Autophagy Activation Protects Ocular Surface from Inflammation in a Dry Eye Model In Vitro

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
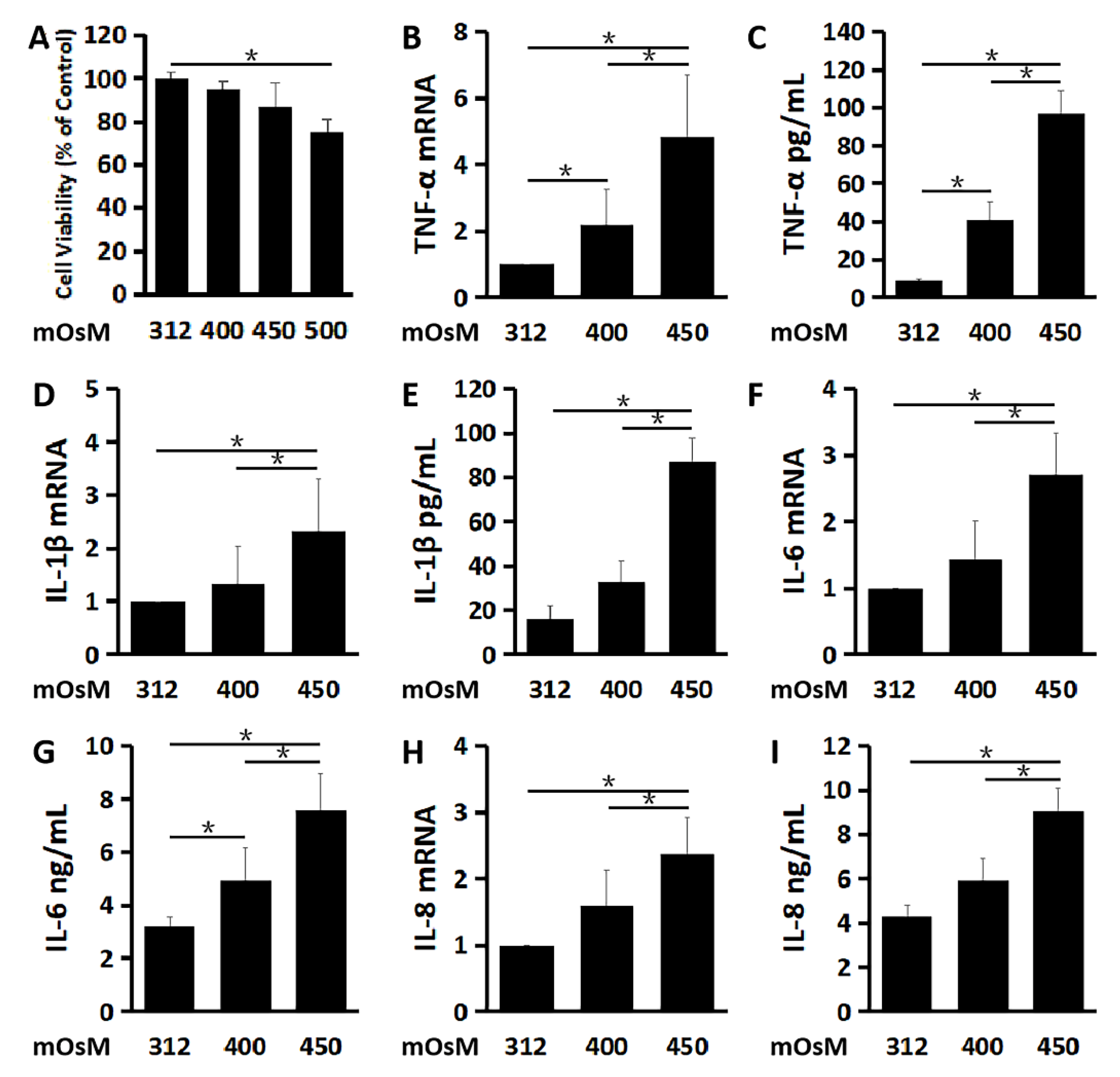
2.1. Inflammatory Mediators Are Largely Stimulatd in Primary HCECs under Hyperosmotic Stress, an In Vitro Dry Eye Model
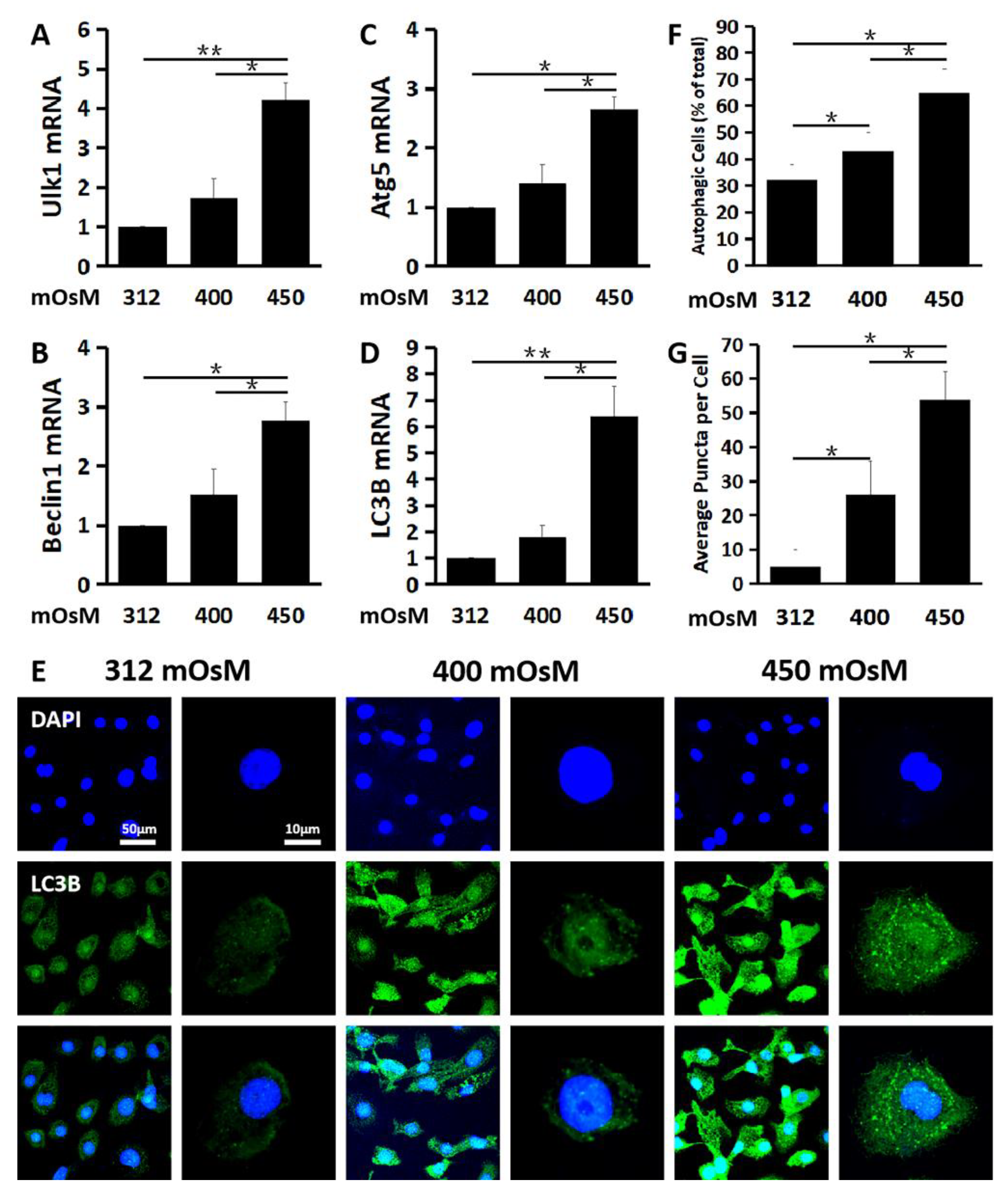
2.2. Hyperosmolarity Induces Autophagosome Formation and Promotes Autophagic Flux in Primary HCECs
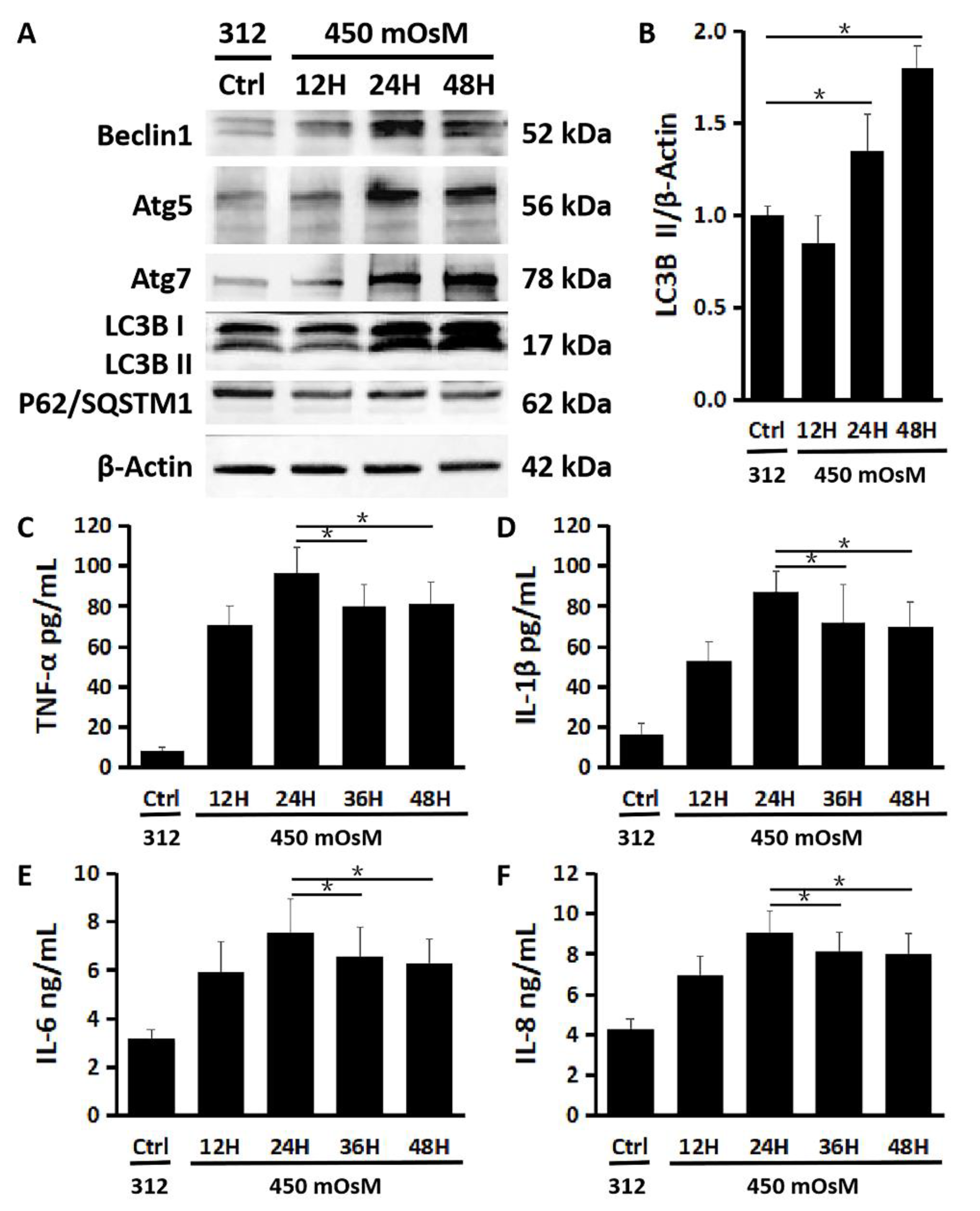
2.3. Autophagy Activation Is a Late Response to Hyperosmolarity after Inflammation in HCECs
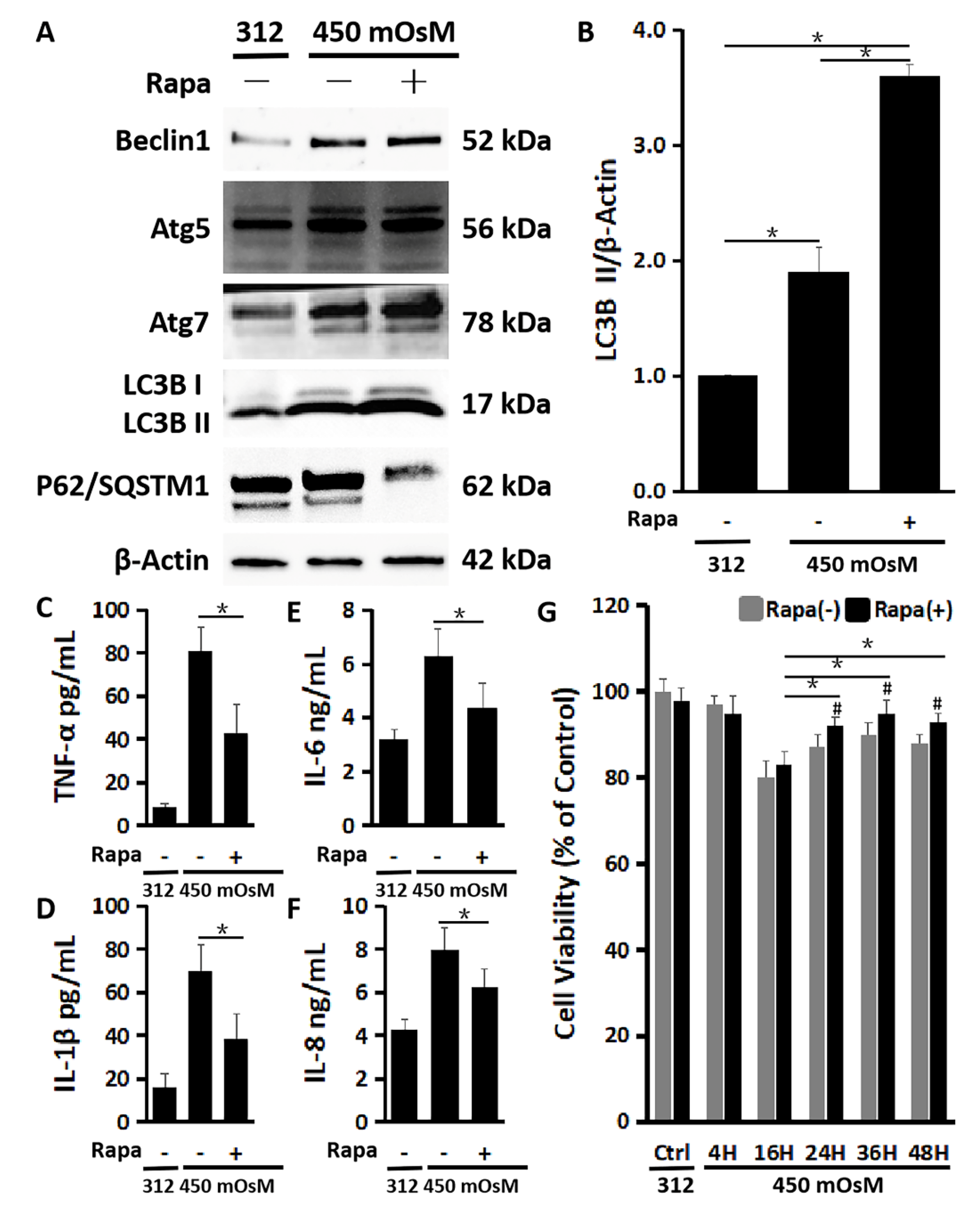
2.4. The Enhanced Autophagy Activation by Rapamycin Suppresses Inflammation and Restores the Cell Viability Damaged by Hyperosmolarity
3. Discussion
4. Materials and Methods
4.1. Cultures of Primary HCECs and Dry Eye Model In Vitro
4.2. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
4.3. Enzyme-Linked Immunosorbent Assay (ELISA)
4.4. Western Blot Analysis
4.5. Immunofluorescent Staining
4.6. MTT Assay
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCECs | human corneal epithelial cells |
| IL | interleukin |
| MMPs | matrix metalloproteinases |
| RAPA | rapamycin |
| SHEM | supplemented hormonal epidermal medium |
| NaCl | sodium chloride |
References
- Stapleton, F.; Alves, M.; Bunya, V.Y.; Jalbert, I.; Lekhanont, K.; Malet, F.; Na, K.S.; Schaumberg, D.; Uchino, M.; Vehof, J.; et al. TFOS DEWS II Epidemiology Report. Ocul. Surf. 2017, 15, 334–365. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.P.; Nichols, K.K.; Akpek, E.K.; Caffery, B.; Dua, H.S.; Joo, C.K.; Liu, Z.; Nelson, J.D.; Nichols, J.J.; Tsubota, K.; et al. TFOS DEWS II Definition and Classification Report. Ocul. Surf 2017, 15, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Su, Z.; Zhang, Z.; Lin, J.; Li, D.Q.; Pflugfelder, S.C. Effects of azithromycin on gene expression profiles of proinflammatory and anti-inflammatory mediators in the eyelid margin and conjunctiva of patients with meibomian gland disease. JAMA Ophthalmol. 2015, 133, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Chi, W.; Hua, X.; Chen, X.; Bian, F.; Yuan, X.; Zhang, L.; Wang, X.; Chen, D.; Deng, R.; Li, Z.; et al. Mitochondrial DNA oxidation induces imbalanced activity of NLRP3/NLRP6 inflammasomes by activation of caspase-8 and BRCC36 in dry eye. J. Autoimmun. 2017, 80, 65–76. [Google Scholar] [CrossRef]
- Wolffsohn, J.S.; Arita, R.; Chalmers, R.; Djalilian, A.; Dogru, M.; Dumbleton, K.; Gupta, P.K.; Karpecki, P.; Lazreg, S.; Pult, H.; et al. TFOS DEWS II Diagnostic Methodology report. Ocul Surf 2017, 15, 539–574. [Google Scholar] [CrossRef] [PubMed]
- Pflugfelder, S.C.; De Paiva, C.S.; Moore, Q.L.; Volpe, E.A.; Li, D.Q.; Gumus, K.; Zaheer, M.L.; Corrales, R.M. Aqueous Tear Deficiency Increases Conjunctival Interferon-gamma (IFN-gamma) Expression and Goblet Cell Loss. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7545–7550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pflugfelder, S.C. Tear dysfunction and the cornea: LXVIII Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 2011, 152, 900–909 e901. [Google Scholar] [CrossRef] [Green Version]
- Baudouin, C.; Aragona, P.; Messmer, E.M.; Tomlinson, A.; Calonge, M.; Boboridis, K.G.; Akova, Y.A.; Geerling, G.; Labetoulle, M.; Rolando, M. Role of hyperosmolarity in the pathogenesis and management of dry eye disease: Proceedings of the OCEAN group meeting. Ocul. Surf. 2013, 11, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Pflugfelder, S.C.; de Paiva, C.S. The Pathophysiology of Dry Eye Disease: What We Know and Future Directions for Research. Ophthalmology 2017, 124, S4–S13. [Google Scholar] [CrossRef]
- Luo, L.; Li, D.Q.; Corrales, R.M.; Pflugfelder, S.C. Hyperosmolar saline is a proinflammatory stress on the mouse ocular surface. Eye Contact Lens 2005, 31, 186–193. [Google Scholar] [CrossRef]
- Zhang, X.; De Paiva, C.S.; Su, Z.; Volpe, E.A.; Li, D.Q.; Pflugfelder, S.C. Topical interferon-gamma neutralization prevents conjunctival goblet cell loss in experimental murine dry eye. Exp Eye Res. 2014, 118, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zhang, X.; Li, J.; Wang, Y.; Chen, Q.; Hou, C.; Garrett, Q. Efficacy of osmoprotectants on prevention and treatment of murine dry eye. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6287–6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.Q.; Chen, Z.; Song, X.J.; Luo, L.; Pflugfelder, S.C. Stimulation of matrix metalloproteinases by hyperosmolarity via a JNK pathway in human corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4302–4311. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Q.; Luo, L.; Chen, Z.; Kim, H.S.; Song, X.J.; Pflugfelder, S.C. JNK and ERK MAP kinases mediate induction of IL-1beta, TNF-alpha and IL-8 following hyperosmolar stress in human limbal epithelial cells. Exp. Eye Res. 2006, 82, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ruzhi, D.; Hua, X.; Zhang, L.; Lu, F.; Coursey, T.G.; Pflugfelder, S.C.; Li, D.Q. Blueberry Component Pterostilbene Protects Corneal Epithelial Cells from Inflammation via Anti-oxidative Pathway. Sci. Rep. 2016, 6, 19408. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Su, Z.; Deng, R.; Lin, J.; Li, D.Q.; Pflugfelder, S.C. Effects of L-carnitine, erythritol and betaine on pro-inflammatory markers in primary human corneal epithelial cells exposed to hyperosmotic stress. Curr. Eye Res. 2015, 40, 657–667. [Google Scholar] [CrossRef]
- Deng, R.; Su, Z.; Hua, X.; Zhang, Z.; Li, D.Q.; Pflugfelder, S.C. Osmoprotectants suppress the production and activity of matrix metalloproteinases induced by hyperosmolarity in primary human corneal epithelial cells. Mol. Vis. 2014, 20, 1243–1252. [Google Scholar]
- Jones, L.; Downie, L.E.; Korb, D.; Benitez-Del-Castillo, J.M.; Dana, R.; Deng, S.X.; Dong, P.N.; Geerling, G.; Hida, R.Y.; Liu, Y.; et al. TFOS DEWS II Management and Therapy Report. Ocul. Surf. 2017, 15, 575–628. [Google Scholar] [CrossRef]
- Boya, P.; Esteban-Martinez, L.; Serrano-Puebla, A.; Gomez-Sintes, R.; Villarejo-Zori, B. Autophagy in the eye: Development, degeneration, and aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef]
- Arias, E.; Cuervo, A.M. Chaperone-mediated autophagy in protein quality control. Curr. Opin. Cell Biol. 2011, 23, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Crisan, T.O.; Plantinga, T.S.; van de Veerdonk, F.L.; Farcas, M.F.; Stoffels, M.; Kullberg, B.J.; van der Meer, J.W.; Joosten, L.A.; Netea, M.G. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS ONE 2011, 6, e18666. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.T.; Li, W.M.; Yao, Y.M. Interactions between Autophagy and Inhibitory Cytokines. Int. J. Biol. Sci. 2016, 12, 884–897. [Google Scholar] [CrossRef]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. Cell Physiol. 2016, 314, C73–C87. [Google Scholar] [CrossRef] [Green Version]
- Lippai, M.; Szatmari, Z. Autophagy-from molecular mechanisms to clinical relevance. Cell Biol. Toxicol. 2017, 33, 145–168. [Google Scholar] [CrossRef]
- Lahm, T.; Petrache, I. LC3 as a potential therapeutic target in hypoxia-induced pulmonary hypertension. Autophagy 2012, 8, 1146–1147. [Google Scholar] [CrossRef] [Green Version]
- Leung, G.K.; Ying, D.; Mak, C.C.; Chen, X.Y.; Xu, W.; Yeung, K.S.; Wong, W.L.; Chu, Y.W.; Mok, G.T.; Chau, C.S.; et al. CFTR founder mutation causes protein trafficking defects in Chinese patients with cystic fibrosis. Mol. Genet. Genom. Med. 2017, 5, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Rubin, D.T.; Feld, L.D.; Goeppinger, S.R.; Margolese, J.; Rosh, J.; Rubin, M.; Kim, S.; Rodriquez, D.M.; Wingate, L. The Crohn’s and Colitis Foundation of America Survey of Inflammatory Bowel Disease Patient Health Care Access. Inflamm. Bowel Dis. 2017, 23, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Byun, Y.S.; Lee, H.J.; Shin, S.; Chung, S.H. Elevation of autophagy markers in Sjogren syndrome dry eye. Sci. Rep. 2017, 7, 17280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Yu, Z.; Feng, S.; Chen, H.; Lu, X. Corneal autophagy and ocular surface inflammation: A new perspective in dry eye. Exp. Eye Res. 2019, 184, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Hua, X.; Li, J.; Chi, W.; Zhang, Z.; Lu, F.; Zhang, L.; Pflugfelder, S.C.; Li, D.Q. Oxidative stress markers induced by hyperosmolarity in primary human corneal epithelial cells. PLoS ONE 2015, 10, e0126561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Li, D.Q.; Pflugfelder, S.C. Hyperosmolarity-induced apoptosis in human corneal epithelial cells is mediated by cytochrome c and MAPK pathways. Cornea 2007, 26, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tong, L.; Li, Z.; Yoon, K.C.; Qi, H.; Farley, W.; Li, D.Q.; Pflugfelder, S.C. Hyperosmolarity-induced cornification of human corneal epithelial cells is regulated by JNK MAPK. Investig. Ophthalmol. Vis. Sci. 2008, 49, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.M.; Jeyabalan, N.; Tripathi, R.; Panigrahi, T.; Johnson, P.J.; Ghosh, A.; Mohan, R.R. Autophagy in corneal health and disease: A concise review. Ocul. Surf. 2019, 17, 186–197. [Google Scholar] [CrossRef]
- Seranova, E.; Ward, C.; Chipara, M.; Rosenstock, T.R.; Sarkar, S. In Vitro Screening Platforms for Identifying Autophagy Modulators in Mammalian Cells. Methods Mol. Biol. 2019, 1880, 389–428. [Google Scholar]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehdashtian, E.; Mehrzadi, S.; Yousefi, B.; Hosseinzadeh, A.; Reiter, R.J.; Safa, M.; Ghaznavi, H.; Naseripour, M. Diabetic retinopathy pathogenesis and the ameliorating effects of melatonin; involvement of autophagy, inflammation and oxidative stress. Life Sci. 2018, 193, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Pflugfelder, S.C.; Bian, F.; de Paiva, C.S. Matrix metalloproteinase-9 in the pathophysiology and diagnosis of dry eye syndrome. Met. Med. 2017, 4, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Deng, R.; Li, J.; Chi, W.; Su, Z.; Lin, J.; Pflugfelder, S.C.; Li, D.Q. Protective Effects of L-Carnitine Against Oxidative Injury by Hyperosmolarity in Human Corneal Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5503–5511. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, H.; Zeng, W.; Wei, J. Edaravone protects against hyperosmolarity-induced oxidative stress and apoptosis in primary human corneal epithelial cells. PLoS ONE 2017, 12, e0174437. [Google Scholar] [CrossRef]
- Kim, H.S.; Jun Song, X.; de Paiva, C.S.; Chen, Z.; Pflugfelder, S.C.; Li, D.Q. Phenotypic characterization of human corneal epithelial cells expanded ex vivo from limbal explant and single cell cultures. Exp. Eye Res. 2004, 79, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Li, D.Q.; Zhou, N.; Zhang, L.; Ma, P.; Pflugfelder, S.C. Suppressive effects of azithromycin on zymosan-induced production of proinflammatory mediators by human corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5623–5629. [Google Scholar] [CrossRef]
- Chen, Z.; de Paiva, C.S.; Luo, L.; Kretzer, F.L.; Pflugfelder, S.C.; Li, D.Q. Characterization of putative stem cell phenotype in human limbal epithelia. Stem Cells 2004, 22, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhou, Q.; Zhu, J.; Xiao, J.; Wan, P.; Zhou, C.; Huang, Z.; Qiang, N.; Zhang, W.; Wu, Z.; et al. Using genipin-crosslinked acellular porcine corneal stroma for cosmetic corneal lens implants. Biomaterials 2012, 33, 7336–7346. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Chen, D.; Chen, X.; Bian, F.; Gao, N.; Li, J.; Pflugfelder, S.C.; Li, D.-Q. Autophagy Activation Protects Ocular Surface from Inflammation in a Dry Eye Model In Vitro. Int. J. Mol. Sci. 2020, 21, 8966. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238966
Liu Z, Chen D, Chen X, Bian F, Gao N, Li J, Pflugfelder SC, Li D-Q. Autophagy Activation Protects Ocular Surface from Inflammation in a Dry Eye Model In Vitro. International Journal of Molecular Sciences. 2020; 21(23):8966. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238966
Chicago/Turabian StyleLiu, Zhao, Ding Chen, Xin Chen, Fang Bian, Ning Gao, Jinmiao Li, Stephen C. Pflugfelder, and De-Quan Li. 2020. "Autophagy Activation Protects Ocular Surface from Inflammation in a Dry Eye Model In Vitro" International Journal of Molecular Sciences 21, no. 23: 8966. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238966