, G.; Vega, J.A.; Tresguerres, I.F. The osteocyte: A multifunctional cell within the bone. Ann. Anat. 2020, 227, 151422. [Google Scholar] [CrossRef]
- Knothe Tate, M.L.; Adamson, J.R.; Tami, A.E.; Bauer, T.W. The osteocyte. Int. J. Biochem. Cell Biol. 2004, 36, 1–8. [Google Scholar] [CrossRef]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell… and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.; Petretto, E.; Gordon, S.; Bassett, J.H.D.; Williams, G.R.; Behmoaras, J. Common signalling pathways in macrophage and osteoclast multinucleation. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Yoshida, H.; Hayashi, S.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef]
- Soe, K.; Delaisse, J.M.; Borggaard, X.G. Osteoclast formation at the bone marrow/bone surface interface: Importance of structural elements, matrix, and intercellular communication. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Hatake, K.; Suzu, S.; Saito, K.; Motoyoshi, K.; Miura, Y. Immunohistochemical identification of proteoglycan form of macrophage colony-stimulating factor on bone surface. Calcif. Tissue Int. 1995, 57, 213–217. [Google Scholar] [CrossRef]
- Collin-Osdoby, P.; Rothe, L.; Anderson, F.; Nelson, M.; Maloney, W.; Osdoby, P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J. Biol. Chem. 2001, 276, 20659–20672. [Google Scholar] [CrossRef] [Green Version]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef]
- Ross, F.P. M-CSF, c-Fms, and signaling in osteoclasts and their precursors. Ann. N. Y. Acad. Sci. 2006, 1068, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Matic, I.; Matthews, B.G.; Wang, X.; Dyment, N.A.; Worthley, D.L.; Rowe, D.W.; Grcevic, D.; Kalajzic, I. Quiescent Bone Lining Cells are a Major Source of Osteoblasts During Adulthood. Stem Cells 2016, 34, 2930–2942. [Google Scholar] [CrossRef] [Green Version]
- Carda, C.; Silvestrini, G.; Gomez de Ferraris, M.E.; Peydro, A.; Bonucci, E. Osteoprotegerin (OPG) and RANKL expression and distribution in developing human craniomandibular joint. Tissue Cell 2005, 37, 247–255. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Piemontese, M.; Onal, M.; Campbell, J.; Goellner, J.J.; Dusevich, V.; Bonewald, L.; Manolagas, S.C.; O’Brien, C.A. Osteocytes, not Osteoblasts or Lining Cells, are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PLoS ONE 2015, 10, e0138189. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin Ligand Is a Cytokine that Regulates Osteoclast Differentiation and Activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Fennen, M.; Pap, T.; Dankbar, B. Smad-dependent mechanisms of inflammatory bone destruction. Arthritis Res. Ther. 2016, 18, 279. [Google Scholar] [CrossRef] [Green Version]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, e8. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar] [CrossRef] [Green Version]
- Ono, T.; Nakashima, T. Recent advances in osteoclast biology. Histochem. Cell Biol. 2018, 149, 325–341. [Google Scholar] [CrossRef]
- Omi, M.; Kaartinen, V.; Mishina, Y. Activin A receptor type 1-mediated BMP signaling regulates RANKL-induced osteoclastogenesis via canonical SMAD-signaling pathway. J. Biol. Chem. 2019, 294, 17818–17836. [Google Scholar] [CrossRef]
- Hsu, H.; Lacey, D.L.; Dunstan, C.R.; Solovyev, I.; Colombero, A.; Timms, E.; Tan, H.L.; Elliott, G.; Kelley, M.J.; Sarosi, I.; et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. USA 1999, 96, 3540–3545. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Yang, Z.; Ma, Y.; Yue, Z.; Lin, H.; Qu, G.; Huang, J.; Dai, W.; Li, C.; Zheng, C.; et al. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat. Med. 2016, 22, 539–546. [Google Scholar] [CrossRef]
- McManus, S.; Chamoux, E.; Bisson, M.; Roux, S. Modulation of tumor necrosis factor related apoptosis-inducing ligand (TRAIL) receptors in a human osteoclast model in vitro. Apoptosis 2012, 17, 121–131. [Google Scholar] [CrossRef]
- Lee, S.H.; Rho, J.; Jeong, D.; Sul, J.Y.; Kim, T.; Kim, N.; Kang, J.S.; Miyamoto, T.; Suda, T.; Lee, S.K.; et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 2006, 12, 1403–1409. [Google Scholar] [CrossRef]
- Koga, T.; Inui, M.; Inoue, K.; Kim, S.; Suematsu, A.; Kobayashi, E.; Iwata, T.; Ohnishi, H.; Matozaki, T.; Kodama, T.; et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 2004, 428, 758–763. [Google Scholar] [CrossRef]
- Humphrey, M.B.; Nakamura, M.C. A Comprehensive Review of Immunoreceptor Regulation of Osteoclasts. Clin. Rev. Allergy Immunol. 2016, 51, 48–58. [Google Scholar] [CrossRef] [Green Version]
- McHugh, K.P.; Hodivala-Dilke, K.; Zheng, M.H.; Namba, N.; Lam, J.; Novack, D.; Feng, X.; Ross, F.P.; Hynes, R.O.; Teitelbaum, S.L. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J. Clin. Investig. 2000, 105, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Sarosi, I.; Yan, X.Q.; Morony, S.; Capparelli, C.; Tan, H.L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef] [Green Version]
- Azuma, Y.; Kaji, K.; Katogi, R.; Takeshita, S.; Kudo, A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 2000, 275, 4858–4864. [Google Scholar] [CrossRef] [Green Version]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Kitaura, H.; Zhou, P.; Kim, H.J.; Novack, D.V.; Ross, F.P.; Teitelbaum, S.L. M-CSF mediates TNF-induced inflammatory osteolysis. J. Clin. Investig. 2005, 115, 3418–3427. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.; Zwerina, J. Positive regulators of osteoclastogenesis and bone resorption in rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, 235. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chung, M.R.; Zhou, S.; Gong, X.; Xu, H.; Hong, Y.; Jin, A.; Huang, X.; Zou, W.; Dai, Q.; et al. STAT3 controls osteoclast differentiation and bone homeostasis by regulating NFATc1 transcription. J. Biol. Chem. 2019, 294, 15395–15407. [Google Scholar] [CrossRef]
- Yoshitake, F.; Itoh, S.; Narita, H.; Ishihara, K.; Ebisu, S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J. Biol. Chem. 2008, 283, 11535–11540. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhou, X.; Huang, D.; Ji, Y.; Kang, F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell. Physiol. Biochem. 2017, 41, 1360–1369. [Google Scholar] [CrossRef]
- Axmann, R.; Bohm, C.; Kronke, G.; Zwerina, J.; Smolen, J.; Schett, G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009, 60, 2747–2756. [Google Scholar] [CrossRef]
- Luxenburg, C.; Winograd-Katz, S.; Addadi, L.; Geiger, B. Involvement of actin polymerization in podosome dynamics. J. Cell Sci. 2012, 125, 1666–1672. [Google Scholar] [CrossRef] [Green Version]
- Georgess, D.; Machuca-Gayet, I.; Blangy, A.; Jurdic, P. Podosome organization drives osteoclast-mediated bone resorption. Cell Adhes. Migr. 2014, 8, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Jurdic, P.; Saltel, F.; Chabadel, A.; Destaing, O. Podosome and sealing zone: Specificity of the osteoclast model. Eur. J. Cell Biol. 2006, 85, 195–202. [Google Scholar] [CrossRef]
- Park, J.H.; Jeong, E.; Lin, J.; Ko, R.; Kim, J.H.; Yi, S.; Choi, Y.; Kang, I.C.; Lee, D.; Lee, S.Y. RACK1 interaction with c-Src is essential for osteoclast function. Exp. Mol. Med. 2019, 51, 86. [Google Scholar] [CrossRef] [Green Version]
- Soysa, N.S.; Alles, N. Osteoclast function and bone-resorbing activity: An overview. Biochem. Biophys. Res. Commun. 2016, 476, 115–120. [Google Scholar] [CrossRef]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.C.; Teitelbaum, S.L.; Tan, H.L.; Koziol, C.M.; Schlesinger, P.H. Passive chloride permeability charge coupled to H(+)-ATPase of avian osteoclast ruffled membrane. Am. J. Physiol. 1991, 260, C1315–C1324. [Google Scholar] [CrossRef]
- Dai, R.; Wu, Z.; Chu, H.Y.; Lu, J.; Lyu, A.; Liu, J.; Zhang, G. Cathepsin K: The Action in and Beyond Bone. Front. Cell Dev. Biol. 2020, 8, 433. [Google Scholar] [CrossRef]
- Christensen, J.; Shastri, V.P. Matrix-metalloproteinase-9 is cleaved and activated by cathepsin K. BMC Res. Notes 2015, 8, 322. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Tang, Y.; Li, X.Y.; Keller, E.T.; Yang, J.; Cho, J.S.; Feinberg, T.Y.; Weiss, S.J. Osteoclast-mediated bone resorption is controlled by a compensatory network of secreted and membrane-tethered metalloproteinases. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Partridge, N.C. Physiological Bone Remodeling: Systemic Regulation and Growth Factor Involvement. Physiology 2016, 31, 233–245. [Google Scholar] [CrossRef]
- Gu, G.; Mulari, M.; Peng, Z.; Hentunen, T.A.; Vaananen, H.K. Death of osteocytes turns off the inhibition of osteoclasts and triggers local bone resorption. Biochem. Biophys. Res. Commun. 2005, 335, 1095–1101. [Google Scholar] [CrossRef]
- Hedgecock, N.L.; Hadi, T.; Chen, A.A.; Curtiss, S.B.; Martin, R.B.; Hazelwood, S.J. Quantitative regional associations between remodeling, modeling, and osteocyte apoptosis and density in rabbit tibial midshafts. Bone 2007, 40, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Batoon, L.; Millard, S.M.; Raggatt, L.J.; Pettit, A.R. Osteomacs and Bone Regeneration. Curr. Osteoporos. Rep. 2017, 15, 385–395. [Google Scholar] [CrossRef]
- Sapir-Koren, R.; Livshits, G. Osteocyte control of bone remodeling: Is sclerostin a key molecular coordinator of the balanced bone resorption-formation cycles? Osteoporos. Int. 2014, 25, 2685–2700. [Google Scholar] [CrossRef]
- Kular, J.; Tickner, J.; Chim, S.M.; Xu, J. An overview of the regulation of bone remodelling at the cellular level. Clin. Biochem. 2012, 45, 863–873. [Google Scholar] [CrossRef]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast-osteoclast interactions. Connect. Tissue Res. 2018, 59, 99–107. [Google Scholar] [CrossRef]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.; Gabay, O.; Salvat, C.; Henrotin, Y.E.; Berenbaum, F. Mechanical loading highly increases IL-6 production and decreases OPG expression by osteoblasts. Osteoarthr. Cartil. 2009, 17, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Kogawa, M.; Khalid, K.A.; Wijenayaka, A.R.; Ormsby, R.T.; Evdokiou, A.; Anderson, P.H.; Findlay, D.M.; Atkins, G.J. Recombinant sclerostin antagonizes effects of ex vivo mechanical loading in trabecular bone and increases osteocyte lacunar size. Am. J. Physiol. Cell Physiol. 2018, 314, C53–C61. [Google Scholar] [CrossRef]
- Hayashi, M.; Nakashima, T.; Taniguchi, M.; Kodama, T.; Kumanogoh, A.; Takayanagi, H. Osteoprotection by semaphorin 3A. Nature 2012, 485, 69–74. [Google Scholar] [CrossRef]
- Negishi-Koga, T.; Shinohara, M.; Komatsu, N.; Bito, H.; Kodama, T.; Friedel, R.H.; Takayanagi, H. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 2011, 17, 1473–1480. [Google Scholar] [CrossRef]
- Tonna, S.; Takyar, F.M.; Vrahnas, C.; Crimeen-Irwin, B.; Ho, P.W.; Poulton, I.J.; Brennan, H.J.; McGregor, N.E.; Allan, E.H.; Nguyen, H.; et al. EphrinB2 signaling in osteoblasts promotes bone mineralization by preventing apoptosis. FASEB J. 2014, 28, 4482–4496. [Google Scholar] [CrossRef]
- Zhao, C.; Irie, N.; Takada, Y.; Shimoda, K.; Miyamoto, T.; Nishiwaki, T.; Suda, T.; Matsuo, K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006, 4, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Gerstenfeld, L.C.; Cullinane, D.M.; Barnes, G.L.; Graves, D.T.; Einhorn, T.A. Fracture healing as a post-natal developmental process: Molecular, spatial, and temporal aspects of its regulation. J. Cell. Biochem. 2003, 88, 873–884. [Google Scholar] [CrossRef]
- Dimitriou, R.; Tsiridis, E.; Giannoudis, P.V. Current concepts of molecular aspects of bone healing. Injury 2005, 36, 1392–1404. [Google Scholar] [CrossRef]
- Burska, A.N.; Giannoudis, P.V.; Tan, B.H.; Ilas, D.; Jones, E.; Ponchel, F. Dynamics of Early Signalling Events during Fracture Healing and Potential Serum Biomarkers of Fracture Non-Union in Humans. J. Clin. Med. 2020, 9, 492. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Bleek, K.; Schell, H.; Kolar, P.; Pfaff, M.; Perka, C.; Buttgereit, F.; Duda, G.; Lienau, J. Cellular composition of the initial fracture hematoma compared to a muscle hematoma: A study in sheep. J. Orthop. Res. 2009, 27, 1147–1151. [Google Scholar] [CrossRef]
- Schmidt-Bleek, K.; Schell, H.; Lienau, J.; Schulz, N.; Hoff, P.; Pfaff, M.; Schmidt, G.; Martin, C.; Perka, C.; Buttgereit, F.; et al. Initial immune reaction and angiogenesis in bone healing. J. Tissue Eng. Regen. Med. 2014, 8, 120–130. [Google Scholar] [CrossRef]
- Stefanowski, J.; Lang, A.; Rauch, A.; Aulich, L.; Kohler, M.; Fiedler, A.F.; Buttgereit, F.; Schmidt-Bleek, K.; Duda, G.N.; Gaber, T.; et al. Spatial Distribution of Macrophages During Callus Formation and Maturation Reveals Close Crosstalk Between Macrophages and Newly Forming Vessels. Front. Immunol. 2019, 10, 2588. [Google Scholar] [CrossRef] [Green Version]
- Einhorn, T.A. The Cell and Molecular Biology of Fracture Healing. Clin. Orthop. Relat. Res. 1998, 355, S7–S21. [Google Scholar] [CrossRef]
- Ahmed, Y.A.; Tatarczuch, L.; Pagel, C.N.; Davies, H.M.; Mirams, M.; Mackie, E.J. Physiological death of hypertrophic chondrocytes. Osteoarthr. Cartil. 2007, 15, 575–586. [Google Scholar] [CrossRef] [Green Version]
- de Larco, J.E.; Todaro, G.J. Growth factors from murine sarcoma virus-transformed cells. Proc. Natl. Acad. Sci. USA 1978, 75, 4001–4005. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B.; Anzano, M.A.; Lamb, L.C.; Smith, J.M.; Sporn, M.B. New class of transforming growth factors potentiated by epidermal growth factor: Isolation from non-neoplastic tissues. Proc. Natl. Acad. Sci. USA 1981, 78, 5339–5343. [Google Scholar] [CrossRef] [Green Version]
- Moses, H.L.; Branum, E.L.; Proper, J.A.; Robinson, R.A. Transforming Growth Factor Production by Chemically Transformed Cells. Cancer Res. 1981, 41, 2842–2848. [Google Scholar]
- Roberts, A.B.; Frolik, C.A.; Anzano, M.A.; Sporn, M.B. Transforming growth factors from neoplastic and nonneoplastic tissues. Fed. Proc. 1983, 42, 2621–2626. [Google Scholar]
- Fujii, D.; Brissenden, J.E.; Derynck, R.; Francke, U. Transforming growth factor beta gene maps to human chromosome 19 long arm and to mouse chromosome 7. Somat. Cell Mol. Genet. 1986, 12, 281–288. [Google Scholar] [CrossRef]
- Barton, D.E.; Foellmer, B.E.; Du, J.; Tamm, J.; Derynck, R.; Francke, U. Chromosomal mapping of genes for transforming growth factors beta 2 and beta 3 in man and mouse: Dispersion of TGF-beta gene family. Oncogene Res. 1988, 3, 323–331. [Google Scholar]
- ten Dijke, P.; Geurts van Kessel, A.H.; Foulkes, J.G.; Le Beau, M.M. Transforming growth factor type beta 3 maps to human chromosome 14, region q23-q24. Oncogene 1988, 3, 721–724. [Google Scholar]
- Gentry, L.E.; Nash, B.W. The pro domain of pre-pro-transforming growth factor beta 1 when independently expressed is a functional binding protein for the mature growth factor. Biochemistry 1990, 29, 6851–6857. [Google Scholar] [CrossRef]
- Derynck, R.; Jarrett, J.A.; Chen, E.Y.; Eaton, D.H.; Bell, J.R.; Assoian, R.K.; Roberts, A.B.; Sporn, M.B.; Goeddel, D.V. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature 1985, 316, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-beta structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Mi, L.Z.; Brown, C.T.; Gao, Y.; Tian, Y.; Le, V.Q.; Walz, T.; Springer, T.A. Structure of bone morphogenetic protein 9 procomplex. Proc. Natl. Acad. Sci. USA 2015, 112, 3710–3715. [Google Scholar] [CrossRef] [Green Version]
- Aluwihare, P.; Mu, Z.; Zhao, Z.; Yu, D.; Weinreb, P.H.; Horan, G.S.; Violette, S.M.; Munger, J.S. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J. Cell Sci. 2009, 122, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, H.; Lioubin, M.N.; Ikeda, T. Complete amino acid sequence of human transforming growth factor type beta 2. J. Biol. Chem. 1987, 262, 12127–12131. [Google Scholar]
- Yue, J.; Mulder, K.M. Transforming growth factor-β signal transduction in epithelial cells. Pharmacol. Ther. 2001, 91, 1–34. [Google Scholar] [CrossRef]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Proetzel, G.; Pawlowski, S.A.; Wiles, M.V.; Yin, M.; Boivin, G.P.; Howles, P.N.; Ding, J.; Ferguson, M.W.; Doetschman, T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995, 11, 409–414. [Google Scholar] [CrossRef]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar]
- Arai, D.; Hayakawa, K.; Ohgane, J.; Hirosawa, M.; Nakao, Y.; Tanaka, S.; Shiota, K. An epigenetic regulatory element of the Nodal gene in the mouse and human genomes. Mech. Dev. 2015, 136, 143–154. [Google Scholar] [CrossRef]
- Bodenstine, T.M.; Chandler, G.S.; Seftor, R.E.; Seftor, E.A.; Hendrix, M.J. Plasticity underlies tumor progression: Role of Nodal signaling. Cancer Metastasis Rev. 2016, 35, 21–39. [Google Scholar] [CrossRef] [Green Version]
- Vale, W.; Rivier, J.; Vaughan, J.; McClintock, R.; Corrigan, A.; Woo, W.; Karr, D.; Spiess, J. Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature 1986, 321, 776–779. [Google Scholar] [CrossRef]
- Pauklin, S.; Vallier, L. Activin/Nodal signalling in stem cells. Development 2015, 142, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Fischer, G.; Hyvonen, M. Structure and activation of pro-activin A. Nat. Commun. 2016, 7, 12052. [Google Scholar] [CrossRef] [Green Version]
- Bloise, E.; Ciarmela, P.; Dela Cruz, C.; Luisi, S.; Petraglia, F.; Reis, F.M. Activin A in Mammalian Physiology. Physiol. Rev. 2019, 99, 739–780. [Google Scholar] [CrossRef]
- Jhaveri, S.; Erzurumlu, R.S.; Chiaia, N.; Kumar, T.R.; Matzuk, M.M. Defective whisker follicles and altered brainstem patterns in activin and follistatin knockout mice. Mol. Cell. Neurosci. 1998, 12, 206–219. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Kumar, T.R.; Vassalli, A.; Bickenbach, J.R.; Roop, D.R.; Jaenisch, R.; Bradley, A. Functional analysis of activins during mammalian development. Nature 1995, 374, 354–356. [Google Scholar] [CrossRef]
- Urist, M.R. Bone: Formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef]
- Wozney, J.M.; Rosen, V.; Celeste, A.J.; Mitsock, L.M.; Whitters, M.J.; Kriz, R.W.; Hewick, R.M.; Wang, E.A. Novel regulators of bone formation: Molecular clones and activities. Science 1988, 242, 1528–1534. [Google Scholar] [CrossRef]
- Ozkaynak, E.; Rueger, D.C.; Drier, E.A.; Corbett, C.; Ridge, R.J.; Sampath, T.K.; Oppermann, H. OP-1 cDNA encodes an osteogenic protein in the TGF-beta family. EMBO J. 1990, 9, 2085–2093. [Google Scholar] [CrossRef]
- Kingsley, D.M. The TGF-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 8, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.A.; Zhao, Q.; Baker, K.A.; Naik, C.; Chen, C.; Pukac, L.; Singh, M.; Tsareva, T.; Parice, Y.; Mahoney, A.; et al. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J. Biol. Chem. 2005, 280, 25111–25118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Luo, Q.; Shu, Y.; Zeng, Z.; Huang, B.; Feng, Y.; Zhang, B.; Wang, X.; Lei, Y.; Ye, Z.; et al. Transcriptomic landscape regulated by the 14 types of bone morphogenetic proteins (BMPs) in lineage commitment and differentiation of mesenchymal stem cells (MSCs). Genes Dis. 2019, 6, 258–275. [Google Scholar] [CrossRef]
- Daluiski, A.; Engstrand, T.; Bahamonde, M.E.; Gamer, L.W.; Agius, E.; Stevenson, S.L.; Cox, K.; Rosen, V.; Lyons, K.M. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat. Genet. 2001, 27, 84–88. [Google Scholar] [CrossRef]
- Wu, F.J.; Lin, T.Y.; Sung, L.Y.; Chang, W.F.; Wu, P.C.; Luo, C.W. BMP8A sustains spermatogenesis by activating both SMAD1/5/8 and SMAD2/3 in spermatogonia. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Chen, H.; Brady Ridgway, J.; Sai, T.; Lai, J.; Warming, S.; Chen, H.; Roose-Girma, M.; Zhang, G.; Shou, W.; Yan, M. Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc. Natl. Acad. Sci. USA 2013, 110, 11887–11892. [Google Scholar] [CrossRef] [Green Version]
- Bidart, M.; Ricard, N.; Levet, S.; Samson, M.; Mallet, C.; David, L.; Subileau, M.; Tillet, E.; Feige, J.J.; Bailly, S. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell. Mol. Life Sci. 2012, 69, 313–324. [Google Scholar] [CrossRef]
- Neugebauer, J.M.; Kwon, S.; Kim, H.S.; Donley, N.; Tilak, A.; Sopory, S.; Christian, J.L. The prodomain of BMP4 is necessary and sufficient to generate stable BMP4/7 heterodimers with enhanced bioactivity in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, E2307–E2316. [Google Scholar] [CrossRef] [Green Version]
- Wohl, A.P.; Troilo, H.; Collins, R.F.; Baldock, C.; Sengle, G. Extracellular Regulation of Bone Morphogenetic Protein Activity by the Microfibril Component Fibrillin-1. J. Biol. Chem. 2016, 291, 12732–12746. [Google Scholar] [CrossRef] [Green Version]
- Gregory, K.E.; Ono, R.N.; Charbonneau, N.L.; Kuo, C.L.; Keene, D.R.; Bachinger, H.P.; Sakai, L.Y. The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. J. Biol. Chem. 2005, 280, 27970–27980. [Google Scholar] [CrossRef] [Green Version]
- Salmon, R.M.; Guo, J.; Wood, J.H.; Tong, Z.; Beech, J.S.; Lawera, A.; Yu, M.; Grainger, D.J.; Reckless, J.; Morrell, N.W.; et al. Molecular basis of ALK1-mediated signalling by BMP9/BMP10 and their prodomain-bound forms. Nat. Commun. 2020, 11, 1621. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Jiang, W.; Phillips, F.M.; Haydon, R.C.; Peng, Y.; Zhou, L.; Luu, H.H.; An, N.; Breyer, B.; Vanichakarn, P.; et al. Osteogenic activity of the fourteen types of human bone morphogenetic proteins (BMPs). J. Bone Joint Surg. Am. 2003, 85, 1544–1552. [Google Scholar] [CrossRef]
- Kang, Q.; Sun, M.H.; Cheng, H.; Peng, Y.; Montag, A.G.; Deyrup, A.T.; Jiang, W.; Luu, H.H.; Luo, J.; Szatkowski, J.P.; et al. Characterization of the distinct orthotopic bone-forming activity of 14 BMPs using recombinant adenovirus-mediated gene delivery. Gene Ther. 2004, 11, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Wutzl, A.; Rauner, M.; Seemann, R.; Millesi, W.; Krepler, P.; Pietschmann, P.; Ewers, R. Bone morphogenetic proteins 2, 5, and 6 in combination stimulate osteoblasts but not osteoclasts in vitro. J. Orthop. Res. 2010, 28, 1431–1439. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, W.; Huang, E.; Wang, J.; Liao, J.; Li, R.; Yu, X.; Zhao, C.; Zeng, Z.; Shu, Y.; et al. BMP9-induced osteoblastic differentiation requires functional Notch signaling in mesenchymal stem cells. Lab. Investig. 2019, 99, 58–71. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W.; Devit, A.; van den Beucken, J. Efficiency of coculture with angiogenic cells or physiological BMP-2 administration on improving osteogenic differentiation and bone formation of MSCs. J. Biomed. Mater. Res. A 2019, 107, 643–653. [Google Scholar] [CrossRef]
- Cao, X.; Chen, D. The BMP signaling and in vivo bone formation. Gene 2005, 357, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Winnier, G.; Blessing, M.; Labosky, P.A.; Hogan, B.L. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995, 9, 2105–2116. [Google Scholar] [CrossRef] [Green Version]
- Lawson, K.A.; Dunn, N.R.; Roelen, B.A.; Zeinstra, L.M.; Davis, A.M.; Wright, C.V.; Korving, J.P.; Hogan, B.L. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999, 13, 424–436. [Google Scholar] [CrossRef]
- Luo, G.; Hofmann, C.; Bronckers, A.L.; Sohocki, M.; Bradley, A.; Karsenty, G. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 1995, 9, 2808–2820. [Google Scholar] [CrossRef] [Green Version]
- Gipson, G.R.; Goebel, E.J.; Hart, K.N.; Kappes, E.C.; Kattamuri, C.; McCoy, J.C.; Thompson, T.B. Structural perspective of BMP ligands and signaling. Bone 2020, 140, 115549. [Google Scholar] [CrossRef]
- Sieber, C.; Kopf, J.; Hiepen, C.; Knaus, P. Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev. 2009, 20, 343–355. [Google Scholar] [CrossRef]
- Greenwald, J.; Groppe, J.; Gray, P.; Wiater, E.; Kwiatkowski, W.; Vale, W.; Choe, S. The BMP7/ActRII Extracellular Domain Complex Provides New Insights into the Cooperative Nature of Receptor Assembly. Mol. Cell 2003, 11, 605–617. [Google Scholar] [CrossRef]
- Yadin, D.; Knaus, P.; Mueller, T.D. Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine Growth Factor Rev. 2016, 27, 13–34. [Google Scholar] [CrossRef]
- Grafe, I.; Alexander, S.; Peterson, J.R.; Snider, T.N.; Levi, B.; Lee, B.; Mishina, Y. TGF-beta Family Signaling in Mesenchymal Differentiation. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- van den Bosch, M.H.; Blom, A.B.; van Lent, P.L.; van Beuningen, H.M.; Davidson, E.N.B.; van der Kraan, P.M.; van den Berg, W.B. Canonical Wnt signaling skews TGF-beta signaling in chondrocytes towards signaling via ALK1 and Smad 1/5/8. Cell Signal. 2014, 26, 951–958. [Google Scholar] [CrossRef]
- Olsen, O.E.; Hella, H.; Elsaadi, S.; Jacobi, C.; Martinez-Hackert, E.; Holien, T. Activins as Dual Specificity TGF-beta Family Molecules: SMAD-Activation via Activin- and BMP-Type 1 Receptors. Biomolecules 2020, 10, 519. [Google Scholar] [CrossRef] [Green Version]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Brazil, D.P.; Church, R.H.; Surae, S.; Godson, C.; Martin, F. BMP signalling: Agony and antagony in the family. Trends Cell Biol. 2015, 25, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Okadome, T.; Yamashita, H.; Franzén, P.; Morén, A.; Heldin, C.H.; Miyazono, K. Distinct roles of the intracellular domains of transforming growth factor-beta type I and type II receptors in signal transduction. J. Biol. Chem. 1994, 269, 30753–30756. [Google Scholar]
- Hinck, A.P. Structural studies of the TGF-betas and their receptors—Insights into evolution of the TGF-beta superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef] [Green Version]
- Fong, D.; Bisson, M.; Laberge, G.; McManus, S.; Grenier, G.; Faucheux, N.; Roux, S. Bone morphogenetic protein-9 activates Smad and ERK pathways and supports human osteoclast function and survival in vitro. Cell Signal. 2013, 25, 717–728. [Google Scholar] [CrossRef]
- Kaneko, H.; Arakawa, T.; Mano, H.; Kaneda, T.; Ogasawara, A.; Nakagawa, M.; Toyama, Y.; Yabe, Y.; Kumegawa, M.; Hakeda, Y. Direct stimulation of osteoclastic bone resorption by bone morphogenetic protein (BMP)-2 and expression of BMP receptors in mature osteoclasts. Bone 2000, 27, 479–486. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [Green Version]
- Gratchev, A. TGF-beta signalling in tumour associated macrophages. Immunobiology 2017, 222, 75–81. [Google Scholar] [CrossRef]
- Ebisawa, T.; Tada, K.; Kitajima, I.; Tojo, K.; Sampath, T.K.; Kawabata, M.; Miyazono, K.; Imamura, T. Characterization of bone morphogenetic protein-6 signaling pathways in osteoblast differentiation. J. Cell Sci. 1999, 112, 3519–3527. [Google Scholar]
- Gratchev, A.; Kzhyshkowska, J.; Kannookadan, S.; Ochsenreiter, M.; Popova, A.; Yu, X.; Mamidi, S.; Stonehouse-Usselmann, E.; Muller-Molinet, I.; Gooi, L.; et al. Activation of a TGF-beta-specific multistep gene expression program in mature macrophages requires glucocorticoid-mediated surface expression of TGF-beta receptor II. J. Immunol. 2008, 180, 6553–6565. [Google Scholar] [CrossRef] [Green Version]
- Ota, K.; Quint, P.; Ruan, M.; Pederson, L.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. TGF-beta induces Wnt10b in osteoclasts from female mice to enhance coupling to osteoblasts. Endocrinology 2013, 154, 3745–3752. [Google Scholar] [CrossRef] [Green Version]
- Meurer, S.K.; Esser, M.; Tihaa, L.; Weiskirchen, R. BMP-7/TGF-beta1 signalling in myoblasts: Components involved in signalling and BMP-7-dependent blockage of TGF-beta-mediated CTGF expression. Eur. J. Cell Biol. 2012, 91, 450–463. [Google Scholar] [CrossRef]
- Suzuki, E.; Ochiai-Shino, H.; Aoki, H.; Onodera, S.; Saito, A.; Saito, A.; Azuma, T. Akt activation is required for TGF-beta1-induced osteoblast differentiation of MC3T3-E1 pre-osteoblasts. PLoS ONE 2014, 9, e112566. [Google Scholar] [CrossRef] [Green Version]
- Sowa, H.; Kaji, H.; Yamaguchi, T.; Sugimoto, T.; Chihara, K. Activations of ERK1/2 and JNK by transforming growth factor beta negatively regulate Smad3-induced alkaline phosphatase activity and mineralization in mouse osteoblastic cells. J. Biol. Chem. 2002, 277, 36024–36031. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S. TGF-beta regulates beta-catenin signaling and osteoblast differentiation in human mesenchymal stem cells. J. Cell. Biochem. 2011, 112, 1651–1660. [Google Scholar] [CrossRef] [Green Version]
- Liyun, G.; Xu, J.; Li, X.; Wang, T.; Wu, W.; Cao, J. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin and TGF-beta3 Mediated-Mouse Embryonic Palatal Mesenchymal Cells. Dose Response 2018, 16. [Google Scholar] [CrossRef]
- Arita, N.A.; Pelaez, D.; Cheung, H.S. Activation of the extracellular signal-regulated kinases 1 and 2 (ERK1/2) is needed for the TGFbeta-induced chondrogenic and osteogenic differentiation of mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2011, 405, 564–569. [Google Scholar] [CrossRef]
- Kajita, T.; Ariyoshi, W.; Okinaga, T.; Mitsugi, S.; Tominaga, K.; Nishihara, T. Mechanisms involved in enhancement of osteoclast formation by activin-A. J. Cell. Biochem. 2018, 119, 6974–6985. [Google Scholar] [CrossRef]
- Murase, Y.; Okahashi, N.; Koseki, T.; Itoh, K.; Udagawa, N.; Hashimoto, O.; Sugino, H.; Noguchi, T.; Nishihara, T. Possible involvement of protein kinases and Smad2 signaling pathways on osteoclast differentiation enhanced by activin A. J. Cell Physiol. 2001, 188, 236–242. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Chen, Y.; Suo, L.; Chen, H.; Zhu, L.; Wan, G.; Han, X. Activin a promotes myofibroblast differentiation of endometrial mesenchymal stem cells via STAT3-dependent Smad/CTGF pathway. Cell Commun. Signal. 2019, 17, 45. [Google Scholar] [CrossRef] [Green Version]
- Broege, A.; Pham, L.; Jensen, E.D.; Emery, A.; Huang, T.H.; Stemig, M.; Beppu, H.; Petryk, A.; O’Connor, M.; Mansky, K.; et al. Bone morphogenetic proteins signal via SMAD and mitogen-activated protein (MAP) kinase pathways at distinct times during osteoclastogenesis. J. Biol. Chem. 2013, 288, 37230–37240. [Google Scholar] [CrossRef] [Green Version]
- Mandal, C.C.; Das, F.; Ganapathy, S.; Harris, S.E.; Choudhury, G.G.; Ghosh-Choudhury, N. Bone Morphogenetic Protein-2 (BMP-2) Activates NFATc1 Transcription Factor via an Autoregulatory Loop Involving Smad/Akt/Ca2+ Signaling. J. Biol. Chem. 2016, 291, 1148–1161. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.; Yao, B.; Zhang, B.D.; Bai, Y.; Sui, W.; Wang, W.; Yu, Q. Stromal-derived Factor-1alpha signaling is involved in bone morphogenetic protein-2-induced odontogenic differentiation of stem cells from apical papilla via the Smad and Erk signaling pathways. Exp. Cell Res. 2019, 381, 39–49. [Google Scholar] [CrossRef]
- Drevelle, O.; Daviau, A.; Lauzon, M.A.; Faucheux, N. Effect of BMP-2 and/or BMP-9 on preosteoblasts attached to polycaprolactone functionalized by adhesive peptides derived from bone sialoprotein. Biomaterials 2013, 34, 1051–1062. [Google Scholar] [CrossRef]
- Lauzon, M.A.; Daviau, A.; Drevelle, O.; Marcos, B.; Faucheux, N. Identification of a growth factor mimicking the synergistic effect of fetal bovine serum on BMP-9 cell response. Tissue Eng. Part A 2014, 20, 2524–2535. [Google Scholar] [CrossRef]
- Lauzon, M.A.; Drevelle, O.; Daviau, A.; Faucheux, N. Effects of BMP-9 and BMP-2 on the PI3K/Akt Pathway in MC3T3-E1 Preosteoblasts. Tissue Eng. Part A 2016, 22, 1075–1085. [Google Scholar] [CrossRef]
- Aoki, H.; Fujii, M.; Imamura, T.; Yagi, K.; Takehara, K.; Kato, M.; Miyazono, K. Synergistic effects of different bone morphogenetic protein type I receptors on alkaline phosphatase induction. J. Cell Sci. 2001, 114, 1483–1489. [Google Scholar]
- Lichtner, B.; Knaus, P.; Lehrach, H.; Adjaye, J. BMP10 as a potent inducer of trophoblast differentiation in human embryonic and induced pluripotent stem cells. Biomaterials 2013, 34, 9789–9802. [Google Scholar] [CrossRef]
- Vijayan, V.; Gupta, S.; Gupta, S. Bone morphogenetic protein-5, a key molecule that mediates differentiation in MC3T3E1 osteoblast cell line. Biofactors 2017, 43, 558–566. [Google Scholar] [CrossRef]
- Moore, R.K.; Otsuka, F.; Shimasaki, S. Molecular basis of bone morphogenetic protein-15 signaling in granulosa cells. J. Biol. Chem. 2003, 278, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Gustafson, A.R.; Schaner Tooley, C.E.; Zhang, M. BMP-9 dependent pathways required for the chondrogenic differentiation of pluripotent stem cells. Differentiation 2016, 92, 298–305. [Google Scholar] [CrossRef]
- Chang, H.M.; Cheng, J.C.; Klausen, C.; Leung, P.C. BMP15 suppresses progesterone production by down-regulating StAR via ALK3 in human granulosa cells. Mol. Endocrinol. 2013, 27, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Shirai, T.; Morishita, S.; Uchida, S.; Saeki-Miura, K.; Makishima, F. p38 mitogen-activated protein kinase functionally contributes to chondrogenesis induced by growth/differentiation factor-5 in ATDC5 cells. Exp. Cell Res. 1999, 250, 351–363. [Google Scholar] [CrossRef]
- Wang, S.S.; Huang, H.Y.; Chen, S.Z.; Li, X.; Zhang, W.T.; Tang, Q.Q. Gdf6 induces commitment of pluripotent mesenchymal C3H10T1/2 cells to the adipocyte lineage. FEBS J. 2013, 280, 2644–2651. [Google Scholar] [CrossRef]
- Andersson, O.; Reissmann, E.; Ibanez, C.F. Growth differentiation factor 11 signals through the transforming growth factor-beta receptor ALK5 to regionalize the anterior-posterior axis. EMBO Rep. 2006, 7, 831–837. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Otsuka, F.; Hino, J.; Miyoshi, T.; Takano, M.; Miyazato, M.; Makino, H.; Kangawa, K. Bone morphogenetic protein-3b (BMP-3b) inhibits osteoblast differentiation via Smad2/3 pathway by counteracting Smad1/5/8 signaling. Mol. Cell Endocrinol. 2012, 350, 78–86. [Google Scholar] [CrossRef]
- Nickel, J.; Ten Dijke, P.; Mueller, T.D. TGF-beta family co-receptor function and signaling. Acta Biochim. Biophys. Sin. 2018, 50, 12–36. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, A.; Lin, H.Y.; Henis, Y.I.; Plamondon, J.; O’Connor-McCourt, M.D.; Lodish, H.F. The transforming growth factor beta receptors types I, II, and III form hetero-oligomeric complexes in the presence of ligand. J. Biol. Chem. 1993, 268, 22215–22218. [Google Scholar]
- Esparza-Lopez, J.; Montiel, J.L.; Vilchis-Landeros, M.M.; Okadome, T.; Miyazono, K.; Lopez-Casillas, F. Ligand binding and functional properties of betaglycan, a co-receptor of the transforming growth factor-beta superfamily. Specialized binding regions for transforming growth factor-beta and inhibin A. J. Biol. Chem. 2001, 276, 14588–14596. [Google Scholar] [CrossRef] [Green Version]
- Bilandzic, M.; Stenvers, K.L. Betaglycan: A multifunctional accessory. Mol. Cell. Endocrinol. 2011, 339, 180–189. [Google Scholar] [CrossRef]
- Cheifetz, S.; Bellón, T.; Calés, C.; Vera, S.; Bernabeu, C.; Massagué, J.; Letarte, M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J. Biol. Chem. 1992, 267, 19027–19030. [Google Scholar]
- Castonguay, R.; Werner, E.D.; Matthews, R.G.; Presman, E.; Mulivor, A.W.; Solban, N.; Sako, D.; Pearsall, R.S.; Underwood, K.W.; Seehra, J.; et al. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011, 286, 30034–30046. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Bokhove, M.; Croci, R.; Zamora-Caballero, S.; Han, L.; Letarte, M.; de Sanctis, D.; Jovine, L. Structural Basis of the Human Endoglin-BMP9 Interaction: Insights into BMP Signaling and HHT1. Cell Rep. 2017, 19, 1917–1928. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Wang, Y.-F.; Jayaraman, L.; Yang, H.; Massagué, J.; Pavletich, N.P. Crystal Structure of a Smad MH1 Domain Bound to DNA. Cell 1998, 94, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Lo, R.S.; Chen, Y.G.; Shi, Y.; Pavletich, N.P.; Massague, J. The L3 loop: A structural motif determining specific interactions between SMAD proteins and TGF-beta receptors. EMBO J. 1998, 17, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Lin, X.; Feng, X.H. Posttranslational Regulation of Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022087. [Google Scholar] [CrossRef] [Green Version]
- Makkar, P.; Metpally, R.P.; Sangadala, S.; Reddy, B.V. Modeling and analysis of MH1 domain of Smads and their interaction with promoter DNA sequence motif. J. Mol. Graph. Model. 2009, 27, 803–812. [Google Scholar] [CrossRef]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [Green Version]
- Kretzschmar, M.; Doody, J.; Massague, J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [CrossRef]
- Hassanisaber, H.; Rouleau, L.; Faucheux, N. Effect of BMP-9 on endothelial cells and its role in atherosclerosis. Front. Biosci. 2019, 24, 994–1023. [Google Scholar] [CrossRef]
- Wu, M.; Chen, G.; Li, Y.P. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef]
- Lawera, A.; Tong, Z.; Thorikay, M.; Redgrave, R.E.; Cai, J.; van Dinther, M.; Morrell, N.W.; Afink, G.B.; Charnock-Jones, D.S.; Arthur, H.M.; et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 17800–17808. [Google Scholar] [CrossRef] [Green Version]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.-G.; Kuriyan, J.; Massagué, J. The TGFβ Receptor Activation Process. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-beta Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Moustakas, A.; Knaus, P.; Wells, R.G.; Henis, Y.I.; Lodish, H.F. The soluble exoplasmic domain of the type II transforming growth factor (TGF)-beta receptor. A heterogeneously glycosylated protein with high affinity and selectivity for TGF-beta ligands. J. Biol. Chem. 1995, 270, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Mathews, L.S. Activin receptors and cellular signaling by the receptor serine kinase family. Endocr. Rev. 1994, 15, 310–325. [Google Scholar] [CrossRef]
- Bernard, D.J.; Lee, K.B.; Santos, M.M. Activin B can signal through both ALK4 and ALK7 in gonadotrope cells. Reprod. Biol. Endocrinol. 2006, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Persson, U.; Izumi, H.; Souchelnytskyi, S.; Itoh, S.; Grimsby, S.; Engström, U.; Heldin, C.-H.; Funa, K.; ten Dijke, P. The L45 loop in type I receptors for TGF-β family members is a critical determinant in specifying Smad isoform activation. FEBS Lett. 1998, 434, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Qin, B.Y.; Chacko, B.M.; Lam, S.S.; de Caestecker, M.P.; Correia, J.J.; Lin, K. Structural Basis of Smad1 Activation by Receptor Kinase Phosphorylation. Mol. Cell 2001, 8, 1303–1312. [Google Scholar] [CrossRef]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE Domain Protein that Recruits Smad2 to the TGFβ Receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Itoh, F.; Divecha, N.; Brocks, L.; Oomen, L.; Janssen, H.; Calafat, J.; Itoh, S.; Dijke Pt, P. The FYVE domain in Smad anchor for receptor activation (SARA) is sufficient for localization of SARA in early endosomes and regulates TGF-beta/Smad signalling. Genes Cells 2002, 7, 321–331. [Google Scholar] [CrossRef]
- Miyazawa, K.; Shinozaki, M.; Hara, T.; Furuya, T.; Miyazono, K. Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells 2002, 7, 1191–1204. [Google Scholar] [CrossRef]
- Hua, X.; Miller, Z.A.; Benchabane, H.; Wrana, J.L.; Lodish, H.F. Synergism between transcription factors TFE3 and Smad3 in transforming growth factor-beta-induced transcription of the Smad7 gene. J. Biol. Chem. 2000, 275, 33205–33208. [Google Scholar] [CrossRef] [Green Version]
- Aragon, E.; Wang, Q.; Zou, Y.; Morgani, S.M.; Ruiz, L.; Kaczmarska, Z.; Su, J.; Torner, C.; Tian, L.; Hu, J.; et al. Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-beta signaling. Genes Dev. 2019, 33, 1506–1524. [Google Scholar] [CrossRef] [Green Version]
- Nohe, A.; Hassel, S.; Ehrlich, M.; Neubauer, F.; Sebald, W.; Henis, Y.I.; Knaus, P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J. Biol. Chem. 2002, 277, 5330–5338. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, M. Endocytosis and trafficking of BMP receptors: Regulatory mechanisms for fine-tuning the signaling response in different cellular contexts. Cytokine Growth Factor Rev. 2016, 27, 35–42. [Google Scholar] [CrossRef]
- Heinecke, K.; Seher, A.; Schmitz, W.; Mueller, T.D.; Sebald, W.; Nickel, J. Receptor oligomerization and beyond: A case study in bone morphogenetic proteins. BMC Biol. 2009, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharpfenecker, M.; van Dinther, M.; Liu, Z.; van Bezooijen, R.L.; Zhao, Q.; Pukac, L.; Lowik, C.W.; ten Dijke, P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007, 120, 964–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Tang, M.; Huang, J.; He, B.C.; Gao, J.L.; Chen, L.; Zuo, G.W.; Zhang, W.; Luo, Q.; Shi, Q.; et al. TGFbeta/BMP type I receptors ALK1 and ALK2 are essential for BMP9-induced osteogenic signaling in mesenchymal stem cells. J. Biol. Chem. 2010, 285, 29588–29598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luther, G.; Wagner, E.R.; Zhu, G.; Kang, Q.; Luo, Q.; Lamplot, J.; Bi, Y.; Luo, X.; Luo, J.; Teven, C.; et al. BMP-9 induced osteogenic differentiation of mesenchymal stem cells: Molecular mechanism and therapeutic potential. Curr. Gene Ther. 2011, 11, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, L. BMP signaling and stem cell regulation. Dev. Biol. 2005, 284, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, T.; Kida, K.; Yamaguchi, A.; Hata, K.; Ichida, F.; Meguro, H.; Aburatani, H.; Nishimura, R.; Yoneda, T. BMP2 regulates Osterix through Msx2 and Runx2 during osteoblast differentiation. J. Biol. Chem. 2008, 283, 29119–29125. [Google Scholar] [CrossRef] [Green Version]
- Onichtchouk, D.; Chen, Y.G.; Dosch, R.; Gawantka, V.; Delius, H.; Massague, J.; Niehrs, C. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999, 401, 480–485. [Google Scholar] [CrossRef]
- Gray, P.C.; Harrison, C.A.; Vale, W. Cripto forms a complex with activin and type II activin receptors and can block activin signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 5193–5198. [Google Scholar] [CrossRef] [Green Version]
- Seemann, P.; Brehm, A.; Konig, J.; Reissner, C.; Stricker, S.; Kuss, P.; Haupt, J.; Renninger, S.; Nickel, J.; Sebald, W.; et al. Mutations in GDF5 reveal a key residue mediating BMP inhibition by NOGGIN. PLoS Genet. 2009, 5, e1000747. [Google Scholar] [CrossRef] [PubMed]
- Feeley, B.T.; Krenek, L.; Liu, N.; Hsu, W.K.; Gamradt, S.C.; Schwarz, E.M.; Huard, J.; Lieberman, J.R. Overexpression of noggin inhibits BMP-mediated growth of osteolytic prostate cancer lesions. Bone 2006, 38, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Rider, C.C. The Bone Morphogenetic Proteins and Their Antagonists. Vitam. Horm. 2015, 99, 63–90. [Google Scholar] [CrossRef] [PubMed]
- Sidis, Y.; Mukherjee, A.; Keutmann, H.; Delbaere, A.; Sadatsuki, M.; Schneyer, A. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 2006, 147, 3586–3597. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-beta Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, Y.; Miyazono, K.; Miyazawa, K. Smad7 inhibits transforming growth factor-beta family type i receptors through two distinct modes of interaction. J. Biol. Chem. 2010, 285, 30804–30813. [Google Scholar] [CrossRef] [Green Version]
- Goto, K.; Kamiya, Y.; Imamura, T.; Miyazono, K.; Miyazawa, K. Selective inhibitory effects of Smad6 on bone morphogenetic protein type I receptors. J. Biol. Chem. 2007, 282, 20603–20611. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, T.; Miyazaki, H.; Hara, T.; Furuya, T.; Imamura, T.; Watabe, T.; Miyazono, K. Roles for the MH2 domain of Smad7 in the specific inhibition of transforming growth factor-beta superfamily signaling. J. Biol. Chem. 2004, 279, 31568–31574. [Google Scholar] [CrossRef] [Green Version]
- Hanyu, A.; Ishidou, Y.; Ebisawa, T.; Shimanuki, T.; Imamura, T.; Miyazono, K. The N domain of Smad7 is essential for specific inhibition of transforming growth factor-beta signaling. J. Cell Biol. 2001, 155, 1017–1027. [Google Scholar] [CrossRef]
- Koganti, P.; Levy-Cohen, G.; Blank, M. Smurfs in Protein Homeostasis, Signaling, and Cancer. Front. Oncol. 2018, 8, 295. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Lin, Z.; Chen, F.; Zhao, X.; Chen, H.; Ning, Y.; Chen, Y.G. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-beta signaling. J. Biol. Chem. 2009, 284, 30097–30104. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, S.; Mizuta, T.; Fujimoto, M.; Ohte, S.; Osawa, K.; Miyamoto, A.; Yoneyama, K.; Murata, E.; Machiya, A.; Jimi, E.; et al. Smad9 is a new type of transcriptional regulator in bone morphogenetic protein signaling. Sci. Rep. 2014, 4, 7596. [Google Scholar] [CrossRef]
- Katagiri, T.; Tsukamoto, S.; Kuratani, M. Heterotopic bone induction via BMP signaling: Potential therapeutic targets for fibrodysplasia ossificans progressiva. Bone 2018, 109, 241–250. [Google Scholar] [CrossRef]
- Bellavia, D.; De Luca, A.; Carina, V.; Costa, V.; Raimondi, L.; Salamanna, F.; Alessandro, R.; Fini, M.; Giavaresi, G. Deregulated miRNAs in bone health: Epigenetic roles in osteoporosis. Bone 2019, 122, 52–75. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Li, B.; Sun, L.; An, C. MicroRNA-153 inhibits osteosarcoma cells proliferation and invasion by targeting TGF-beta2. PLoS ONE 2015, 10, e0119225. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hassan, M.Q.; Jafferji, M.; Aqeilan, R.I.; Garzon, R.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Biological functions of miR-29b contribute to positive regulation of osteoblast differentiation. J. Biol. Chem. 2009, 284, 15676–15684. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Xiusheng, H.; Xiao, X.; Wang, Y. Overexpression of miR-422a inhibits cell proliferation and invasion, and enhances chemosensitivity in osteosarcoma cells. Oncol. Rep. 2016, 36, 3371–3378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Zhou, H.; Hong, Y.; Li, J.; Jiang, X.; Huang, H. miR-30 family members negatively regulate osteoblast differentiation. J. Biol. Chem. 2012, 287, 7503–7511. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Hassan, M.Q.; Volinia, S.; van Wijnen, A.J.; Stein, J.L.; Croce, C.M.; Lian, J.B.; Stein, G.S. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc. Natl. Acad. Sci. USA 2008, 105, 13906–13911. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.F.; Fu, W.M.; He, M.L.; Xie, W.D.; Lv, Q.; Wan, G.; Li, G.; Wang, H.; Lu, G.; Hu, X.; et al. MiRNA-20a promotes osteogenic differentiation of human mesenchymal stem cells by co-regulating BMP signaling. RNA Biol. 2011, 8, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, S.; Iguchi, M.; Watanabe, K.; Hoshino, R.; Tsujimoto, M.; Kohno, M. Specific activation of the p38 mitogen-activated protein kinase signaling pathway and induction of neurite outgrowth in PC12 cells by bone morphogenetic protein-2. J. Biol. Chem. 1999, 274, 26503–26510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guicheux, J.; Lemonnier, J.; Ghayor, C.; Suzuki, A.; Palmer, G.; Caverzasio, J. Activation of p38 mitogen-activated protein kinase and c-Jun-NH2-terminal kinase by BMP-2 and their implication in the stimulation of osteoblastic cell differentiation. J. Bone Miner. Res. 2003, 18, 2060–2068. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Zhang, X.; Sanjay, A.; Owen, T.A.; Smock, S.L.; Rehman, S.; DeLong, W.G.; Safadi, F.F.; Popoff, S.N. Molecular requirements for induction of CTGF expression by TGF-beta1 in primary osteoblasts. Bone 2008, 42, 871–885. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, D.; Wang, S.; Ding, J.; Zhao, L. Bone morphogenetic protein9 promotes the differentiation of mouse spleen macrophages into osteoclasts via the ALK1 receptor and ERK 1/2 pathways in vitro. Mol. Med. Rep. 2016, 14, 4545–4550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Hong, S.H.; Bae, S.C. Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-beta and bone morphogenetic protein. Oncogene 2002, 21, 7156–7163. [Google Scholar] [CrossRef] [Green Version]
- Celil, A.B.; Campbell, P.G. BMP-2 and insulin-like growth factor-I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. J. Biol. Chem. 2005, 280, 31353–31359. [Google Scholar] [CrossRef] [Green Version]
- Ge, C.; Xiao, G.; Jiang, D.; Yang, Q.; Hatch, N.E.; Roca, H.; Franceschi, R.T. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J. Biol. Chem. 2009, 284, 32533–32543. [Google Scholar] [CrossRef] [Green Version]
- Ulsamer, A.; Ortuno, M.J.; Ruiz, S.; Susperregui, A.R.; Osses, N.; Rosa, J.L.; Ventura, F. BMP-2 induces Osterix expression through up-regulation of Dlx5 and its phosphorylation by p38. J. Biol. Chem. 2008, 283, 3816–3826. [Google Scholar] [CrossRef] [Green Version]
- Ortuno, M.J.; Ruiz-Gaspa, S.; Rodriguez-Carballo, E.; Susperregui, A.R.; Bartrons, R.; Rosa, J.L.; Ventura, F. p38 regulates expression of osteoblast-specific genes by phosphorylation of osterix. J. Biol. Chem. 2010, 285, 31985–31994. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.F.; Lin, J.J.; Lin, C.H.; Su, Y.; Hung, S.C. c-Jun N-terminal kinase 1 negatively regulates osteoblastic differentiation induced by BMP2 via phosphorylation of Runx2 at Ser104. J. Bone Miner. Res. 2012, 27, 1093–1105. [Google Scholar] [CrossRef]
- Aubin, J.; Davy, A.; Soriano, P. In vivo convergence of BMP and MAPK signaling pathways: Impact of differential Smad1 phosphorylation on development and homeostasis. Genes Dev. 2004, 18, 1482–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, J.; Mueller, T.D. Specification of BMP Signaling. Cells 2019, 8, 1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Liu, W.; Tan, T.; Dong, C.Q.; Lin, D.Y.; Zhao, J.; Yu, C.; Luo, X.J. Notch signaling negatively regulates BMP9-induced osteogenic differentiation of mesenchymal progenitor cells by inhibiting JunB expression. Oncotarget 2017, 8, 109661–109674. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-beta: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef]
- LaFoya, B.; Munroe, J.A.; Mia, M.M.; Detweiler, M.A.; Crow, J.J.; Wood, T.; Roth, S.; Sharma, B.; Albig, A.R. Notch: A multi-functional integrating system of microenvironmental signals. Dev. Biol. 2016, 418, 227–241. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Rijsewijk, F.; Schuermann, M.; Wagenaar, E.; Parren, P.; Weigel, D.; Nusse, R. The Drosophila homology of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 1987, 50, 649–657. [Google Scholar] [CrossRef]
- Kikuchi, A.; Yamamoto, H.; Sato, A.; Matsumoto, S. New insights into the mechanism of Wnt signaling pathway activation. Int. Rev. Cell Mol. Biol. 2011, 291, 21–71. [Google Scholar] [CrossRef]
- Ng, L.F.; Kaur, P.; Bunnag, N.; Suresh, J.; Sung, I.C.H.; Tan, Q.H.; Gruber, J.; Tolwinski, N.S. WNT Signaling in Disease. Cells 2019, 8, 826. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Yost, C.; Torres, M.; Miller, J.R.; Huang, E.; Kimelman, D.; Moon, R.T. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996, 10, 1443–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, M.; Concordet, J.P.; Lassot, I.; Albert, I.; del los Santos, R.; Durand, H.; Perret, C.; Rubinfeld, B.; Margottin, F.; Benarous, R.; et al. The F-box protein β-TrCP associates with phosphorylated β-catenin and regulates its activity in the cell. Curr. Biol. 1999, 9, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, V.; Bryant, H.U.; Macdougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Investig. 2006, 116, 1202–1209. [Google Scholar] [CrossRef]
- Zhao, C.; Yu, T.; Dou, Q.; Guo, Y.; Yang, X.; Chen, Y. Knockout of TLR4 promotes fracture healing by activating Wnt/beta-catenin signaling pathway. Pathol. Res. Pract. 2020, 216, 152766. [Google Scholar] [CrossRef]
- Morsczeck, C.; Reck, A.; Reichert, T.E. WNT3A and the induction of the osteogenic differentiation in adipose tissue derived mesenchymal stem cells. Tissue Cell 2017, 49, 489–494. [Google Scholar] [CrossRef]
- Glass, D.A., 2nd; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Inkson, C.A.; Ono, M.; Kuznetsov, S.A.; Fisher, L.W.; Robey, P.G.; Young, M.F. TGF-beta1 and WISP-1/CCN-4 can regulate each other’s activity to cooperatively control osteoblast function. J. Cell. Biochem. 2008, 104, 1865–1878. [Google Scholar] [CrossRef] [Green Version]
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.; Komm, B.S.; Javed, A.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 2005, 280, 33132–33140. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Ramirez, A.; Waddell, D.S.; Li, Z.; Liu, X.; Wang, X.F. Axin and GSK3- control Smad3 protein stability and modulate TGF- signaling. Genes Dev. 2008, 22, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Fuentealba, L.C.; Eivers, E.; Ikeda, A.; Hurtado, C.; Kuroda, H.; Pera, E.M.; De Robertis, E.M. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 2007, 131, 980–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Song, W.X.; Luo, J.; Luo, X.; Chen, J.; Sharff, K.A.; Bi, Y.; He, B.C.; Huang, J.Y.; Zhu, G.H.; et al. BMP-9-induced osteogenic differentiation of mesenchymal progenitors requires functional canonical Wnt/beta-catenin signalling. J. Cell Mol. Med. 2009, 13, 2448–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Carballo, E.; Ulsamer, A.; Susperregui, A.R.; Manzanares-Cespedes, C.; Sanchez-Garcia, E.; Bartrons, R.; Rosa, J.L.; Ventura, F. Conserved regulatory motifs in osteogenic gene promoters integrate cooperative effects of canonical Wnt and BMP pathways. J. Bone Miner. Res. 2011, 26, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Rawadi, G.; Vayssiere, B.; Dunn, F.; Baron, R.; Roman-Roman, S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J. Bone Miner. Res. 2003, 18, 1842–1853. [Google Scholar] [CrossRef]
- Strutt, D.I.; Weber, U.; Mlodzik, M. The role of RhoA in tissue polarity and Frizzled signalling. Nature 1997, 387, 292–295. [Google Scholar] [CrossRef]
- Schambony, A.; Wedlich, D. Wnt-5A/Ror2 regulate expression of XPAPC through an alternative noncanonical signaling pathway. Dev. Cell 2007, 12, 779–792. [Google Scholar] [CrossRef] [Green Version]
- Uehara, S.; Udagawa, N.; Kobayashi, Y. Non-canonical Wnt signals regulate cytoskeletal remodeling in osteoclasts. Cell. Mol. Life Sci. 2018, 75, 3683–3692. [Google Scholar] [CrossRef] [Green Version]
- Uehara, S.; Udagawa, N.; Mukai, H.; Ishihara, A.; Maeda, K.; Yamashita, T.; Murakami, K.; Nishita, M.; Nakamura, T.; Kato, S.; et al. Protein kinase N3 promotes bone resorption by osteoclasts in response to Wnt5a-Ror2 signaling. Sci. Signal. 2017, 10, eaan0023. [Google Scholar] [CrossRef] [Green Version]
- Sheldahl, L.C.; Park, M.; Malbon, C.C.; Moon, R.T. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in aG-protein-dependent manner. Curr. Biol. 1999, 9, 695–698, S1. [Google Scholar] [CrossRef] [Green Version]
- Dejmek, J.; Safholm, A.; Kamp Nielsen, C.; Andersson, T.; Leandersson, K. Wnt-5a/Ca2+-induced NFAT activity is counteracted by Wnt-5a/Yes-Cdc42-casein kinase 1alpha signaling in human mammary epithelial cells. Mol. Cell. Biol. 2006, 26, 6024–6036. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xu, P.; Lamouille, S.; Xu, J.; Derynck, R. TACE-mediated ectodomain shedding of the type I TGF-beta receptor downregulates TGF-beta signaling. Mol. Cell 2009, 35, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopan, R. Notch signaling. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Regan, J.; Long, F. Notch signaling and bone remodeling. Curr. Osteoporos. Rep. 2013, 11, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Pereira, R.M.; Delany, A.M.; Durant, D.; Canalis, E. Cortisol regulates the expression of Notch in osteoblasts. J. Cell. Biochem. 2002, 85, 252–258. [Google Scholar] [CrossRef]
- Ashley, J.W.; Ahn, J.; Hankenson, K.D. Notch signaling promotes osteoclast maturation and resorptive activity. J. Cell. Biochem. 2015, 116, 2598–2609. [Google Scholar] [CrossRef] [Green Version]
- Sekine, C.; Koyanagi, A.; Koyama, N.; Hozumi, K.; Chiba, S.; Yagita, H. Differential regulation of osteoclastogenesis by Notch2/Delta-like 1 and Notch1/Jagged1 axes. Arthritis Res. Ther. 2012, 14, R45. [Google Scholar] [CrossRef] [Green Version]
- Tezuka, K.; Yasuda, M.; Watanabe, N.; Morimura, N.; Kuroda, K.; Miyatani, S.; Hozumi, N. Stimulation of osteoblastic cell differentiation by Notch. J. Bone Miner. Res. 2002, 17, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Yamazaki, H.; Yamane, T.; Yoshino, M.; Okuyama, H.; Tsuneto, M.; Kurino, T.; Hayashi, S.; Sakano, S. Regulation of osteoclast development by Notch signaling directed to osteoclast precursors and through stromal cells. Blood 2003, 101, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zou, L. Notch-1 inhibition reduces proliferation and promotes osteogenic differentiation of bone marrow mesenchymal stem cells. Exp. Ther. Med. 2019, 18, 1884–1890. [Google Scholar] [CrossRef]
- AlMuraikhi, N.; Ali, D.; Vishnubalaji, R.; Manikandan, M.; Atteya, M.; Siyal, A.; Alfayez, M.; Aldahmash, A.; Kassem, M.; Alajez, N.M. Notch Signaling Inhibition by LY411575 Attenuates Osteoblast Differentiation and Decreased Ectopic Bone Formation Capacity of Human Skeletal (Mesenchymal) Stem Cells. Stem Cells Int. 2019, 2019, 3041262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, P.N.; Moharrer, Y.; Hebb, J.H.; Egol, A.J.; Kaur, G.; Hankenson, K.D.; Ahn, J.; Ashley, J.W. Suppression of Notch Signaling in Osteoclasts Improves Bone Regeneration and Healing. J. Orthop. Res. 2019, 37, 2089–2103. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wei, Y.; Lian, J.; Yang, L.; Zhang, X.; Xie, J.; Liu, Q.; Luo, J.; He, B.; Tang, M. Notch signaling pathway promotes osteogenic differentiation of mesenchymal stem cells by enhancing BMP9/Smad signaling. Int. J. Mol. Med. 2017, 40, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Khorsand, B.; Elangovan, S.; Hong, L.; Dewerth, A.; Kormann, M.S.; Salem, A.K. A Comparative Study of the Bone Regenerative Effect of Chemically Modified RNA Encoding BMP-2 or BMP-9. AAPS J. 2017, 19, 438–446. [Google Scholar] [CrossRef]
- Alinejad, Y.; Lauzon, M.A.; Grenier, G.; Balg, F.; Faucheux, N. Both Human Hematoma Punctured from Pelvic Fractures and Serum Increase Muscle Resident Stem Cells Response to BMP9: A Multivariate Statistical Approach. J. Clin. Med. 2020, 9, 1175. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Wu, G.; Zhao, J.; Wang, L.; Sun, P.; Gu, Z. rhBMP2/7 heterodimer: An osteoblastogenesis inducer of not higher potency but lower effective concentration compared with rhBMP2 and rhBMP7 homodimers. Tissue Eng. Part A 2010, 16, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Loozen, L.D.; Vandersteen, A.; Kragten, A.H.; Oner, F.C.; Dhert, W.J.; Kruyt, M.C.; Alblas, J. Bone formation by heterodimers through non-viral gene delivery of BMP-2/6 and BMP-2/7. Eur. Cell Mater. 2018, 35, 195–208. [Google Scholar] [CrossRef]
- Kaito, T.; Morimoto, T.; Mori, Y.; Kanayama, S.; Makino, T.; Takenaka, S.; Sakai, Y.; Otsuru, S.; Yoshioka, Y.; Yoshikawa, H. BMP-2/7 heterodimer strongly induces bone regeneration in the absence of increased soft tissue inflammation. Spine J. 2018, 18, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Qin, D.; Cao, P.; Lu, P.; Xia, Y.; Li, M.; Sun, M.; Zhang, W.; Yang, F.; Zhang, Y.; et al. BMP2/7 heterodimer enhances osteogenic differentiation of rat BMSCs via ERK signaling compared with respective homodimers. J. Cell Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, S.; Liu, Y.; Zhang, P.; Ge, Y.; Guo, J.; Wu, G.; Zhou, Y. Heterodimeric BMP-2/7 exhibits different osteoinductive effects in human and murine cells. Growth Factors 2018, 36, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Oh, Y.; Jo, S.; Kim, T.H.; Ji, J.D. A dual role of TGF-beta in human osteoclast differentiation mediated by Smad1 versus Smad3 signaling. Immunol. Lett. 2019, 206, 33–40. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, L.; Zhang, X.; Zhang, X.; Gu, Z.; Wu, G. BMP2/7 heterodimer can modulate all cellular events of the in vitro RANKL-mediated osteoclastogenesis, respectively, in different dose patterns. Tissue Eng. Part A 2012, 18, 621–630. [Google Scholar] [CrossRef]
- Yan, T.; Riggs, B.L.; Boyle, W.J.; Khosla, S. Regulation of osteoclastogenesis and RANK expression by TGF-beta1. J. Cell. Biochem. 2001, 83, 320–325. [Google Scholar] [CrossRef]
- Quinn, J.M.; Itoh, K.; Udagawa, N.; Hausler, K.; Yasuda, H.; Shima, N.; Mizuno, A.; Higashio, K.; Takahashi, N.; Suda, T.; et al. Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J. Bone Miner. Res. 2001, 16, 1787–1794. [Google Scholar] [CrossRef]
- Karst, M.; Gorny, G.; Galvin, R.J.; Oursler, M.J. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulation of osteoclast differentiation. J. Cell. Physiol. 2004, 200, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houde, N.; Chamoux, E.; Bisson, M.; Roux, S. Transforming growth factor-beta1 (TGF-beta1) induces human osteoclast apoptosis by up-regulating Bim. J. Biol. Chem. 2009, 284, 23397–23404. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.D.; Pham, L.; Billington, C.J., Jr.; Espe, K.; Carlson, A.E.; Westendorf, J.J.; Petryk, A.; Gopalakrishnan, R.; Mansky, K. Bone morphogenic protein 2 directly enhances differentiation of murine osteoclast precursors. J. Cell. Biochem. 2010, 109, 672–682. [Google Scholar] [CrossRef] [Green Version]
- Dankbar, B.; Fennen, M.; Brunert, D.; Hayer, S.; Frank, S.; Wehmeyer, C.; Beckmann, D.; Paruzel, P.; Bertrand, J.; Redlich, K.; et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat. Med. 2015, 21, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Udagawa, N.; Katagiri, T.; Iemura, S.; Ueno, N.; Yasuda, H.; Higashio, K.; Quinn, J.M.; Gillespie, M.T.; Martin, T.J.; et al. Bone morphogenetic protein 2 stimulates osteoclast differentiation and survival supported by receptor activator of nuclear factor-kappaB ligand. Endocrinology 2001, 142, 3656–3662. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.J.; Gerstenfeld, L.C.; Einhorn, T.A. Differential temporal expression of members of the transforming growth factor beta superfamily during murine fracture healing. J. Bone Miner. Res. 2002, 17, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.A., Jr.; Meyer, M.H.; Tenholder, M.; Wondracek, S.; Wasserman, R.; Garges, P. Gene Expression in Older Rats with Delayed Union of Femoral Fractures. JBJS 2003, 85, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, J.A.; Keane, O.; Sutton Lin, S.; O’Connor, J.P. BMP-2 modulates expression of other growth factors in a rat fracture healing model. J. Appl. Biomed. 2014, 12, 127–135. [Google Scholar] [CrossRef]
- Murata, M.; Huang, B.-Z.; Shibata, T.; Imai, S.; Nagal, N.; Arisue, M. Bone augmentation by recombinant human BMP-2 and collagen on adult rat parietal bone. Int. J. Oral Maxillofac. Surg. 1999, 28, 232–237. [Google Scholar] [CrossRef]
- Antebi, Y.E.; Linton, J.M.; Klumpe, H.; Bintu, B.; Gong, M.; Su, C.; McCardell, R.; Elowitz, M.B. Combinatorial Signal Perception in the BMP Pathway. Cell 2017, 170, 1184–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasi Ghaemi, S.; Delalat, B.; Ceto, X.; Harding, F.J.; Tuke, J.; Voelcker, N.H. Synergistic influence of collagen I and BMP 2 drives osteogenic differentiation of mesenchymal stem cells: A cell microarray analysis. Acta Biomater. 2016, 34, 41–52. [Google Scholar] [CrossRef]
- Sarahrudi, K.; Thomas, A.; Mousavi, M.; Kaiser, G.; Kottstorfer, J.; Kecht, M.; Hajdu, S.; Aharinejad, S. Elevated transforming growth factor-beta 1 (TGF-beta1) levels in human fracture healing. Injury 2011, 42, 833–837. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, G.; Henle, P.; Kusswetter, M.; Moghaddam, A.; Wentzensen, A.; Richter, W.; Weiss, S. TGF-beta1 as a marker of delayed fracture healing. Bone 2005, 36, 779–785. [Google Scholar] [CrossRef]
- Onishi, T.; Ishidou, Y.; Nagamine, T.; Yone, K.; Imamura, T.; Kato, M.; Sampath, T.K.; ten Dijke, P.; Sakou, T. Distinct and Overlapping Patterns of Localization of Bone Morphogenetic Protein (BMP) Family Members and a BMP Type II Receptor During Fracture Healing in Rats. Bone 1998, 22, 605–612. [Google Scholar] [CrossRef]
- Kloen, P.; Di Paola, M.; Borens, O.; Richmond, J.; Perino, G.; Helfet, D.L.; Goumans, M.J. BMP signaling components are expressed in human fracture callus. Bone 2003, 33, 362–371. [Google Scholar] [CrossRef]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 2008, 1782, 197–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeman, E. Pathogenesis of bone fragility in women and men. Lancet 2002, 359, 1841–1850. [Google Scholar] [CrossRef]
- Hughes, D.E.; Dai, A.; Tiffee, J.C.; Li, H.H.; Mundy, G.R.; Boyce, B.F. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-b. Nat. Med. 1996, 2, 1132–1136. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Boyle, W.J.; Penninger, J.M. Osteoprotegerin ligand: A regulator of immune responses and bone physiology. Immunol. Today 2000, 21, 495–502. [Google Scholar] [CrossRef]
- Walsh, M.C.; Choi, Y. Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef] [Green Version]
- Janssens, K.; ten Dijke, P.; Janssens, S.; Van Hul, W. Transforming growth factor-beta1 to the bone. Endocr. Rev. 2005, 26, 743–774. [Google Scholar] [CrossRef] [Green Version]
- Ralston, S.H.; de Crombrugghe, B. Genetic regulation of bone mass and susceptibility to osteoporosis. Genes Dev. 2006, 20, 2492–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grafe, I.; Yang, T.; Alexander, S.; Homan, E.P.; Lietman, C.; Jiang, M.M.; Bertin, T.; Munivez, E.; Chen, Y.; Dawson, B.; et al. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 2014, 20, 670–675. [Google Scholar] [CrossRef] [Green Version]
- Tauer, J.T.; Abdullah, S.; Rauch, F. Effect of Anti-TGF-beta Treatment in a Mouse Model of Severe Osteogenesis Imperfecta. J. Bone Miner. Res. 2019, 34, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijs, J.T.; Stayrook, K.R.; Guise, T.A. The role of TGF-beta in bone metastasis: Novel therapeutic perspectives. Bonekey Rep. 2012, 1, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clezardin, P. Pathophysiology of bone metastases from solid malignancies. Joint Bone Spine 2017, 84, 677–684. [Google Scholar] [CrossRef]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef] [Green Version]
- Roux, S. Myeloma bone disease: Pathophysiology. Rev. Rhum. 2017, 84, 181–186. [Google Scholar]
- Matsumoto, T.; Abe, M. TGF-beta-related mechanisms of bone destruction in multiple myeloma. Bone 2011, 48, 129–134. [Google Scholar] [CrossRef]
- Lu, A.; Pallero, M.A.; Lei, W.; Hong, H.; Yang, Y.; Suto, M.J.; Murphy-Ullrich, J.E. Inhibition of Transforming Growth Factor-beta Activation Diminishes Tumor Progression and Osteolytic Bone Disease in Mouse Models of Multiple Myeloma. Am. J. Pathol. 2016, 186, 678–690. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Kastritis, E.; Christoulas, D.; Gkotzamanidou, M.; Eleutherakis-Papaiakovou, E.; Kanellias, N.; Papatheodorou, A.; Dimopoulos, M.A. Circulating activin-A is elevated in patients with advanced multiple myeloma and correlates with extensive bone involvement and inferior survival; no alterations post-lenalidomide and dexamethasone therapy. Ann. Oncol. 2012, 23, 2681–2686. [Google Scholar] [CrossRef]
- Vallet, S.; Mukherjee, S.; Vaghela, N.; Hideshima, T.; Fulciniti, M.; Pozzi, S.; Santo, L.; Cirstea, D.; Patel, K.; Sohani, A.R.; et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc. Natl. Acad. Sci. USA 2010, 107, 5124–5129. [Google Scholar] [CrossRef] [Green Version]
- Ruckle, J.; Jacobs, M.; Kramer, W.; Pearsall, A.E.; Kumar, R.; Underwood, K.W.; Seehra, J.; Yang, Y.; Condon, C.H.; Sherman, M.L. Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J. Bone Miner. Res. 2009, 24, 744–752. [Google Scholar] [CrossRef]
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, E. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Demirhan, O.; Turkmen, S.; Schwabe, G.C.; Soyupak, S.; Akgul, E.; Tastemir, D.; Karahan, D.; Mundlos, S.; Lehmann, K. A homozygous BMPR1B mutation causes a new subtype of acromesomelic chondrodysplasia with genital anomalies. J. Med. Genet. 2005, 42, 314–317. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, L.; Stange, K.; Deichsel, A.; Gossen, M.; Seemann, P. The Fibrodysplasia Ossificans Progressiva (FOP) mutation p.R206H in ACVR1 confers an altered ligand response. Cell Signal. 2017, 29, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, F.S.; Al Mukaddam, M.; Stanley, A.; Towler, O.W.; Shore, E.M. Fibrodysplasia ossificans progressiva (FOP): A disorder of osteochondrogenesis. Bone 2020, 140, 115539. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, T.; Kohda, M.; Kanomata, K.; Nojima, J.; Nakamura, A.; Kamizono, J.; Noguchi, Y.; Iwakiri, K.; Kondo, T.; Kurose, J.; et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J. Biol. Chem. 2009, 284, 7149–7156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiori, J.L.; Billings, P.C.; de la Pena, L.S.; Kaplan, F.S.; Shore, E.M. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 2006, 21, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Haupt, J.; Stanley, A.; McLeod, C.M.; Cosgrove, B.D.; Culbert, A.L.; Wang, L.; Mourkioti, F.; Mauck, R.L.; Shore, E.M. ACVR1(R206H) FOP mutation alters mechanosensing and tissue stiffness during heterotopic ossification. Mol. Biol. Cell 2019, 30, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.A.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206Hreceptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.M., Jr. The new bone biology: Pathologic, molecular, and clinical correlates. Am. J. Med. Genet. A 2006, 140, 2646–2706. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Jha, S.; Ivovic, A.; Fratzl-Zelman, N.; Deng, Z.; Mitra, A.; Cabral, W.A.; Hanson, E.P.; Lange, E.; Cowen, E.W.; et al. Somatic SMAD3-activating mutations cause melorheostosis by up-regulating the TGF-beta/SMAD pathway. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Fratzl-Zelman, N.; Roschger, P.; Papadakis, G.Z.; Cowen, E.W.; Kang, H.; Lehky, T.J.; Alter, K.; Deng, Z.; Ivovic, A.; et al. Distinct Clinical and Pathological Features of Melorheostosis Associated With Somatic MAP2K1 Mutations. J. Bone Miner. Res. 2019, 34, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Wozney, J.M. Overview of bone morphogenetic proteins. Spine 2002, 27, S2–S8. [Google Scholar] [CrossRef]
- Maruoka, Y.; Oida, S.; Iimura, T.; Takeda, K.; Asahina, I.; Enomoto, S.; Sasaki, S. Production of functional human bone morphogenetic protein-2 using a baculovirus/Sf-9 insect cell system. Biochem. Mol. Biol. Int. 1995, 35, 957–963. [Google Scholar]
- Karyagina, A.S.; Boksha, I.S.; Grunina, T.M.; Demidenko, A.V.; Poponova, M.S.; Sergienko, O.V.; Lyashchuk, A.M.; Galushkina, Z.M.; Soboleva, L.A.; Osidak, E.O.; et al. Two Variants of Recombinant Human Bone Morphogenetic Protein-2 (rhBMP-2) with Additional Protein Domains: Synthesis in an Escherichia coli Heterologous Expression System. Biochemistry 2017, 82, 613–624. [Google Scholar] [CrossRef]
- Bustos-Valenzuela, J.C.; Halcsik, E.; Bassi, E.J.; Demasi, M.A.; Granjeiro, J.M.; Sogayar, M.C. Expression, purification, bioactivity, and partial characterization of a recombinant human bone morphogenetic protein-7 produced in human 293T cells. Mol. Biotechnol. 2010, 46, 118–126. [Google Scholar] [CrossRef]
- Brooks, S.A. Appropriate Glycosylation of Recombinant Proteins for Human Use: Implications of Choice of Expression System. Mol. Biotechnol. 2004, 28, 241–256. [Google Scholar] [CrossRef]
- Sola, R.J.; Griebenow, K. Glycosylation of therapeutic proteins: An effective strategy to optimize efficacy. BioDrugs 2010, 24, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Carreira, A.C.; Lojudice, F.H.; Halcsik, E.; Navarro, R.D.; Sogayar, M.C.; Granjeiro, J.M. Bone morphogenetic proteins: Facts, challenges, and future perspectives. J. Dent. Res. 2014, 93, 335–345. [Google Scholar] [CrossRef]
- Ratko, T.A.; Belinson, S.E.; Samson, D.J.; Bonnell, C.; Ziegler, K.M.; Aronson, N. AHRQ Technology Assessments. In Bone Morphogenetic Protein: The State of the Evidence of On-Label and Off-Label Use; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2010. [Google Scholar]
- Medtronic Sofamor Danek USA Inc. INFUSE Bone Graft Product Information: Lumbar; FDA gov: White Oak, MD, USA, 2002.
- Medtronic Sofamor Danek USA Inc. INFUSE Bone Graft Product Information: Tibial; FDA gov: White Oak, MD, USA, 2004.
- Medtronic Sofamor Danek USA Inc. INFUSE Bone Graft Product Information: Oral/Facial; FDA gov: White Oak, MD, USA, 2006.
- Ong, K.L.; Villarraga, M.L.; Lau, E.; Carreon, L.Y.; Kurtz, S.M.; Glassman, S.D. Off-label use of bone morphogenetic proteins in the United States using administrative data. Spine 2010, 35, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.R.; Anderson, D.G.; Patel, T.; Fischgrund, J.; Truumees, E.; Herkowitz, H.N.; Phillips, F.; Hilibrand, A.; Albert, T.J.; Wetzel, T.; et al. Comparison of OP-1 Putty (rhBMP-7) to iliac crest autograft for posterolateral lumbar arthrodesis: A minimum 2-year follow-up pilot study. Spine 2005, 30, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Faundez, A.; Tournier, C.; Garcia, M.; Aunoble, S.; Le Huec, J.C. Bone morphogenetic protein use in spine surgery-complications and outcomes: A systematic review. Int. Orthop. 2016, 40, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zeng, Z.; Wang, J.; Liu, H.; Wang, H.; Zheng, Z. Comparison of the use of rhBMP-7 versus iliac crest autograft in single-level lumbar fusion: A meta-analysis of randomized controlled trials. J. Bone Miner. Metab. 2018, 36, 119–127. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Y.; Liang, Z.; Zhou, M.; Chen, C. Comparative Clinical Effectiveness and Safety of Bone Morphogenetic Protein Versus Autologous Iliac Crest Bone Graft in Lumbar Fusion: A Meta-analysis and Systematic Review. Spine 2020, 45, E729–E741. [Google Scholar] [CrossRef]
- Dai, J.; Li, L.; Jiang, C.; Wang, C.; Chen, H.; Chai, Y. Bone Morphogenetic Protein for the Healing of Tibial Fracture: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2015, 10, e0141670. [Google Scholar] [CrossRef] [Green Version]
- Cahill, K.S.; Chi, J.H.; Day, A.; Claus, E.B. Prevalence, complications, and hospital charges associated with use of bone-morphogenetic proteins in spinal fusion procedures. JAMA 2009, 302, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.J.; Smith, J.S.; Fu, K.M.; Hamilton, D.K.; Polly, D.W., Jr.; Ames, C.P.; Berven, S.H.; Perra, J.H.; Knapp, D.R., Jr.; McCarthy, R.E.; et al. Does bone morphogenetic protein increase the incidence of perioperative complications in spinal fusion? A comparison of 55,862 cases of spinal fusion with and without bone morphogenetic protein. Spine 2011, 36, 1685–1691. [Google Scholar] [CrossRef]
- Fu, R.; Selph, S.; McDonagh, M.; Peterson, K.; Tiwari, A.; Chou, R.; Helfand, M. Effectiveness and harms of recombinant human bone morphogenetic protein-2 in spine fusion: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 158, 890–902. [Google Scholar] [CrossRef] [Green Version]
- Carragee, E.J.; Hurwitz, E.L.; Weiner, B.K. A critical review of recombinant human bone morphogenetic protein-2 trials in spinal surgery: Emerging safety concerns and lessons learned. Spine J. 2011, 11, 471–491. [Google Scholar] [CrossRef]
- James, A.W.; LaChaud, G.; Shen, J.; Asatrian, G.; Nguyen, V.; Zhang, X.; Ting, K.; Soo, C. A Review of the Clinical Side Effects of Bone Morphogenetic Protein-2. Tissue Eng. Part B Rev. 2016, 22, 284–297. [Google Scholar] [CrossRef]
- Vaidya, R.; Weir, R.; Sethi, A.; Meisterling, S.; Hakeos, W.; Wybo, C.D. Interbody fusion with allograft and rhBMP-2 leads to consistent fusion but early subsidence. J. Bone Joint Surg. Br. 2007, 89, 342–345. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Aruwajoye, O.; Du, J.; Kamiya, N. Local administration of bone morphogenetic protein-2 and bisphosphonate during non-weight-bearing treatment of ischemic osteonecrosis of the femoral head: An experimental investigation in immature pigs. J. Bone Joint Surg. Am. 2014, 96, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Dumanian, Z.P.; Tollemar, V.; Ye, J.; Lu, M.; Zhu, Y.; Liao, J.; Ameer, G.A.; He, T.C.; Reid, R.R. Repair of critical sized cranial defects with BMP9-transduced calvarial cells delivered in a thermoresponsive scaffold. PLoS ONE 2017, 12, e0172327. [Google Scholar] [CrossRef]
- Vhora, I.; Lalani, R.; Bhatt, P.; Patil, S.; Misra, A. Lipid-nucleic acid nanoparticles of novel ionizable lipids for systemic BMP-9 gene delivery to bone-marrow mesenchymal stem cells for osteoinduction. Int. J. Pharm. 2019, 563, 324–336. [Google Scholar] [CrossRef]
- Chiari, C.; Grgurevic, L.; Bordukalo-Niksic, T.; Oppermann, H.; Valentinitsch, A.; Nemecek, E.; Staats, K.; Schreiner, M.; Trost, C.; Kolb, A.; et al. Recombinant Human BMP6 Applied Within Autologous Blood Coagulum Accelerates Bone Healing: Randomized Controlled Trial in High Tibial Osteotomy Patients. J. Bone Miner. Res. 2020. [Google Scholar] [CrossRef]
- Krishnakumar, G.S.; Roffi, A.; Reale, D.; Kon, E.; Filardo, G. Clinical application of bone morphogenetic proteins for bone healing: A systematic review. Int. Orthop. 2017, 41, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, M.C.; Brown, J.V.; Heirs, M.K.; Higgins, J.P.; Mannion, R.J.; Rodgers, M.A.; Stewart, L.A. Safety and effectiveness of recombinant human bone morphogenetic protein-2 for spinal fusion: A meta-analysis of individual-participant data. Ann. Intern. Med. 2013, 158, 877–889. [Google Scholar] [CrossRef]
- Latzman, J.M.; Kong, L.; Liu, C.; Samadani, U. Administration of human recombinant bone morphogenetic protein-2 for spine fusion may be associated with transient postoperative renal insufficiency. Spine 2010, 35, E231–E237. [Google Scholar] [CrossRef]
- Devine, J.G.; Dettori, J.R.; France, J.C.; Brodt, E.; McGuire, R.A. The use of rhBMP in spine surgery: Is there a cancer risk? Evid. Based Spine Care J. 2012, 3, 35–41. [Google Scholar] [CrossRef] [Green Version]

| Type I Receptor | Type II Receptor | TGF-β ligands | Signaling Pathway Activation in Osteoclast Precursors and Mature Osteoclasts | Signaling Pathway Activation in Stem Cells and Osteoblast |
|---|---|---|---|---|
| TGF-β/Nodal/Activin family | ||||
| TβRI (ALK5); ALK1 | TβRII | TGF-β1 | ↑ pSmad2/3 (human M2 monocyte-derived macrophages; 10 ng/mL) [176]; ↑ Smad1/5 (human M2 monocyte-derived macrophages; concentration is not specified) [174]; ↑ Wnt10b and crosstalk between Smad2/3 and canonical Wnt signaling (murine osteoclasts; 2 ng/mL) [177] | ↑ pSmad2/3 (L6E9 myoblasts; < 0.01 ng/mL) [178]; ↑ pSmad1/5 (L6E9 myoblasts; <1 ng/mL) [178]; Crosstalk with Akt (early phase of osteoblast differentiation MC3T3-E1; 0.1 ng/mL) [179]; MAPK: ↑ pERK1/2, ↑pp38, ↑pJNK (MC3T3-E1; 2.5 ng/mL) [180]; Canonical Wnt: ↑ β-catenin via ALK5, Smad3 receptor and PI3K (hMSC; 1 ng/mL) [181] |
| TGF-β3 | N.A. | ↑ pSmad2/3 (mouse embryonic palatal mesenchymal cells; 10 ng/mL) [182]; MAPK: ↑ pERK1/2 (human mesenchymal stem cells; 10 ng/mL) [183] | ||
| ActRIb (ALK4) | ActRIIA; ActRIIB | Activin A | ↑ c-fos (murine macrophages RAW264.7; 50 ng/mL of activin A with 40 ng/mL of RANKL) [184]; ↑ pSmad2/3 (RAW264.7; 50 ng/mL) [184]; ↑ pSmad2/3 (murine bone marrow macrophages; 100 ng/mL) [185]; MAPK: ↑ pp38 and ↑ pERK1/2 (murine bone marrow macrophages; 100 ng/mL of activin A with 50 ng/mL of M-CSF) [185]. | ↑ pSmad2/3 (human endometrial stromal cells; <20 ng/mL) [186] |
| BMP/GDF family | ||||
| BMPRIA (ALK3); BMPRIB (ALK6); ActRI | BMPRII; ActRIIA; ActRIIB | BMP-2 | ↑ Smad1/5/9 (murine bone marrow mononuclear cells; 100 ng/mL) [59]; MAPK: ↑ pERK and ↑ pp38 (minimal) (murine bone marrow mononuclear cells; 100 ng/mL) [59]; ↑ pAkt (murine bone marrow mononuclear cells; 100 ng/mL) [59]; ↑ pSmad1/5 (osteoclasts precursor fusion; 30 ng/mL) [187]; MAPK: ↑ pp38 (osteoclasts precursor fusion; 30 ng/mL) [187] | ↑ pSmad1/5 (C2C12 cells; 100 ng/mL) [188]; ↑ pSmad1/5 (human stem cells from the apical papilla; 100 ng/mL) [189]; ↑ pERK1/2 (human stem cells from the apical papilla; 100 ng/mL) [189]; ↑ pSmad1/5 (MC3T3-E1 preosteoblasts; 0.38 nM) [190,191]; MAPK: ↑ pERK1/2, ↑ pp38 (MC3T3-E1 preosteoblasts; 0.38 nM) [190,191]; ↑ pAkt (MC3T3-E1 cells; < 0.1 nM) [192]. |
| BMP-4 | N.A. | ↑ pSmad1/5 (C2C12 cells; 200 ng/mL) [193] | ||
| BMPRIA (ALK3); BMPRIB (ALK6); ALK2; ALK1 | BMPRII; ActRIIA; ActRIIB | BMP-5 | N.A. | ↑ pSmad1/5 (human embryonic stem cells; 100 ng/mL) [194]; MAPK: ↑ pp38 (human embryonic stem cells; 100 ng/mL) [194]; MAPK: ↓ pp38 (murine osteoblasts—differentiated MC3T3-E1 cells; BMP-5 siRNA 40 nmol/mL) [195] |
| BMP-6 | ↑ pSmad1/5 (rat and human granulosa cells; 100 ng/mL) [196]. | ↑ pSmad1/5 (C2C12 cells; 200 ng/mL) [193]; ↑ pSmad1/5 (human embryonic stem cells; 100 ng/mL) [194]; ↑ pSmad1/5 (MC3T3-E1 cells; 300 ng/mL) [175]. | ||
| BMP-7 | ↑ pSmad1/5/9 (murine bone marrow mononuclear cells; 100 ng/mL) [59]; MAPK: ↑ pp38 (murine bone marrow mononuclear cells; 100 ng/mL) [59]; ↑ pSmad1/5 (rat and human granulosa cells; 100 ng/mL) [196]. | ↑ pSmad1/5 (C2C12 cells; 1000 ng/mL) [193]; ↑ pSmad1/5 (human embryonic stem cells; 100 ng/mL) [194]; MAPK: ↑ pp38 (human embryonic stem cells; 100 ng/mL) [194]. | ||
| ALK1 ALK2 | BMPRII; ActRIIA; ActRIIB | BMP-9 (GDF-2) | ↑ pSmad1/5 (human cord blood monocyte as osteoclast precursor; 150 ng/mL) [171]; MAPK: ↑ pERK1/2 (human cord blood monocyte as osteoclast precursor; 150 ng/mL) [171] | ↑ pSmad1/5 (MC3T3-E1 cells; 0.38 nM) [190,191]; MAPK: ↓ pERK1/2, ↑ pp38, ↑ pJNK (MC3T3-E1 cells; 0.38 nM) [190,191]; ↑ pAkt (MC3T3-E1 cells; < 0.1 nM) [192]; ↑ pSmad1/5 (murine multipotent stem C3H10T1/2 cells; 10 ng/mL) [197]; MAPK: ↑ pp38 (C3H10T1/2 cells; 10 ng/mL) [197]; |
| BMP-10 | N.A. | ↑ pSmad1/5 (human embryonic stem cells; 100 ng/mL) [194]; MAPK: ↑ pp38 (human embryonic stem cells; 100 ng/mL) [194]. | ||
| BMPRIA (ALK3); BMPRIB (ALK6) | BMPRII | BMP-15 | ↑ pSmad1/5 (immortalized human granulosa cells and human granulosa cell tumor cells; 100 ng/mL) [198]; ↑ pSmad1/5 (rat and human granulosa cells; 100 ng/mL) [196]. | N.A. |
| BMPRIA (ALK3); BMPRIB (ALK6) | BMPRII; ActRIIA; ActRIIB | GDF-5/-6/-7 | N.A. | MAPK: ↑ pp38 and ↑ pERK1/2 (chondrogenic mouse carcinoma cell line ATDC5; <10 ng/mL and 1000 ng/mL, respectively) [199]; ↑pSmad1/5 (C3H10T1/2 cells; Ad-GDF6) [200]; MAPK: ↑ pp38 (C3H10T1/2 cells; Ad-GDF6) [200]. |
| ActRIb (ALK4) | ActRIIA; ActRIIB | GDF-8(myostatin)/-11 | ↑ pSmad2/3 (human hepatocellular carcinoma; Ad-GDF11) [201] | N.A. |
| ActRIb (ALK4) | ActRIIA; ActRIIB | GDF-10/BMP-3 | N.A. | ↑ pSmad 2/3 (murine C2C12 cells; 100 ng/mL) [202]. |
Publisher‘s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jann, J.; Gascon, S.; Roux, S.; Faucheux, N. Influence of the TGF-β Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions. Int. J. Mol. Sci. 2020, 21, 7597. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207597
Jann J, Gascon S, Roux S, Faucheux N. Influence of the TGF-β Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions. International Journal of Molecular Sciences. 2020; 21(20):7597. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207597
Chicago/Turabian StyleJann, Jessica, Suzanne Gascon, Sophie Roux, and Nathalie Faucheux. 2020. "Influence of the TGF-β Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions" International Journal of Molecular Sciences 21, no. 20: 7597. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207597