Understanding and Treating Niemann–Pick Type C Disease: Models Matter


1
Biomedical and Technology Science Section, Department of Science, Roma Tre University, Viale Marconi 446, 00146 Rome, Italy
2
Centre National de la Recherche Scientifique, Institut des Neurosciences Cellulaires et Intégratives, Université de Strasbourg, 8 Allée General Rouvillois, 67000 Strasbourg, France
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(23), 8979; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238979
Submission received: 6 October 2020
/
Revised: 20 November 2020
/
Accepted: 23 November 2020
/
Published: 26 November 2020
(This article belongs to the Special Issue Advances in Knowledge in Niemann-Pick Disease Type C: Facts and Perspectives- 2nd Edition)
Abstract
:Biomedical research aims to understand the molecular mechanisms causing human diseases and to develop curative therapies. So far, these goals have been achieved for a small fraction of diseases, limiting factors being the availability, validity, and use of experimental models. Niemann–Pick type C (NPC) is a prime example for a disease that lacks a curative therapy despite substantial breakthroughs. This rare, fatal, and autosomal-recessive disorder is caused by defects in NPC1 or NPC2. These ubiquitously expressed proteins help cholesterol exit from the endosomal–lysosomal system. The dysfunction of either causes an aberrant accumulation of lipids with patients presenting a large range of disease onset, neurovisceral symptoms, and life span. Here, we note general aspects of experimental models, we describe the line-up used for NPC-related research and therapy development, and we provide an outlook on future topics.
1. Niemann–Pick Type C Disease
The prime purpose of biomedical research is to understand the molecular underpinnings of human diseases enabling the development of curative therapies. Unfortunately, these goals have been reached merely for a minuscule fraction of diseases. The large majority of ailments—affecting from just a handful of patients to millions worldwide—awaits a treatment [1,2,3]. There are numerous reasons for the slow progress such as rare occurrence, molecular complexity, and variability of symptoms. However, a decisive factor is the availability, quality, and use of experimental models [4,5,6,7,8,9,10,11,12,13].
NPC is a prime example for a disease that lacks a curative therapy despite impressive breakthroughs within the last decades [14,15,16,17] and rapidly growing publication counts (Appendix A, Figure 1). At first sight, the disease seems relatively easy to study and to understand: previous research showed that it is monogenic with autosomal-recessive inheritance and caused by mutations in either of two genes, NPC1 (OMIM #257220) [18] or NPC2 (OMIM #607625) [19,20]. The structures of the corresponding proteins [21,22,23,24,25] together with a wealth of cell-based data indicate that this duo collaborates to pilot unesterified cholesterol out of the endosomal–lysosomal system [26,27,28]. If the activity of the membrane-resident NPC1 or its intralumenal partner NPC2 is diminished or absent, unesterified cholesterol accumulates in compartments of the endosome-lysosome [29,30] together with other molecules [31,32]. How can this—at first sight well-defined—cellular problem cause havoc in humans presenting an enormous variability in disease onset, symptoms, and life span? In fact, NPC disease comprises several forms based on the age at which patients present neurological symptoms [14,20,33,34,35,36,37,38]. Rare peri- and neonatal cases present hepatosplenomegaly, jaundice, and fetal hydrops with rapid death often due to hepatic and respiratory failure [39,40,41,42,43,44]. Most patients show infantile forms presenting hypotonia and delayed motor development (early: <2 years) as well as clumsiness, speech delay, and cataplexy (late: 2–6 years) reaching life spans of several years [20,44,45]. The second largest group of patients shows the juvenile form (6–15 years) presenting cognitive impairment, ataxia, and dystonia [20,38,40,41,44]. The adolescent/adult form (>15 years) is characterized by cognitive impairment and psychiatric symptoms such as hallucinations and schizophrenia; the number of these patients is probably underestimated [14,20,46,47,48,49,50]. Notably, there is considerable overlap between the groups with respect to symptoms; many patients present common signs such as ataxia, dysphagia, and vertical supranuclear gaze palsy [20,38,41,44]. However, siblings bearing the same mutations can show distinct forms of the disease [20,33].
The diagnosis of NPC disease is complicated by the heterogeneous clinical presentation and therefore depends on laboratory tests. This includes the so-called filipin test, the detection of plasma biomarkers [51,52,53,54,55], and genetic analyses [56,57,58]. For decades, the filipin test represented the sine qua non to diagnose NPC. It requires primary cultures of fibroblasts from patient-derived skin biopsies followed by the staining of chemically fixed cells with filipin. This bacteria-derived, fluorescent complex of molecules binds unesterified cholesterol, thus allowing the visualization of its intracellular distribution [59]. Therapeutic options are limited to symptomatic treatment [14]. The only disease-modifying drug approved for NPC in many countries, except for the USA, is Miglustat/Zavesca (N-butyldeoxynojirimycin), which decelerates disease progression in some patients [38,60,61]. The drug also serves as FDA-approved substrate reduction therapy for Gaucher disease [62,63].
Understanding the somewhat mysterious links between cellular damage and the unpredictable outcome in patients, and the development of diagnostic tests and of efficient therapies require appropriate experimental approaches and models. In the following, we will make some general remarks, we will present currently available models for NPC research, and we will highlight crucial points.
2. The Use of Experimental Models in Biomedicine
The use of experimental models in research has a long history. The first “publication” dates to the 17th century, when William Harvey described physiologic experiments with animals such as shrimp, eel, chick, and pigeon to understand blood circulation [64]. For centuries, it was believed that animals are unable to feel pain and that they resemble machines [65]. These views changed during the age of enlightenment: In 1789, the philosopher and jurist Jeremy Bentham was one of the first to raise the issue of animal protection by stating: “The question is not, Can they reason? or Can they talk? but, Can they suffer?” [66]. In 1876, the parliament of the United Kingdom passed the “Cruelty to Animals/Anti-Vivisection Act” that updated previous legislation and imposed rules on experiments with animals. The 20th century saw the establishment of three rules, named replacement, reduction, and refinement (the 3Rs) to match “the intimate relationship between humanity and efficiency in experimentation” [67]. These rules have become a key element of laws regulating the scientific use of animals worldwide [68].
Today, biomedical research on human diseases depends entirely on experimental models that range from single cells to non-human primates. Disease models may emerge from spontaneous changes. A famous example is the nude mouse (Mus musculus) introduced by Flanagan [69] and used extensively to create models requiring immunodeficiency, for example for patient-derived xenografts [70]. Models can also be based on healthy animals, in which a disease-like state is induced experimentally. Examples include pharmacologically-induced diabetes in rodents and rabbits [71,72], Parkinson-like symptoms in non-human primates [73] and autism-like behavior in rats (Rattus norvegicus) [74]. Other pathologic conditions such as stroke and retinal ischemia can be provoked by an artificial interruption of blood supply [75] and increase of intraocular pressure [76,77], respectively. Loss of bone mass mimicking osteoporosis occurs after tail immobilization in rats [78]. Meanwhile, most experimental disease models are generated by powerful genetic tools. Not surprisingly, oncology was the first area profiting from genetically modified mice with transgenic expression of oncogenes [79]. Mice are not the only species used to mimic human pathologies. The nematode Caenorhabditis elegans has been genetically modified to generate models of Parkinson’s [80], Alzheimer’s disease [81], PolyQ disease [82], and lysosomal disorders [83]. The fruit fly Drosophila melanogaster serves as disease model for different organs including the brain [84,85,86], kidney [87], and pancreas [88].
The usefulness of a model depends on the specific question. Ideally, the model accurately recapitulates key aspects of the disease of interest, for example pathologic changes in cells or symptoms of patients. Furthermore, it should allow extrapolating results to the target organism. Interestingly, history teaches that extrapolability does not necessarily scale with evolutionary kinship: closer may not be better. For example, thalidomide and aspirin are well tolerated by mammalian species but not by pregnant women [89], and chimpanzee have proven inappropriate for studies on AIDS [90]. Similarly, body size and metabolic rates do not always scale with disease processes. For example, some drugs can be effective at different dosages in different animal models [91,92] and humans [93]. In addition, some animals simply do not show specific symptoms: rats cannot really cough [94], rabbits (Oryctolagus cuniculus) and rats do not show some symptoms of cystic fibrosis [95], and no animal except for non-human primates displays endometriosis symptoms [96]. On the other hand, exotic species can serve as important models for human diseases. Examples are the armadillo Dasypus novemcinctus for research on leprosy [97], the turtle Trachemys scripta to study brain hypoxia and anoxia [98], and the pet Chinchilla lanigera to investigate hearing loss [99]. Diurnal rodents represent unique models of cone-related retinal diseases [100].
Disease models based on cultured cells have seen a remarkable renaissance due to the possibility of generating specific human cells from patient-derived induced pluripotent stem cells [101]. A recent article exemplifies this new approach going from in vitro data to retrospective analysis of clinical data exposing a possible treatment [102].
3. Experimental Models for Niemann–Pick Type C1 and C2 Disease
Numerous experimental models are available to study NPC disease [103], probably because the disease is monogenic, the transmission is recessive, and orthologues of the causative genes are present in many phyla ranging from plants to mammals [104] (Table 1). The models have driven the enormous progress in the field during the last decades. Most of them concern NPC1, which is mutated in 95% of patients. Only a few experimental models are available to study mutant NPC2. The presence of multiple isoforms in specific phyla suggests important and so far undiscovered functions of these proteins. Figure 2 indicates the use of the different models based on the number of publications (Appendix A). Clearly, the mouse has become the preferred workhorse in the NPC disease field.
3.1. Non-Mammalian Models
The knock-out of NPC1 orthologues in plants (At1g42470, At4g38350; Arabidopsis thaliana) [105] and yeast (NCR1; Saccharomyces cerevisiae) [106] have been generated. Because both showed changes in sphingolipid, but not sterol metabolism, and because NPC2 orthologues were not known [107], it was assumed that the proteins have distinct roles across phyla. However, yeast cells bear a homologue of NPC2, which can replace the human version [108]. Moreover, yeast cells have been used to screen for pathways influencing the outcome of NPC1 deficiency [109,110,111] and to explore the molecular mechanism of sterol transfer based on structural data [25]. The knock-down of NPC1 and NPC2 homologues in the sterol auxotroph pathogen Entamoeba histolytica revealed their contribution to cholesterol uptake (Ehnpc1, Ehnpc2) [112]. The genome of Caenorhabditis elegans contains two homologues of mammalian NPC1 (ncr-1, ncr-2). Elimination of both forms stalls a specific phase of larval development, which is probably due to defects in the intracellular transport of cholesterol and the production of essential steroid hormones [113,114,115] (Table 1). The defects can be rescued by human NPC1L1 and NPC1 proteins [116] and by specific glycosphingolipids and endocannabinoids [117,118].
The elimination of Npc1a, one of two NPC1 homologues from Drosophila melanogaster, causes larval lethality (Table 1), which can be rescued by dietary supply of the steroid hormone ecdysone or by local expression of Npc1a in the ring gland [119,120]. The elimination of Npc1b also causes larval lethality due to defects in sterol absorption in the midgut (Table 1), which cannot be rescued by ecdysone [121]. The fruit fly bears a family of eight genes resembling NPC2. The simultaneous elimination of two of these genes, Npc2a and Npc2b, causes larval lethality and neurodegeneration, which again can be rescued by dietary cholesterol or ecdysone [122,123]. A genetic screen for pathways mediating cholesterol trafficking and steroidogenesis in Drosophila revealed that the activation of autophagy can overcome cholesterol accumulation due to NPC1 deficiency [124].
Induced models of NPC were created in the zebrafish Danio rerio (Table 1) using anti-oligonucleotide-based knock-down of npc1 [125,126]. These manipulations interfered with gastrulation and led to the premature death of embryos, which could be rescued by mouse Npc1 and in part by steroids [125]. Moreover, morpholino-based knock-down mimicked thrombopenia observed in human patients possibly due to defects in myeloid development [126]. CRISPR-CAS-induced null alleles of npc1 caused premature death with only a few animals surviving into adulthood. Mutant animals showed massive cholesterol accumulation and defects in the liver, cerebellum, and lateral line organ causing disturbed balance and motor control [127,128].
The non-mammalian models clearly matter, as they reveal how functions of NPC-related proteins evolved, they enable screens to identify NPC1- or NPC2-related pathways and processes, and they help to explore new therapeutic approaches. Up to now, their publication counts are lower than those of mammalian models (Figure 2).
3.2. Mammalian Models
Of all mammalian species serving biomedical research, only mice and cats (Felis catus) are currently used to study NPC (Table 1). No rat or large animal model has been established for this disease. A single case report described NPC-like symptoms in a Boxer dog [149], and a recent study described calfs (Bos taurus) with progressive neurologic symptoms due to mutant NPC1, suggesting the possibility of a bovine model [144] (Table 1).
The first mouse strains to study NPC carried spontaneous mutations in Npc1, namely the insertion of a retroposon (Nih allele, further referred to as Npc1Nih) [18,129] and of a 43 base-pair (spm allele, Npc1spm) [130,132] in BALB/c and C57BLKS/J colonies, respectively each causing a de facto Npc1 knock-out. These mice present a relatively early onset of the disease, which is characterized by hepatomegaly, weight loss, disturbed motor coordination, tremor, and ataxia. The mice die prematurely between 11 and 13 weeks of age (Table 1). Cells show an accumulation of unesterified cholesterol, gangliosides, and other lipids in different organs and tissues [132,150,151,152,153,154]. A similar phenotype was observed in a genetically modified mouse from the Goldstein/Brown lab. In this line (Npc1pf), a double mutation (P202A/F203A) abolishes cholesterol binding by NPC1 and invalidates its function, but it leaves its level and localization unaffected [131] (Table 1). The mice discussed so far represent one end of the model spectrum as they lack the NPC1 function completely and irreversibly. The complete absence of NPC1 occurs only in a small fraction of patients [45]. Nevertheless, these models mattered, as they enabled important discoveries including the gene responsible for the disease [18], the progressive neurodegeneration in the cerebellum [155,156,157], and links to autophagy [158,159,160] and Alzheimer’s disease [161]. Moreover, they were used extensively to explore new therapies (Table 2).
More common NPC1 mutations in humans induce errors in the structure of the protein leading to its degradation but leave its function more or less intact. Mouse models mimicking these changes have appeared on the scene within the last ten years (Table 1). Maue and colleagues described a mouse line with a D1005G variant that was generated by ethyl nitrosourea mutagenesis (Npc1nmf164) [132]. Praggastis and colleagues presented a knock-in of the human I1061T version of NPC1 (Npc1I1061T) [133]. This model matters as it represents approximately 20% of all NPC cases [211,212]. The mouse strains bear misfolded NPC1, causing a partial loss of function. The onset of the disease is delayed, its progress is less severe, and the life span is extended to 17 weeks compared to the complete loss-of-function mutants [133] (Table 1). In 2017, two mouse strains bearing specific human mutations were presented together with a thorough characterization of their behavioral phenotypes [134]. The strains carry either an intronic point mutation (c.1554-1009G > A) generating a pseudoexon due to aberrant splicing (Npc1Imagine) or the c.1920delG mutation, generating a truncated protein (Npc1Pioneer) (Table 1). Homozygous Npc1Imagine mice and compound heterozygous animals Npc1Imagine/Pioneer displayed symptoms similar to those reported in other NPC animal models with an onset of first neurologic symptoms between 7 weeks and an average life span of 9 weeks. Notably, most homozygous Npc1Pioneer mice died during the embryonic stage; the few surviving mice (1–2%) were predominantly female [134]. The latest entry in the defilé of models bears a mutant Npc1 allele generated by the CRISPR-Cas technique (Npc1em1Pav) [135] (Table 1). These mice help address a key question in the field: which factors determine the enormous phenotypic variability observed in patients? Humans with the same mutation can present completely different disease onsets, progress, and life spans [213,214]. In mice, the outcome of a given mutation varies with the genetic background of strains [37,135,215,216,217,218]. Numerous double mutant mice have been created to test whether and how specific candidate genes impact the disease [163,173,177,190,191,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235]. Sex-dependent differences in behavior [236], life span [37,134], and responses to immune activation [237] and to potential therapies [171,238] were reported in some NPC1 mutant mice, raising the question of whether sex is a modifying factor in NPC disease [37] as in other cholesterol-related pathologies [239,240,241,242] and normal cholesterol homeostasis [243,244].
Several mouse models were established to study the relevance of NPC1 in specific cell types or tissues (Table 1). Using morula aggregation, so-called chimeric mouse lines were generated, in which distinct ratios of cells harbor the wild-type or the mutant allele [158]. Mice for the cell-specific elimination of Npc1 were based on the Cre/loxP technique (Npc1tm1.1Apl) [136,245,246] (Table 1). A first study showed that the elimination of Npc1 from Purkinje cells induces their degeneration but leaves the life span of mice unaffected [136]. A mouse model to study NPC1 deficiency in the liver forgoing neurologic complications was established by intra-peritoneal injections of antisense oligonucleotides in healthy BALB/c mice [137,186]. The over-expression of Npc1 in specific cell types has been accomplished using classic transgenic mice to target GFAP-expressing cells [138], the inducible TetOn/Off system, which was used to target neurons [139], and the Cre/loxP system allowing the cell-specific reversal of a Npc1 knock-out [140] (Table 1). These mice enable a cell- or tissue-specific rescue of NPC1 deficiency [218,247,248]. For example, the re-establishment of Npc1 expression in the liver rescued liver disease, but it did not prevent progressive neurodegeneration and premature death [140]. The use of cell-specific promoters requires a thorough validation of their expression patterns [249,250]. Moreover, the observation that NPC1 deficiency in neurons is sufficient to induce their death [158,245] does not exclude a demise-provoking contribution by non-neuronal cells such as microglia or astrocytes [251,252,253], serving potentially as therapeutic targets.
Compared to Npc1, the line-up of mouse models targeting Npc2 is much smaller. The first mouse line was created by gene targeting, resulting in 4% of normal protein levels. These animals showed a similar phenotype as NPC1-deficient mice and as mice lacking both proteins. The latter finding provided first evidence for the functional cooperation between NPC1 and NPC2 in vivo [145]. Additional lines targeting Npc2 have been generated using the gene trap approach [146,147] (Table 1). The over-expression of Npc2 in the liver was accomplished using transgenic mice and specific promoter elements [148]. More mutant alleles of mouse Npc1 and Npc2 are listed on the MGI website.
NPC-like symptoms in a domestic cat (Felis catus) were first reported by Lowenthal and collaborators [141] (Table 1). A colony was subsequently established, and the cats were further characterized. They develop neurologic symptoms such as ataxia and vestibular defects at juvenile age similar to humans, and they show neuroaxonal dystrophy [141,142,254,255,256,257,258]. In 2003, the genetic defect was uncovered: a single base substitution (2864G-C) in NPC1 causes an amino acid change (C955S) [143]. Two case reports described cats with distinct mutant alleles of NPC1 [259] and NPC2 [260], indicating that more feline NPC models could be established.
3.3. In Vitro Models
Cultured cells are instrumental to uncover basic protein functions and molecular disease mechanisms and to test potential therapeutic approaches at the cellular level [12]. The use of cell cultures to study NPC disease dates back to the 1960s, when the Fredrickson group prepared primary fibroblasts from skin and bone marrow of patients with different forms of Niemann–Pick disease, including type C [261]. This pioneering publication initiated a decades-long series of studies based on patient-derived fibroblasts (Figure 3), enabling ground-breaking discoveries. Examples are the defect in cholesterol esterification and the accumulation of unesterified cholesterol [262,263,264], the functional validation of NPC1-encoding cDNA [265] and of secreted NPC2 [19], and the degradation of the misfolded p.I1061T NPC1 variant [266].
An alternative method to induce the cellular hallmark of NPC, an accumulation of unesterified cholesterol, relies on hydrophobic amines such as U18666A [267,268,269,270]. Originally, this molecule was developed as an inhibitor of cholesterol synthesis [271], and it was later shown to inhibit NPC1 activity directly [272].
3.3.1. Cell-Lines
The first cell lines to study NPC disease were established from patient-derived blood lymphocytes, which were immortalized through transformation by the Epstein–Barr virus [273]. A similar approach was used to immortalize lymphoid cells from NPC2 patients [153]. A fibroblast cell line based on the Npc1spm mouse was generated using a spontaneous immortalization (3T3) protocol [274,275]. Immortalized mouse embryonic fibroblasts from NPC1-deficient mice were transduced with different constructs to monitor autophagy [276]. A mouse embryonic fibroblast cell line from NPC2-deficient mice expressing a NPC2–crmCherry fusion protein was established to track the intracellular distribution of the protein [277]. A line of NPC2-deficient patient human fibroblasts showed a down-regulation of NPC1 upon infection with HIV [278]. Several models were derived from Chinese hamster ovary (CHO) cells, the workhorse of cell biology: NPC1-deficient CHO cells were generated using chemical or gene trap mutagenesis and assays to detect cholesterol transport-deficiency [279,280,281]. Other CHO lines stably over-express NPC1 [282,283], myc-tagged NPC2 [284], as well as NPC1-EGFP or -RFP fusion proteins [285,286,287], allowing for example to track the movement of NPC1-containing organelles [285]. CRISPR-Cas technology [288] or transfection with short interfering RNA constructs were used to generate NPC1- and NPC2-deficient HeLa [289,290,291,292] and Hek-293T cells [293]. The knock-down of NPC1 in a neuroblastoma cell line (SH-SY5Y) was achieved by stable transfection with short hairpin RNA [294]. Immortalized human hepatocytes and hepatic stellate cells with stable knock-down of NPC1 or NPC2 were obtained by transduction with lentivirus and short hairpin RNAs [295,296]. The artificial expression of NPC1 in Escherichia coli has been used to study its transport function [297]. In the context of Alzheimer disease research, NPC1 was stably down-regulated in a neuron-like Neuro-2a line that over-expresses a specific form of the amyloid precursor protein [298]. Schwann cell lines were derived using dorsal root ganglia and peripheral nerves of the Npc1spm mouse [299]. Knock-down in an oligodendroglial cell line was accomplished using short interfering RNA [200]. The first NPC model based on a haploid human cell line has been introduced recently [300].
Cell line-based models matter to uncover basic molecular functions of NPC1 [21] or NPC2 [301] and NPC1-dependent signaling pathways [293], to perform comparative studies at the cellular level [302], and to identify disease-relevant genes [289]. Cell lines helped to identify NPC1 as a receptor mediating Ebola virus infection [303,304] and to investigate its involvement in hepatitis C virus replication [305]. However, they cannot inform about cell-type specific dependency on NPC1 and consequences of its dysfunction. Moreover, it is not clear whether NPC1- or NPC2-related cellular processes observed in cell lines occur also in specialized cells in vivo. Another caveat derives from the fact that cell lines are per definitionem mitotic, whereas most differentiated cells in the body are post-mitotic. Cell division may modify how NPC1- or NPC2-deficiency affects cells.
3.3.2. Primary Cultures of Brain Cells
An alternative to cell lines are primary cultures, where cells are isolated from the organism and used after different periods of culture without immortalization. Cultured cells retain their in vivo properties to degrees that depend on the cell type and the culture conditions, namely the artificial exposure to chemically undefined serum [306,307,308,309,310].
Most NPC patients suffer from debilitating neurologic symptoms and therefore, it appears imperative to study the impact of dysfunctional NPC1 or NPC2 on cells in the brain (Figure 3). The first studies using primary cultures of central nervous systen (CNS) cells investigated the expression and distribution of NPC1 in cerebellar neurons and glial cells [311] and reported defects in cholesterol metabolism and neurotrophin signaling in striatal neurons [312]. Thereafter, sympathetic [313], cortical [314], hippocampal [315,316] and retinal neurons [317] (Figure 4) as well as purified cerebellar Purkinje cells [318] have been studied in vitro. These models matter, as they revealed neuron-specific defects caused by NPC1 deficiency such as impaired synaptic function [316,318,319], depletion of cholesterol from axons, and an accumulation of cholesterol independently from lipoprotein uptake [313,317]. They also helped to identify lamellar inclusions as the site of cholesterol accumulation [317]. Cultured astrocytes [320], oligodendrocytes [321,322,323], and microglial cells [324,325] have rarely been studied, despite the potential glial involvement in neurodegeneration [251], evidence for myelination defects [246], and signs of neuroinflammation in NPC disease [326]. Organotypic cultures represent a more integrated preparation to study neurons, but they have been used only sporadically in this field [325,327].
3.3.3. Primary Cultures of Other Cells
Predominant in vitro models of NPC research are the above-mentioned patient-derived skin fibroblasts, which are mitotic primary cells, but not cell lines unless they have been immortalized. Only very few differentiated cell types are studied in the field (Figure 3). Liver and spleen are affected in many NPC patients, but few reports used primary hepatocytes [199,328,329] and hepatic stellate (Ito) cells [330] from NPC1-deficient mice, splenocytes from NPC2-deficient mice [146], and NPC1-deficient splenic B cells [331]. Acutely isolated Kupffer cells were examined in chimeric mice following bone marrow transplantation [332]. With respect to lung defects, one report studied primary type 2 pneumocytes treated with U18666A [333]. With respect to immune cells, studies used NPC1-deficient macrophages [328,332,334], invariant Natural Killer T cells and human B cell lines [335], lymphoblasts [275], monocyte-derived dendritic cells [336], and T cells [146,337,338]. To date, no studies on cultured leukocytes or granulocytes have been published. Among other cells, the effects of NPC2 knock-down on adipocyte differentiation and function were studied using primary cultures [339], and spermatozoa from NPC2-deficient mice were isolated and analyzed [340].
3.3.4. Stem Cell-Derived Models
The differentiation of specific cell types from embryonic or induced stem cells has become popular, because this technology allows studying cells from patients and producing them in large quantities. Consequently, the number of publications related to these models in the NPC field is increasing (Figure 3). A first report showed the impaired self-renewal and differentiation of neural stem cells from embryonic brains of NPC1-deficient mice [341]. Ordonez and colleagues created a short hairpin RNA-based knock-down of NPC1 in human embryonic stem cells and differentiated these cells to neurons [342]. These neurons recapitulated the pathologic hallmark of NPC, the accumulation of unesterified cholesterol, and showed impaired mitochondrial function and defective autophagy. Multipotent adult stem cells were isolated from skin biopsies of NPC patients and control subjects and differentiated to neurons showing an accumulation of cholesterol [343]. These cells were selected by specific culture conditions. An alternative and meanwhile standard approach is the reprogramming of cells from adult tissues to create induced pluripotent stem cells and their subsequent differentiation to specialized, often postmitotic cells. Several studies used this approach to generate neurons from NPC patients and healthy donors [344,345,346,347,348,349]. Maetzel and colleagues also generated stem cell-derived hepatic cells and isogenic control lines to avoid confounding effects by distinct genetic backgrounds of patients and donors [346]. The stem cell-derived models matter: they enable studying the impact of NPC1 or NPC2 deficiency on differentiated human cells, notably neurons, and to explore new therapeutic strategies [347,350,351]. However, the protocols for reprogramming and differentiation need to be standardized to allow for comparison of results.
4. Models Mattering for Therapy Development
Experimental models are indispensable for the preclinical exploration of therapeutic approaches. In the NPC field, cell-based screens for targets and drugs used yeast [109], immortalized embryonic fibroblasts [276] or ovarian granulosa cells from mutant mice [352], human stem cell-derived neurons [347,348,350,351,353,354], mutant CHO lines [355], and patient-derived fibroblasts [356,357]. Numerous therapeutic approaches were tested in vivo using NPC mice and cats. Table 2 lists studies where the impact of treatments on disease progression was assessed with proper controls.
Few studies have delivered an approved drug or treatments reaching clinical development. The disease-modifying N-butyldeoxynojirimycin inhibits glucosylceramide synthase [358]. Curiously, a first in vitro study on CHO cells showed that the compound does not revert cholesterol accumulation in NPC1-deficient cells [359]. This was also observed in stem cell-derived neurons in vitro [347], arguing against a therapeutic effect. However, in vivo studies showed that the drug slows down neurologic disease progression and prolongs the life span of NPC1-deficient BALB/c mice and NPC1 mutant cats [184,360], providing preclinical evidence for its therapeutic use (Table 2).
A potential treatment is based on 2-hydroxypropyl-beta-cyclodextrin (CD) that chelates cholesterol and other components [361] (Table 2). Curiously, the exploration of this compound started with in vivo experiments—again with discouraging results. A first study using intra-peritoneal or intra-thecal injection in NPC1-deficient mice failed to show a positive effect [362]. However, subsequent reports revealed that CD prolongs the life span, slows down neurologic disease progression, and halts the degeneration of Purkinje cells in the mouse and cat model [177,178,179,180,181,363,364]. Intra-thecal injections were required, as CD cannot pass the blood–brain barrier [365]. NPC1-deficient mice were also used to study ototoxicity of CD [366,367] and its effects on microglial cells [368] and the liver [369]. Effects of CD on NPC1-deficient cells were explored in lateral line neuromast cells in vivo [128], siRNA-treated HeLa cells [370], liver-derived cell lines [371], cultured fibroblasts [372,373,374], and primary [317,375,376] or stem cell-derived neurons [342,353,377]. First clinical data showed that CD decelerates disease progression in patients [378,379].
Histone deacetylases (HDACs) emerged as a possible therapeutic target for NPC from a genetic screen in yeast [109] and from in vitro studies of NPC1-deficient neuronal stem cells [380], patient- and mutant mouse-derived fibroblasts [133,357,374,381,382,383,384], cell lines [384,385], and U18666A-treated hippocampal neurons [386]. A first in vivo study using Npc1 mutant mice claimed that repeated intra-peritoneal injections of vorinostat, an HDAC inhibitor, together with polyetheylene–glycol and CD slow down neurologic disease progression, but some controls were missing [387]. A subsequent report on mice attributed the effects on neurologic symptoms to CD [363]. Repeated intra-peritoneal injections of vorinostat in NPC1 mutant mice improved liver function but did not slow down weight loss or increase life span [194] probably because the drug cannot enter the central nervous system [363]. A comparison of drug effects using different mouse models revealed that drug effects on liver function were not mediated by proteostatic effects on NPC1 [194] (Table 2).
Evidence from Npc1 mutant mice that heat-shock proteins protect Purkinje cells from degeneration suggested these components as new drug targets in NPC [157,227]. The idea was supported by in vitro studies on patient-derived fibroblasts [193,227,388] and U18666A-treated neurons [227] and in vivo studies exploring the over-expression or knock-down of heat shock protein beta-1 in NPC1-deficient mice [227]. A corresponding disease-modifying therapy may be based on arimoclomol, a small molecule enhancer of heat shock proteins, whose effects were explored in patient-derived fibroblasts and NPC1-deficient mice [193] (Table 2).
Within the last years, NPC1-deficient mice also helped to explore gene therapy for NPC (Table 2). First support for this approach came from two observations. The over-expression of NPC1 in brain cells was achieved following the intra-cerebral injection of an adenoviral construct in vivo [389]. The cell-specific over-expression of NPC1 in transgenic mice rescued pathologic changes due to NPC1 deficiency [139,390]. Within the last few years, a series of studies showed that the progress of neurologic disease in NPC1-deficient mice is slowed down by intra-cardiac [195], intra-cisternal [196], and intra-cerebroventricular [197] injection of vectors based on adeno-associated virus 9 (AAV9). Similar improvements were found in mice lacking NPC2 following intra-cisternal injections of AAVrh.10 carrying NPC2 [198].
5. Conclusions and Outlook
The diversity and validity of experimental models and their pertinence to topics of interest are key to advance biomedical research. Over the last decades, the NPC field has developed a gang of models that matter as they revealed the origin of the disease, provided important insight in disease mechanisms, and helped to explore new diagnostic and therapeutic approaches. Moreover, these models are used extensively outside the NPC field to understand fundamental aspects of cholesterol homeostasis [391] in different organs, notably the brain [392], and mechanisms of other cholesterol-related diseases [393,394,395].
The publication record indicates a clear preference for NPC1, mice, and fibroblasts as gene, animal, and cell of choice, respectively. A few points should be considered with respect to future developments and advances. The focus on NPC1 is understandable given that most patients bear mutations in this gene. However, new models targeting NPC2 are of high interest, as they can help for example to discern NPC1- and NPC2-dependent genetic, epigenetic, and sex-dependent disease modifiers. The identification of modifiers remains a top priority in the field. The predominance of mouse models in NPC research is readily explained by the increasing ease of genetic manipulations and the relative cost efficacy. However, mice impose several limitations, notably with respect to their small size and their limited behavioral repertoire [396]. Therefore, new models based on larger mammals including rats are highly desirable last but not least to enable the successful translation of therapeutic approaches into the clinic [6]. There is also a clear demand for inducible/reversible pharmacologic models based on highly selective small molecule inhibitors of NPC1 or NPC2. These approaches would allow for before/after studies and thereby help to discern within-subject variability. The surprising discovery that NPC1 serves as receptor for filovirus entry into cells [303,304] will help to develop such inhibitors and new models.
The focus on fibroblasts originates from their availability through skin biopsies, their ease of maintenance, and their long-standing use as a diagnostic tool. However, studies of patient-derived fibroblasts cannot inform about the outcome of NPC1 dysfunction in highly specialized postmitotic cells such as neurons. Therefore, it is imperative to elucidate how specific cells, namely the most vulnerable, react to defects in NPC1 and NPC2. This will require a combination of preparations allowing to study the same type of cells in vivo, ex vivo, and in vitro (Figure 4) as well as new approaches to analyze mRNA, protein, and lipid content of defined cell types replacing transcriptomic, proteomic, and lipidomic studies of entire organs or tissues. As an example, acutely isolated cells combined with single cell transcriptomics [231] represent a first step that needs to be refined and extended with a focus on vulnerable cells in most affected organs, including the brain, liver, and lung. Cells differentiated from induced pluripotent stem cells represent an alternative although with caveats [397]. Whatever the source of cells, advanced culture systems preserving their three-dimensional arrangement should be considered as well [398,399]. The development of therapeutic approaches for neurologic and psychiatric symptoms faces fundamental hurdles with respect to diagnosis and model validity that are not specific to NPC [400,401].
Clearly, the establishment of new models requires substantial investments and bears risks, but ultimately, all that matters are the models: they are indispensable to expose molecular mechanisms underlying the disease and to develop efficient therapies.
Author Contributions
Conceptualization, V.P. and F.W.P.; methodology, F.W.P.; software, F.W.P.; visualization, F.W.P; writing—original draft preparation, V.P. and F.W.P.; writing—review and editing, V.P. and F.W.P. All authors have read and agreed to the published version of the manuscript.
Funding
The APC for this review was funded by the Niemann-Pick Selbsthilfegruppe e.V. (Germany). The authors’ research is funded by Together Strong NPC Foundation (V.P.), Niemann-Pick Selbsthilfegruppe e.V., Ara Parseghian Medical Research Foundation and Bild hilft e.V. “Ein Herz für Kinder” (F.W.P.).
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
To obtain quantitative data on the publication output in the field, Boolean queries were performed in PubMed and records were downloaded in csv format. Data analysis and visualization were accomplished using the open source software R [402] and selected R packages (data.table [403], ggplot2 [404], readr [405].
References
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef]
- Ruz, C.; Alcantud, J.L.; Vives Montero, F.; Duran, R.; Bandres-Ciga, S. Proteotoxicity and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5646. [Google Scholar] [CrossRef]
- Tanner, L.; Single, A.B. Animal Models Reflecting Chronic Obstructive Pulmonary Disease and Related Respiratory Disorders: Translating Pre-Clinical Data into Clinical Relevance. J. Innate Immun. 2020, 12, 203–225. [Google Scholar] [CrossRef]
- Justice, M.J.; Dhillon, P. Using the mouse to model human disease: Increasing validity and reproducibility. Dis. Model. Mech. 2016, 9, 101–103. [Google Scholar] [CrossRef] [Green Version]
- Cacheiro, P.; Haendel, M.A.; Smedley, D. New models for human disease from the International Mouse Phenotyping Consortium. Mamm. Genome 2019, 30, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Gurda, B.L.; Vite, C.H. Large animal models contribute to the development of therapies for central and peripheral nervous system dysfunction in patients with lysosomal storage diseases. Hum. Mol. Genet. 2019, 28, R119–R131. [Google Scholar] [CrossRef] [Green Version]
- Madeja, Z.E.; Pawlak, P.; Piliszek, A. Beyond the mouse: Non-rodent animal models for study of early mammalian development and biomedical research. Int. J. Dev. Biol. 2019, 63, 187–201. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.R.; Bolton, E.R.; Dwinell, M.R. The Rat: A Model Used in Biomedical Research. Methods Mol. Biol. 2019, 2018, 1–41. [Google Scholar]
- Breuer, M.; Patten, S.A. A Great Catch for Investigating Inborn Errors of Metabolism-Insights Obtained from Zebrafish. Biomolecules 2020, 10, 1352. [Google Scholar] [CrossRef]
- Favret, J.M.; Weinstock, N.I.; Feltri, M.L.; Shin, D. Pre-clinical Mouse Models of Neurodegenerative Lysosomal Storage Diseases. Front. Mol. Biosci. 2020, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Kruczek, K.; Swaroop, A. Pluripotent stem cell-derived retinal organoids for disease modeling and development of therapies. Stem. Cells 2020, 38, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, I.; Nagai, Y.; Seki, K. Generation of Common Marmoset Model Lines of Spinocerebellar Ataxia Type 3. Front. Neurosci. 2020, 14, 548002. [Google Scholar] [CrossRef] [PubMed]
- Geberhiwot, T.; Moro, A.; Dardis, A.; Ramaswami, U.; Sirrs, S.; Marfa, M.P.; Vanier, M.T.; Walterfang, M.; Bolton, S.; Dawson, C.; et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J. Rare Dis. 2018, 13, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bräuer, A.U.; Kuhla, A.; Holzmann, C.; Wree, A.; Witt, M. Current Challenges in Understanding the Cellular and Molecular Mechanisms in Niemann-Pick Disease Type C1. Int. J. Mol. Sci. 2019, 20, 4392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, N.; Munkacsi, A.B.; Sturley, S.L. The complexity of a monogenic neurodegenerative disease: More than two decades of therapeutic driven research into Niemann-Pick type C disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.; Sillence, D.J. Niemann-Pick type C disease: Cellular pathology and pharmacotherapy. J. Neurochem. 2020, 153, 674–692. [Google Scholar] [CrossRef] [Green Version]
- Loftus, S.K.; Morris, J.A.; Carstea, E.D.; Gu, J.Z.; Cummings, C.; Brown, A.; Ellison, J.; Ohno, K.; Rosenfeld, M.A.; Tagle, D.A.; et al. Murine model of Niemann-Pick C disease: Mutation in a cholesterol homeostasis gene. Science 1997, 277, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Naureckiene, S.; Sleat, D.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the Second Gene of Niemann-Pick C Disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M. Niemann-Pick disease type C. Mol. Chem. Neuropathol. 1996, 27, 70–72. [Google Scholar] [CrossRef]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.; Qian, H.; Zhou, X.; Wu, J.; Wan, T.; Cao, P.; Huang, W.; Zhao, X.; Wang, X.; Wang, P.; et al. Structural Insights into the Niemann-Pick C1 (NPC1)-Mediated Cholesterol Transfer and Ebola Infection. Cell 2016, 165, 1467–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, J.; Coutavas, E.; Shi, H.; Hao, Q.; Blobel, G. Structure of human Niemann-Pick C1 protein. Proc. Natl. Acad. Sci. USA 2016, 113, 8212–8217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lu, F.; Trinh, M.N.; Schmiege, P.; Seemann, J.; Wang, J.; Blobel, G. 3.3 Å structure of Niemann-Pick C1 protein reveals insights into the function of the C-terminal luminal domain in cholesterol transport. Proc. Natl. Acad. Sci. USA 2017, 114, 9116–9121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, M.B.L.; Kidmose, R.T.; Szomek, M.; Thaysen, K.; Rawson, S.; Muench, S.P.; Wüstner, D.; Pedersen, B.P. Structural Insight into Eukaryotic Sterol Transport through Niemann-Pick Type C Proteins. Cell 2019, 179, 485–497.e418. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, S.G.; Peränen, J.; Ikonen, E. LDL-cholesterol transport to the endoplasmic reticulum: Current concepts. Curr. Opin. Lipidol. 2016, 27, 282–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.R. NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem. 2019, 294, 1706–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Heybrock, S.; Neculai, D.; Saftig, P. Cholesterol Handling in Lysosomes and Beyond. Trends Cell Biol. 2020, 30, 452–466. [Google Scholar] [CrossRef]
- Sokol, J.; Blanchette-Mackie, J.; Kruth, H.S.; Dwyer, N.K.; Amende, L.M.; Butler, J.D.; Robinson, E.; Patel, S.; Brady, R.O.; Comly, M.E.; et al. Type C Niemann-Pick disease. Lysosomal accumulation and defective intracellular mobilization of low density lipoprotein cholesterol. J. Biol. Chem. 1988, 263, 3411–3417. [Google Scholar] [PubMed]
- Liscum, L.; Ruggiero, R.M.; Faust, J.R. The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann-Pick type C fibroblasts. J.Cell Biol. 1989, 108, 1625–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breiden, B.; Sandhoff, K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int. J. Mol. Sci. 2020, 21, 2566. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Patterson, M.C.; Hendriksz, C.J.; Walterfang, M.; Sedel, F.; Vanier, M.T.; Wijburg, F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: An update. Mol. Genet. Metab. 2012, 106, 330–344. [Google Scholar] [CrossRef]
- Patterson, M.C.; Mengel, E.; Wijburg, F.A.; Muller, A.; Schwierin, B.; Drevon, H.; Vanier, M.T.; Pineda, M. Disease and patient characteristics in NP-C patients: Findings from an international disease registry. Orphanet J. Rare Dis. 2013, 8, 12. [Google Scholar] [CrossRef]
- Mengel, E.; Klünemann, H.H.; Lourenço, C.M.; Hendriksz, C.J.; Sedel, F.; Walterfang, M.; Kolb, S.A. Niemann-Pick disease type C symptomatology: An expert-based clinical description. Orphanet J. Rare Dis. 2013, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Mengel, E.; Pineda, M.; Hendriksz, C.J.; Walterfang, M.; Torres, J.V.; Kolb, S.A. Differences in Niemann-Pick disease Type C symptomatology observed in patients of different ages. Mol. Genet. Metab. 2017, 120, 180–189. [Google Scholar] [CrossRef]
- Bianconi, S.E.; Hammond, D.I.; Farhat, N.Y.; Dang Do, A.; Jenkins, K.; Cougnoux, A.; Martin, K.; Porter, F.D. Evaluation of age of death in Niemann-Pick disease, type C: Utility of disease support group websites to understand natural history. Mol. Genet. Metab. 2019, 126, 466–469. [Google Scholar] [CrossRef]
- Patterson, M.C.; Garver, W.S.; Giugliani, R.; Imrie, J.; Jahnova, H.; Meaney, F.J.; Nadjar, Y.; Vanier, M.T.; Moneuse, P.; Morand, O.; et al. Long-term survival outcomes of patients with Niemann-Pick disease type C receiving miglustat treatment: A large retrospective observational study. J. Inherit. Metab. Dis. 2020, 43, 1060–1069. [Google Scholar] [CrossRef]
- Spiegel, R.; Raas-Rothschild, A.; Reish, O.; Regev, M.; Meiner, V.; Bargal, R.; Sury, V.; Meir, K.; Nadjari, M.; Hermann, G.; et al. The clinical spectrum of fetal Niemann-Pick type C. Am. J. Med. Genet. A 2009, 149, 446–450. [Google Scholar] [CrossRef]
- Jahnova, H.; Dvorakova, L.; Vlaskova, H.; Hulkova, H.; Poupetova, H.; Hrebicek, M.; Jesina, P. Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: A surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J. Rare Dis. 2014, 9, 140. [Google Scholar] [CrossRef] [Green Version]
- Imrie, J.; Heptinstall, L.; Knight, S.; Strong, K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: A 5-year update from the UK clinical database. BMC. Neurol. 2015, 15, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumus, E.; Haliloglu, G.; Karhan, A.N.; Demir, H.; Gurakan, F.; Topcu, M.; Yuce, A. Niemann-Pick disease type C in the newborn period: A single-center experience. Eur. J. Pediatric 2017, 176, 1669–1676. [Google Scholar] [CrossRef] [PubMed]
- Staretz-Chacham, O.; Aviram, M.; Morag, I.; Goldbart, A.; Hershkovitz, E. Pulmonary involvement in Niemann-Pick C type 1. Eur. J. Pediatric 2018, 177, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Juríčková, K.; Karimzadeh, P.; Kolnikova, M.; Malinova, V.; Insua, J.L.; Velten, C.; Kolb, S.A. Disease characteristics, prognosis and miglustat treatment effects on disease progression in patients with Niemann-Pick disease Type C: An international, multicenter, retrospective chart review. Orphanet J. Rare Dis. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, B.S.; Baruteau, J.; Rahim, A.A.; Gissen, P. Clinical and Molecular Features of Early Infantile Niemann Pick Type C Disease. Int. J. Mol. Sci. 2020, 21, 5059. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Marano, M.; Florio, L.; De Santis, S. Niemann-Pick type C: Focus on the adolescent/adult onset form. Int. J. Neurosci. 2016, 126, 963–971. [Google Scholar] [CrossRef]
- Wassif, C.A.; Cross, J.L.; Iben, J.; Sanchez-Pulido, L.; Cougnoux, A.; Platt, F.M.; Ory, D.S.; Ponting, C.P.; Bailey-Wilson, J.E.; Biesecker, L.G.; et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet. Med. 2016, 18, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Bonnot, O.; Klünemann, H.H.; Velten, C.; Torres Martin, J.V.; Walterfang, M. Systematic review of psychiatric signs in Niemann-Pick disease type C. World J. Biol. Psychiatry 2019, 20, 320–332. [Google Scholar] [CrossRef]
- Nadjar, Y.; Hütter-Moncada, A.L.; Latour, P.; Ayrignac, X.; Kaphan, E.; Tranchant, C.; Cintas, P.; Degardin, A.; Goizet, C.; Laurencin, C.; et al. Adult Niemann-Pick disease type C in France: Clinical phenotypes and long-term miglustat treatment effect. Orphanet J. Rare Dis. 2018, 13, 175. [Google Scholar] [CrossRef]
- Rego, T.; Farrand, S.; Goh, A.M.Y.; Eratne, D.; Kelso, W.; Mangelsdorf, S.; Velakoulis, D.; Walterfang, M. Psychiatric and Cognitive Symptoms Associated with Niemann-Pick Type C Disease: Neurobiology and Management. CNS Drugs 2019, 33, 125–142. [Google Scholar] [CrossRef]
- Porter, F.D.; Scherrer, D.E.; Lanier, M.H.; Langmade, S.J.; Molugu, V.; Gale, S.E.; Olzeski, D.; Sidhu, R.; Dietzen, D.J.; Fu, R.; et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci. Transl. Med. 2010, 2, 56ra81. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.K.; Mascher, H.; Grittner, U.; Eichler, S.; Kramp, G.; Lukas, J.; Te Vruchte, D.; Al Eisa, N.; Cortina-Borja, M.; Porter, F.D.; et al. A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease. Orphanet J. Rare Dis. 2015, 10, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Sidhu, R.; Mydock-McGrane, L.; Hsu, F.F.; Covey, D.F.; Scherrer, D.E.; Earley, B.; Gale, S.E.; Farhat, N.Y.; Porter, F.D.; et al. Development of a bile acid-based newborn screen for Niemann-Pick disease type C. Sci. Transl. Med. 2016, 8, 337ra363. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Jinnoh, I.; Matsumoto, Y.; Narita, A.; Mashima, R.; Takahashi, H.; Iwahori, A.; Saigusa, D.; Fujii, K.; Abe, A.; et al. Structural Determination of Lysosphingomyelin-509 and Discovery of Novel Class Lipids from Patients with Niemann-Pick Disease Type C. Int. J. Mol. Sci. 2019, 20, 5018. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, R.; Kell, P.; Dietzen, D.J.; Farhat, N.Y.; Do, A.N.D.; Porter, F.D.; Berry-Kravis, E.; Vite, C.H.; Reunert, J.; Marquardt, T.; et al. Application of N-palmitoyl-O-phosphocholineserine for diagnosis and assessment of response to treatment in Niemann-Pick type C disease. Mol. Genet. Metab. 2020, 129, 292–302. [Google Scholar] [CrossRef]
- Vanier, M.T.; Gissen, P.; Bauer, P.; Coll, M.J.; Burlina, A.; Hendriksz, C.J.; Latour, P.; Goizet, C.; Welford, R.W.; Marquardt, T.; et al. Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Mol. Genet. Metab. 2016, 118, 244–254. [Google Scholar] [CrossRef]
- Patterson, M.C.; Clayton, P.; Gissen, P.; Anheim, M.; Bauer, P.; Bonnot, O.; Dardis, A.; Dionisi-Vici, C.; Klünemann, H.H.; Latour, P.; et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: An update. Neurol. Clin. Pract. 2017, 7, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Sitarska, D.; Ługowska, A. Laboratory diagnosis of the Niemann-Pick type C disease: An inherited neurodegenerative disorder of cholesterol metabolism. Metab. Brain Dis. 2019, 34, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanier, M.T.; Latour, P. Laboratory diagnosis of Niemann-Pick disease type C: The filipin staining test. Methods Cell Biol. 2015, 126, 357–375. [Google Scholar]
- Patterson, M.C.; Vecchio, D.; Prady, H.; Abel, L.; Wraith, J.E. Miglustat for treatment of Niemann-Pick C disease: A randomised controlled study. Lancet Neurol. 2007, 6, 765–772. [Google Scholar] [CrossRef]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef]
- Harvey, W.; Leake, C.D. Exercitatio anatomica de motu cordis et sanguinis in animalibus. Am. J. Med. Sci. 1929, 177, 578. [Google Scholar] [CrossRef] [Green Version]
- Descartes, R. Discours de la Methode Pour Bien Conduire la Raison et Chercher la Verité Dans les Sciences; De l’imprimérie de Ian Maire: Ian Maire, Leyden, 1637. [Google Scholar]
- Bentham, J. An Introduction to the Principles of Morals and Legislation; T. Payne and Sons: London, UK, 1789. [Google Scholar]
- Russell, W.M.S.B. The Principles of Humane Experimental Technique; Universities Federation for Animal Welfare: Wheathampstead, UK, 1959. [Google Scholar]
- Tannenbaum, J.; Bennett, B.T. Russell and Burch’s 3Rs then and now: The need for clarity in definition and purpose. J. Am. Assoc. Lab. Anim. Sci. 2015, 54, 120–132. [Google Scholar]
- Flanagan, S.P. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet. Res. 1966, 8, 295–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Wei, X.; Lin, S.; Qin, L.; Cheng, L.; Li, P. Current status and perspectives of patient-derived xenograft models in cancer research. J. Hematol. Oncol. 2017, 10, 106. [Google Scholar] [CrossRef]
- Arison, R.N.; Ciaccio, E.I.; Glitzer, M.S.; Cassaro, J.A.; Pruss, M.P. Light and Electron Microscopy of Lesions in Rats Rendered Diabetic with Streptozotocin. Diabetes 1967, 16, 51–56. [Google Scholar] [CrossRef]
- Lazarus, S.S.; Shapiro, S.H. Serial morphologic changes in rabbit pancreatic islet cells after streptozotocin. Lab. Investig. 1972, 27, 174–183. [Google Scholar]
- Burns, R.S.; Chiueh, C.C.; Markey, S.P.; Ebert, M.H.; Jacobowitz, D.M.; Kopin, I.J. A primate model of parkinsonism: Selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc. Natl. Acad. Sci. USA 1983, 80, 4546–4550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Servadio, M.; Vanderschuren, L.J.; Trezza, V. Modeling autism-relevant behavioral phenotypes in rats and mice. Behav. Pharmacol. 2015, 26, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Lunardi Baccetto, S.; Lehmann, C. Microcirculatory Changes in Experimental Models of Stroke and CNS-Injury Induced Immunodepression. Int. J. Mol. Sci. 2019, 20, 5184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, L.; Pannicke, T.; Rupprecht, V.; Frommherz, I.; Volz, C.; Illes, P.; Hirrlinger, J.; Jägle, H.; Egger, V.; Haydon, P.G.; et al. Suppression of SNARE-dependent exocytosis in retinal glial cells and its effect on ischemia-induced neurodegeneration. Glia 2017, 65, 1059–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamian, P.; Burford, J.; Farzad, S.; Blair, N.P.; Shahidi, M. Alterations in Retinal Oxygen Delivery, Metabolism, and Extraction Fraction During Bilateral Common Carotid Artery Occlusion in Rats. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3247–3253. [Google Scholar] [CrossRef]
- Fiorentino, S.; Melillo, G.; Fedele, G.; Clavenna, G.; D’Agostino, C.; Mainetti, E.; Caselli, G.F. Ketoprofen lysine salt inhibits disuse-induced osteopenia in a new non-traumatic immobilization model in the rat. Pharmacol. Res. 1996, 33, 277–281. [Google Scholar] [CrossRef]
- Hanahan, D.; Wagner, E.F.; Palmiter, R.D. The origins of oncomice: A history of the first transgenic mice genetically engineered to develop cancer. Genes Dev. 2007, 21, 2258–2270. [Google Scholar] [CrossRef] [Green Version]
- Cooper, J.F.; Van Raamsdonk, J.M. Modeling Parkinson’s Disease in C. elegans. J. Park. Dis. 2018, 8, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [Green Version]
- de Voer, G.; Peters, D.; Taschner, P.E. Caenorhabditis elegans as a model for lysosomal storage disorders. Biochim. Biophys. Acta 2008, 1782, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Jeibmann, A.; Paulus, W. Drosophila melanogaster as a Model Organism of Brain Diseases. Int. J. Mol. Sci. 2009, 10, 407–440. [Google Scholar] [CrossRef] [PubMed]
- Prüßing, K.; Voigt, A.; Schulz, J.B. Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.; Arias, R.; Sonti, S.; Odgerel, Z.; Santa-Maria, I.; McCabe, B.D.; Tsaneva-Atanasova, K.; Louis, E.D.; Hodge, J.J.L.; Clark, L.N. A Drosophila Model of Essential Tremor. Sci. Rep. 2018, 8, 7664. [Google Scholar] [CrossRef] [PubMed]
- Rodan, A.R. The Drosophila Malpighian tubule as a model for mammalian tubule function. Curr. Opin. Nephrol. Hypertens. 2019, 28, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.W.; Nilsson, K.P.; Westermark, G.T. Drosophila melanogaster as a model system for studies of islet amyloid polypeptide aggregation. PLoS ONE 2011, 6, e20221. [Google Scholar] [CrossRef] [Green Version]
- Mann, R.D. Modern Drug Use: An Enquiry on Historical Principles; MTP Press: Lancaster, UK, 1984; p. 769. [Google Scholar]
- King, N.W. Simian Models of Acquired Immunodeficiency Syndrome (AIDS): A Review. Vet. Pathol. 1986, 23, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Yamawaki, I.; Geppetti, P.; Bertrand, C.; Huber, O.; Daffonchio, L.; Omini, C.; Nadel, J.A. Levodropropizine reduces capsaicin- and substance P-induced plasma extravasation in the rat trachea. Eur. J. Pharmacol. 1993, 243, 1–6. [Google Scholar] [CrossRef]
- Luo, Y.L.; Li, P.B.; Zhang, C.C.; Zheng, Y.F.; Wang, S.; Nie, Y.C.; Zhang, K.J.; Su, W.W. Effects of four antitussives on airway neurogenic inflammation in a guinea pig model of chronic cough induced by cigarette smoke exposure. Inflamm. Res. 2013, 62, 1053–1061. [Google Scholar] [CrossRef]
- Banderali, G.; Riva, E.; Fiocchi, A.; Cordaro, C.I.; Giovannini, M. Efficacy and tolerability of levodropropizine and dropropizine in children with non-productive cough. J. Int. Med. Res. 1995, 23, 175–183. [Google Scholar] [CrossRef]
- Bolser, D.C. Experimental models and mechanisms of enhanced coughing. Pulm. Pharmacol. Ther. 2004, 17, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Rosen, B.H.; Chanson, M.; Gawenis, L.R.; Liu, J.; Sofoluwe, A.; Zoso, A.; Engelhardt, J.F. Animal and model systems for studying cystic fibrosis. J. Cyst. Fibros. 2018, 17, S28–S34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, J.M.; Fazleabas, A.T. A baboon model for endometriosis: Implications for fertility. Reprod. Biol. Endocrinol. 2006, 4, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balamayooran, G.; Pena, M.; Sharma, R.; Truman, R.W. The armadillo as an animal model and reservoir host for Mycobacterium leprae. Clin. Dermatol. 2015, 33, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Milton, S.L.; Prentice, H.M. Beyond anoxia: The physiology of metabolic downregulation and recovery in the anoxia-tolerant turtle. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2007, 147, 277–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevino, M.; Lobarinas, E.; Maulden, A.C.; Heinz, M.G. The chinchilla animal model for hearing science and noise-induced hearing loss. J. Acoust. Soc. Am. 2019, 146, 3710. [Google Scholar] [CrossRef]
- Verra, D.M.; Sajdak, B.S.; Merriman, D.K.; Hicks, D. Diurnal rodents as pertinent animal models of human retinal physiology and pathology. Prog. Retin. Eye Res. 2020, 74, 100776. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy-Promise and Challenges. Cell Stem. Cell 2020, 27, 523–531. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Fog, C.K.; Kirkegaard, T. Animal models for Niemann-Pick type C: Implications for drug discovery & development. Expert Opin. Drug Discov. 2019, 14, 499–509. [Google Scholar]
- Higaki, K.; Almanzar-Paramio, D.; Sturley, S.L. Metazoan and microbial models of Niemann-Pick Type C disease. Biochim. Biophys. Acta 2004, 1685, 38–47. [Google Scholar] [CrossRef]
- Feldman, M.J.; Poirier, B.C.; Lange, B.M. Misexpression of the Niemann-Pick disease type C1 (NPC1)-like protein in Arabidopsis causes sphingolipid accumulation and reproductive defects. Planta 2015, 242, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Higaki, K.; Tinkelenberg, A.H.; Balderes, D.A.; Almanzar-Paramio, D.; Wilcox, L.J.; Erdeniz, N.; Redican, F.; Padamsee, M.; Liu, Y.; et al. Mutagenesis of the putative sterol-sensing domain of yeast Niemann Pick C-related protein reveals a primordial role in subcellular sphingolipid distribution. J. Cell Biol. 2004, 164, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, G.; Pelosi, P. Plant transcriptomes reveal hidden guests. Biochem. Biophys. Res. Commun. 2016, 474, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Vanderford, T.H.; Gernert, K.M.; Nichols, J.W.; Faundez, V.; Corbett, A.H. Saccharomyces cerevisiae Npc2p is a functionally conserved homologue of the human Niemann-Pick disease type C 2 protein, hNPC2. Eukaryot. Cell 2005, 4, 1851–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munkacsi, A.B.; Chen, F.W.; Brinkman, M.A.; Higaki, K.; Gutiérrez, G.D.; Chaudhari, J.; Layer, J.V.; Tong, A.; Bard, M.; Boone, C.; et al. An “exacerbate-reverse” strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease. J. Biol. Chem. 2011, 286, 23842–23851. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, T.; Fujimoto, M.; Tatematsu, T.; Cheng, J.; Orii, M.; Takatori, S.; Fujimoto, T. Niemann-Pick type C proteins promote microautophagy by expanding raft-like membrane domains in the yeast vacuole. eLife 2017, 6, e25960. [Google Scholar] [CrossRef]
- Colaco, A.; Fernández-Suárez, M.E.; Shepherd, D.; Gal, L.; Bibi, C.; Chuartzman, S.; Diot, A.; Morten, K.; Eden, E.; Porter, F.D.; et al. Unbiased yeast screens identify cellular pathways affected in Niemann-Pick disease type C. Life Sci. Alliance 2020, 3, e201800253. [Google Scholar] [CrossRef]
- Bolaños, J.; Betanzos, A.; Javier-Reyna, R.; García-Rivera, G.; Huerta, M.; Pais-Morales, J.; González-Robles, A.; Rodríguez, M.A.; Schnoor, M.; Orozco, E. EhNPC1 and EhNPC2 Proteins Participate in Trafficking of Exogenous Cholesterol in Entamoeba histolytica Trophozoites: Relevance for Phagocytosis. PLoS Pathog. 2016, 12, e1006089. [Google Scholar] [CrossRef]
- Sym, M.; Basson, M.; Johnson, C. A model for Niemann–Pick type C disease in the nematode Caenorhabditis elegans. Curr. Biol. 2000, 10, 527–530. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Brown, G.; Ailion, M.; Lee, S.; Thomas, J.H. NCR-1 and NCR-2, the C. elegans homologs of the human Niemann-Pick type C1 disease protein, function upstream of DAF-9 in the dauer formation pathways. Development 2004, 131, 5741–5752. [Google Scholar] [CrossRef] [Green Version]
- Wüstner, D.; Landt Larsen, A.; Faergeman, N.J.; Brewer, J.R.; Sage, D. Selective visualization of fluorescent sterols in Caenorhabditis elegans by bleach-rate-based image segmentation. Traffic 2010, 11, 440–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.M.; Levitan, D.J. Human NPC1L1 and NPC1 can functionally substitute for the ncr genes to promote reproductive development in C. elegans. Biochim. Biophys. Acta 2007, 1770, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Boland, S.; Schmidt, U.; Zagoriy, V.; Sampaio, J.L.; Fritsche, R.F.; Czerwonka, R.; Lübken, T.; Reimann, J.; Penkov, S.; Knölker, H.J.; et al. Phosphorylated glycosphingolipids essential for cholesterol mobilization in Caenorhabditis elegans. Nat. Chem. Biol. 2017, 13, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Galles, C.; Prez, G.M.; Penkov, S.; Boland, S.; Porta, E.O.J.; Altabe, S.G.; Labadie, G.R.; Schmidt, U.; Knölker, H.J.; Kurzchalia, T.V.; et al. Endocannabinoids in Caenorhabditis elegans are essential for the mobilization of cholesterol from internal reserves. Sci. Rep. 2018, 8, 6398. [Google Scholar] [CrossRef]
- Huang, X.; Suyama, K.; Buchanan, J.; Zhu, A.J.; Scott, M.P. A Drosophila model of the Niemann-Pick type C lysosome storage disease: Dnpc1a is required for molting and sterol homeostasis. Development 2005, 132, 5115–5124. [Google Scholar] [CrossRef] [Green Version]
- Fluegel, M.L.; Parker, T.J.; Pallanck, L.J. Mutations of a Drosophila NPC1 gene confer sterol and ecdysone metabolic defects. Genetics 2006, 172, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Voght, S.P.; Fluegel, M.L.; Andrews, L.A.; Pallanck, L.J. Drosophila NPC1b promotes an early step in sterol absorption from the midgut epithelium. Cell Metab. 2007, 5, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Warren, J.T.; Buchanan, J.; Gilbert, L.I.; Scott, M.P. Drosophila Niemann-Pick Type C-2 genes control sterol homeostasis and steroid biosynthesis: A model of human neurodegenerative disease. Development 2007, 134, 3733–3742. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.E.; Woodruff, E.A., III; Liang, P.; Patten, M.; Broadie, K. Neuronal loss of Drosophila NPC1a causes cholesterol aggregation and age-progressive neurodegeneration. J. Neurosci. 2008, 28, 6569–6582. [Google Scholar] [CrossRef]
- Danielsen, E.T.; Moeller, M.E.; Yamanaka, N.; Ou, Q.; Laursen, J.M.; Soenderholm, C.; Zhuo, R.; Phelps, B.; Tang, K.; Zeng, J.; et al. A Drosophila Genome-Wide Screen Identifies Regulators of Steroid Hormone Production and Developmental Timing. Dev. Cell 2016, 37, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Schwend, T.; Loucks, E.J.; Snyder, D.; Ahlgren, S.C. Requirement of Npc1 and availability of cholesterol for early embryonic cell movements in zebrafish. J. Lipid Res. 2011, 52, 1328–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louwette, S.; Régal, L.; Wittevrongel, C.; Thys, C.; Vandeweeghde, G.; Decuyper, E.; Leemans, P.; De Vos, R.; Van Geet, C.; Jaeken, J.; et al. NPC1 defect results in abnormal platelet formation and function: Studies in Niemann-Pick disease type C1 patients and zebrafish. Hum. Mol. Genet. 2013, 22, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Cai, X.; Wang, G.; Ouyang, G.; Cao, H. Model construction of Niemann-Pick type C disease in zebrafish. Biol. Chem. 2018, 399, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.-C.; Loeb, H.E.; Pei, W.; Tsai-Morris, C.-H.; Xu, L.; Cluzeau, C.V.; Wassif, C.A.; Feldman, B.; Burgess, S.M.; Pavan, W.J.; et al. Modeling Niemann-Pick disease type C1 in zebrafish: A robust platform for in vivo screening of candidate therapeutic compounds. Dis. Model. Mech. 2018, 11, dmm034165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentchev, P.G.; Gal, A.E.; Booth, A.D.; Omodeo-Sale, F.; Fours, J.; Neumeyer, B.A.; Quirk, J.M.; Dawson, G.; Brady, R.O. A lysosomal storage disorder in mice characterized by a dual deficiency of sphingomyelinase and glucocerebrosidase. Biochim. Biophys. Acta 1980, 619, 669–679. [Google Scholar] [CrossRef]
- Miyawaki, S.; Mitsuoka, S.; Sakiyama, T.; Kitagawa, T. Sphingomyelinosis, a new mutation in the mouse: A model of Niemann-Pick disease in humans. J. Hered. 1982, 73, 257–263. [Google Scholar] [CrossRef]
- Xie, X.; Brown, M.S.; Shelton, J.M.; Richardson, J.A.; Goldstein, J.L.; Liang, G. Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 15330–15335. [Google Scholar] [CrossRef] [Green Version]
- Maue, R.A.; Burgess, R.W.; Wang, B.; Wooley, C.M.; Seburn, K.L.; Vanier, M.T.; Rogers, M.A.; Chang, C.C.; Chang, T.Y.; Harris, B.T.; et al. A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet. 2012, 21, 730–750. [Google Scholar] [CrossRef] [Green Version]
- Praggastis, M.; Tortelli, B.; Zhang, J.; Fujiwara, H.; Sidhu, R.; Chacko, A.; Chen, Z.; Chung, C.; Lieberman, A.P.; Sikora, J.; et al. A murine Niemann-Pick C1 I1061T knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 2015, 35, 8091–8106. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Grau, M.; Albaigès, J.; Casas, J.; Auladell, C.; Dierssen, M.; Vilageliu, L.; Grinberg, D. New murine Niemann-Pick type C models bearing a pseudoexon-generating mutation recapitulate the main neurobehavioural and molecular features of the disease. Sci. Rep. 2017, 7, 41931. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Gil, J.L.; Watkins-Chow, D.E.; Baxter, L.L.; Elliot, G.; Harper, U.L.; Wincovitch, S.M.; Wedel, J.C.; Incao, A.A.; Huebecker, M.; Boehm, F.; et al. Genetic background modifies phenotypic severity and longevity in a mouse model of Niemann-Pick disease type C1. Dis. Model. Mech. 2020, 13, dmm042614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrick, M.J.; Pacheco, C.D.; Yu, T.; Dadgar, N.; Shakkottai, V.G.; Ware, C.; Paulson, H.L.; Lieberman, A.P. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet. 2010, 19, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimkunas, V.M.; Graham, M.J.; Crooke, R.M.; Liscum, L. In vivo antisense oligonucleotide reduction of NPC1 expression as a novel mouse model for Niemann Pick type C-associated liver disease. Hepatology 2008, 47, 1504–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Strnatka, D.; Donohue, C.; Hallows, J.L.; Vincent, I.; Erickson, R.P. Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1-/- mice. J. Neurosci. Res. 2008, 86, 2848–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, M.E.; Klein, A.D.; Dimbil, U.J.; Scott, M.P. Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J. Neurosci. 2011, 31, 4367–4378. [Google Scholar] [CrossRef] [Green Version]
- Bosch, M.; Fajardo, A.; Alcalá-Vida, R.; Fernández-Vidal, A.; Tebar, F.; Enrich, C.; Cardellach, F.; Pérez-Navarro, E.; Pol, A. Hepatic Primary and Secondary Cholesterol Deposition and Damage in Niemann-Pick Disease. Am. J. Pathol. 2016, 186, 517–523. [Google Scholar] [CrossRef] [Green Version]
- Lowenthal, A.C.; Cummings, J.F.; Wenger, D.A.; Thrall, M.A.; Wood, P.A.; de Lahunta, A. Feline sphingolipidosis resembling Niemann-Pick disease type C. Acta Neuropathol. 1990, 81, 189–197. [Google Scholar] [CrossRef]
- Muñana, K.R.; Luttgen, P.J.; Thrall, M.A.; Mitchell, T.W.; Wenger, D.A. Neurological manifestations of Niemann-Pick disease type C in cats. J. Vet. Intern. Med. 1994, 8, 117–121. [Google Scholar] [CrossRef]
- Somers, K.L.; Royals, M.A.; Carstea, E.D.; Rafi, M.A.; Wenger, D.A.; Thrall, M.A. Mutation analysis of feline Niemann-Pick C1 disease. Mol. Genet. Metab. 2003, 79, 99–103. [Google Scholar] [CrossRef]
- Woolley, S.A.; Tsimnadis, E.R.; Lenghaus, C.; Healy, P.J.; Walker, K.; Morton, A.; Khatkar, M.S.; Elliott, A.; Kaya, E.; Hoerner, C.; et al. Molecular basis for a new bovine model of Niemann-Pick type C disease. PLoS ONE 2020, 15, e0238697. [Google Scholar] [CrossRef]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Price, S.M.; Verot, L.; Shen, M.M.; Tint, G.S.; Vanier, M.T.; Walkley, S.U.; Lobel, P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. USA 2004, 101, 5886–5891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrantz, N.; Sagiv, Y.; Liu, Y.; Savage, P.B.; Bendelac, A.; Teyton, L. The Niemann-Pick type C2 protein loads isoglobotrihexosylceramide onto CD1d molecules and contributes to the thymic selection of NKT cells. J. Exp. Med. 2007, 204, 841–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, G.K.; Dagnaes-Hansen, F.; Holm, I.E.; Meaney, S.; Symula, D.; Andersen, N.T.; Heegaard, C.W. Protein Replacement Therapy Partially Corrects the Cholesterol-Storage Phenotype in a Mouse Model of Niemann-Pick Type C2 Disease. PLoS ONE 2011, 6, e27287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuña, M.; González-Hódar, L.; Amigo, L.; Castro, J.; Morales, M.G.; Cancino, G.I.; Groen, A.K.; Young, J.; Miquel, J.F.; Zanlungo, S. Transgenic overexpression of Niemann-Pick C2 protein promotes cholesterol gallstone formation in mice. J. Hepatol. 2016, 64, 361–369. [Google Scholar] [CrossRef]
- Kuwamura, M.; Awakura, T.; Shimada, A.; Umemura, T.; Kagota, K.; Kawamura, N.; Naiki, M. Type C Niemann-Pick disease in a boxer dog. Acta Neuropathol. 1993, 85, 345–348. [Google Scholar] [CrossRef]
- Zervas, M.; Dobrenis, K.; Walkley, S.U. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropathol. Exp. Neurol. 2001, 60, 49–64. [Google Scholar] [CrossRef] [Green Version]
- Reid, P.C.; Sakashita, N.; Sugii, S.; Ohno-Iwashita, Y.; Shimada, Y.; Hickey, W.F.; Chang, T.Y. A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J. Lipid Res. 2004, 45, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Beltroy, E.P.; Richardson, J.A.; Horton, J.D.; Turley, S.D.; Dietschy, J.M. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology 2005, 42, 886–893. [Google Scholar] [CrossRef]
- Li, H.; Repa, J.J.; Valasek, M.A.; Beltroy, E.P.; Turley, S.D.; German, D.C.; Dietschy, J.M. Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick type C disease. J. Neuropathol. Exp. Neurol. 2005, 64, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Claudepierre, T.; Paques, M.; Simonutti, M.; Buard, I.; Sahel, J.; Maue, R.A.; Picaud, S.; Pfrieger, F.W. Lack of Niemann Pick type C1 induces age-related degeneration in the mouse retina. Mol. Cell Neurosci. 2010, 43, 164–176. [Google Scholar] [CrossRef]
- Tanaka, J.; Nakamura, H.; Miyawaki, S. Cerebellar involvement in murine sphingomyelinosis: A new model of Niemann-Pick disease. J. Neuropathol. Exp. Neurol. 1988, 47, 291–300. [Google Scholar] [CrossRef]
- Higashi, Y.; Murayama, S.; Pentchev, P.G.; Suzuki, K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 1993, 85, 175–184. [Google Scholar] [CrossRef]
- Sarna, J.R.; Larouche, M.; Marzban, H.; Sillitoe, R.V.; Rancourt, D.E.; Hawkes, R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol. 2003, 456, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.C.; Milenkovic, L.; Beier, S.M.; Manuel, H.; Buchanan, J.; Scott, M.P. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS. Genet. 2005, 1, 81–95. [Google Scholar] [PubMed]
- Liao, G.; Yao, Y.; Liu, J.; Yu, Z.; Cheung, S.; Xie, A.; Liang, X.; Bi, X. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am. J. Pathol. 2007, 171, 962–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, C.D.; Kunkel, R.; Lieberman, A.P. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007, 16, 1495–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, M.; Gaynor, K.; Olm, V.; Mercken, M.; LaFrancois, J.; Wang, L.; Mathews, P.M.; Noble, W.; Matsuoka, Y.; Duff, K. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J. Neurosci. 2003, 23, 5645–5649. [Google Scholar] [CrossRef] [Green Version]
- Erickson, R.P.; Garver, W.S.; Camargo, F.; Hossain, G.S.; Heidenreich, R.A. Pharmacological and genetic modifications of somatic cholesterol do not substantially alter the course of CNS disease in Niemann-Pick C mice. J. Inherit. Metab. Dis 2000, 23, 54–62. [Google Scholar] [CrossRef]
- Erickson, R.P.; Bernard, O. Studies on neuronal death in the mouse model of Niemann-Pick C disease. J. Neurosci. Res. 2002, 68, 738–744. [Google Scholar] [CrossRef]
- Zhang, M.; Hallows, J.L.; Wang, X.; Bu, B.; Wang, W.; Vincent, I. Mitogen-activated protein kinase activity may not be necessary for the neuropathology of Niemann-Pick type C mice. J. Neurochem. 2008, 107, 814–822. [Google Scholar] [CrossRef]
- Somers, K.L.; Brown, D.E.; Fulton, R.; Schultheiss, P.C.; Hamar, D.; Smith, M.O.; Allison, R.; Connally, H.E.; Just, C.; Mitchell, T.W.; et al. Effects of dietary cholesterol restriction in a feline model of Niemann-Pick type C disease. J. Inherit. Metab. Dis. 2001, 24, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Hunter, R.E.; Flax, J.D.; Snyder, E.Y.; Erickson, R.P. Neural stem cell implantation extends life in Niemann-Pick C1 mice. J. Appl Genet. 2007, 48, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Han, H.S.; Youn, D.H.; Carter, J.E.; Modo, M.; Schuchman, E.H.; Jin, H.K. Bone marrow-derived mesenchymal stem cells promote neuronal networks with functional synaptic transmission after transplantation into mice with neurodegeneration. Stem. Cells 2007, 25, 1307–1316. [Google Scholar] [CrossRef]
- Lee, H.; Bae, J.S.; Jin, H.K. Human umbilical cord blood-derived mesenchymal stem cells improve neurological abnormalities of Niemann-Pick type C mouse by modulation of neuroinflammatory condition. J. Vet. Med. Sci. 2010, 72, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, J.S.; Carter, J.E.; Jin, H.K. Adipose tissue-derived stem cells rescue Purkinje neurons and alleviate inflammatory responses in Niemann-Pick disease type C mice. Cell Tissue Res. 2010, 340, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Wallom, K.L.; Williams, I.M.; Jeyakumar, M.; Platt, F.M. Beneficial effects of anti-inflammatory therapy in a mouse model of Niemann-Pick disease type C1. Neurobiol. Dis. 2009, 36, 242–251. [Google Scholar] [CrossRef]
- Bascuñan-Castillo, E.C.; Erickson, R.P.; Howison, C.M.; Hunter, R.J.; Heidenreich, R.H.; Hicks, C.; Trouard, T.P.; Gillies, R.J. Tamoxifen and vitamin E treatments delay symptoms in the mouse model of Niemann-Pick C. J. Appl. Genet. 2004, 45, 461–467. [Google Scholar]
- Marín, T.; Contreras, P.; Castro, J.F.; Chamorro, D.; Balboa, E.; Bosch-Morató, M.; Muñoz, F.J.; Alvarez, A.R.; Zanlungo, S. Vitamin E dietary supplementation improves neurological symptoms and decreases c-Abl/p73 activation in Niemann-Pick C mice. Nutrients 2014, 6, 3000–3017. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Li, H.; Frank-Cannon, T.C.; Valasek, M.A.; Turley, S.D.; Tansey, M.G.; Dietschy, J.M. Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J. Neurosci. 2007, 27, 14470–14480. [Google Scholar] [CrossRef] [Green Version]
- Langmade, S.J.; Gale, S.E.; Frolov, A.; Mohri, I.; Suzuki, K.; Mellon, S.H.; Walkley, S.U.; Covey, D.F.; Schaffer, J.E.; Ory, D.S. Pregnane X receptor (PXR) activation: A mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc. Natl. Acad. Sci. USA 2006, 103, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Li, H.M.; Chen, Y.R.; Gu, X.S.; Duan, S. Decreased estradiol release from astrocytes contributes to the neurodegeneration in a mouse model of Niemann-Pick disease type C. GLIA 2007, 55, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.R.; Klein, A.; Castro, J.; Cancino, G.I.; Amigo, J.; Mosqueira, M.; Vargas, L.M.; Yévenes, L.F.; Bronfman, F.C.; Zanlungo, S. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 2008, 22, 3617–3627. [Google Scholar] [CrossRef]
- Liu, B.; Li, H.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 2008, 49, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Turley, S.D.; Burns, D.K.; Miller, A.M.; Repa, J.J.; Dietschy, J.M. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl. Acad. Sci. USA 2009, 106, 2377–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, C.D.; Ali, N.F.; Micsenyi, M.C.; Stephney, G.; Renault, S.; Dobrenis, K.; Ory, D.S.; Vanier, M.T.; Walkley, S.U. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 2009, 4, e6951. [Google Scholar] [CrossRef] [Green Version]
- Aqul, A.; Liu, B.; Ramirez, C.M.; Pieper, A.A.; Estill, S.J.; Burns, D.K.; Liu, B.; Repa, J.J.; Turley, S.D.; Dietschy, J.M. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 2011, 31, 9404–9413. [Google Scholar] [CrossRef]
- Vite, C.H.; Bagel, J.H.; Swain, G.P.; Prociuk, M.; Sikora, T.U.; Stein, V.M.; O’Donnell, P.; Ruane, T.; Ward, S.; Crooks, A.; et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci. Transl. Med. 2015, 7, 276ra226. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Pan, D.; Zhang, M.; Xu, J.; Li, L.; Wei, J.; Wang, X. The neuroprotective effects of cyclin-dependent kinase-5 inhibition in mice with Niemann-Pick disease type C. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2009, 29, 324–329. [Google Scholar] [CrossRef]
- Borbon, I.A.; Hillman, Z.; Duran, E., Jr.; Kiela, P.R.; Frautschy, S.A.; Erickson, R.P. Lack of efficacy of curcumin on neurodegeneration in the mouse model of Niemann-Pick C1. Pharmacol. Biochem. Behav. 2012, 101, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Stein, V.M.; Crooks, A.; Ding, W.; Prociuk, M.; O’Donnell, P.; Bryan, C.; Sikora, T.; Dingemanse, J.; Vanier, M.T.; Walkley, S.U.; et al. Miglustat improves purkinje cell survival and alters microglial phenotype in feline niemann-pick disease type C. J. Neuropathol. Exp. Neurol. 2012, 71, 434–448. [Google Scholar] [CrossRef] [Green Version]
- Nietupski, J.B.; Pacheco, J.J.; Chuang, W.L.; Maratea, K.; Li, L.; Foley, J.; Ashe, K.M.; Cooper, C.G.; Aerts, J.M.; Copeland, D.P.; et al. Iminosugar-based inhibitors of glucosylceramide synthase prolong survival but paradoxically increase brain glucosylceramide levels in Niemann-Pick C mice. Mol. Genet. Metab. 2012, 105, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Wassif, C.A.; Yanjanin, N.M.; Watkins-Chow, D.E.; Baxter, L.L.; Incao, A.; Liscum, L.; Sidhu, R.; Firnkes, S.; Graham, M.; et al. Efficacy of N-acetylcysteine in phenotypic suppression of mouse models of Niemann-Pick disease, type C1. Hum. Mol. Genet. 2013, 22, 3508–3523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argüello, G.; Martinez, P.; Peña, J.; Chen, O.; Platt, F.; Zanlungo, S.; González, M. Hepatic metabolic response to restricted copper intake in a Niemann-Pick C murine model. Metallomics 2014, 6, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Shin, Y.; Kim, H.S.; Kang, I.; Hong, I.S.; Choi, S.W.; Yu, K.R.; Kang, K.S. Donepezil enhances Purkinje cell survival and alleviates motor dysfunction by inhibiting cholesterol synthesis in a murine model of Niemann Pick disease type C. J. Neuropathol. Exp. Neurol. 2014, 73, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Williams, I.M.; Wallom, K.L.; Smith, D.A.; Al Eisa, N.; Smith, C.; Platt, F.M. Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiol. Dis. 2014, 67, 9–17. [Google Scholar] [CrossRef]
- Marques, A.R.; Aten, J.; Ottenhoff, R.; van Roomen, C.P.; Herrera Moro, D.; Claessen, N.; Vinueza Veloz, M.F.; Zhou, K.; Lin, Z.; Mirzaian, M.; et al. Reducing GBA2 Activity Ameliorates Neuropathology in Niemann-Pick Type C Mice. PLoS ONE 2015, 10, e0135889. [Google Scholar] [CrossRef] [Green Version]
- Cougnoux, A.; Clifford, S.; Salman, A.; Ng, S.L.; Bertin, J.; Porter, F.D. Necroptosis inhibition as a therapy for Niemann-Pick disease, type C1: Inhibition of RIP kinases and combination therapy with 2-hydroxypropyl-β-cyclodextrin. Mol. Genet. Metab. 2018, 125, 345–350. [Google Scholar] [CrossRef]
- Cougnoux, A.; Cluzeau, C.; Mitra, S.; Li, R.; Williams, I.; Burkert, K.; Xu, X.; Wassif, C.A.; Zheng, W.; Porter, F.D. Necroptosis in Niemann-Pick disease, type C1: A potential therapeutic target. Cell Death Dis. 2016, 7, e2147. [Google Scholar] [CrossRef] [Green Version]
- Kirkegaard, T.; Gray, J.; Priestman, D.A.; Wallom, K.L.; Atkins, J.; Olsen, O.D.; Klein, A.; Drndarski, S.; Petersen, N.H.; Ingemann, L.; et al. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 2016, 8, 355ra118. [Google Scholar] [CrossRef]
- Munkacsi, A.B.; Hammond, N.; Schneider, R.T.; Senanayake, D.S.; Higaki, K.; Lagutin, K.; Bloor, S.J.; Ory, D.S.; Maue, R.A.; Chen, F.W.; et al. Normalization of Hepatic Homeostasis in the Npc1(nmf164) Mouse Model of Niemann-Pick Type C Disease Treated with the Histone Deacetylase Inhibitor Vorinostat. J. Biol Chem. 2017, 292, 4395–4410. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Gong, X.M.; Luo, J.; Li, B.L.; Song, B.L. AAV9-NPC1 significantly ameliorates Purkinje cell death and behavioral abnormalities in mouse NPC disease. J. Lipid Res. 2017, 58, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, R.J.; Williams, I.M.; Gibson, A.L.; Davidson, C.D.; Incao, A.A.; Hubbard, B.T.; Porter, F.D.; Pavan, W.J.; Venditti, C.P. Systemic AAV9 gene therapy improves the lifespan of mice with Niemann-Pick disease, type C1. Hum. Mol. Genet. 2017, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.P.; Smith, D.A.; Morris, L.; Fletcher, C.; Colaco, A.; Huebecker, M.; Tordo, J.; Palomar, N.; Massaro, G.; Henckaerts, E.; et al. AAV9 intracerebroventricular gene therapy improves lifespan, locomotor function and pathology in a mouse model of Niemann-Pick type C1 disease. Hum. Mol. Genet. 2018, 27, 3079–3098. [Google Scholar] [CrossRef] [PubMed]
- Markmann, S.; J, J.C.-R.; Rosenberg, J.B.; De, B.P.; Kaminsky, S.M.; Crystal, R.G.; Sondhi, D. Attenuation of the Niemann-Pick type C2 disease phenotype by intracisternal administration of an AAVrh.10 vector expressing Npc2. Exp. Neurol. 2018, 306, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Matías, N.; Baulies, A.; Nuñez, S.; Alarcon-Vila, C.; Martinez, L.; Nuño, N.; Fernandez, A.; Caballeria, J.; Levade, T.; et al. Mitochondrial GSH replenishment as a potential therapeutic approach for Niemann Pick type C disease. Redox Biol. 2017, 11, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, A.; Pezzola, A.; Matteucci, A.; Di Biase, A.; Attorri, L.; Armida, M.; Martire, A.; Chern, Y.; Popoli, P. The adenosine A(2A) receptor agonist T1-11 ameliorates neurovisceral symptoms and extends the lifespan of a mouse model of Niemann-Pick type C disease. Neurobiol. Dis. 2018, 110, 1–11. [Google Scholar] [CrossRef]
- Kulkarni, A.; Caporali, P.; Dolas, A.; Johny, S.; Goyal, S.; Dragotto, J.; Macone, A.; Jayaraman, R.; Fiorenza, M.T. Linear Cyclodextrin Polymer Prodrugs as Novel Therapeutics for Niemann-Pick Type C1 Disorder. Sci. Rep. 2018, 8, 9547. [Google Scholar] [CrossRef] [Green Version]
- Houben, T.; Magro Dos Reis, I.; Oligschlaeger, Y.; Steinbusch, H.; Gijbels, M.J.J.; Hendrikx, T.; Binder, C.J.; Cassiman, D.; Westerterp, M.; Prickaerts, J.; et al. Pneumococcal Immunization Reduces Neurological and Hepatic Symptoms in a Mouse Model for Niemann-Pick Type C1 Disease. Front. Immunol. 2018, 9, 3089. [Google Scholar] [CrossRef]
- Moreau, D.; Vacca, F.; Vossio, S.; Scott, C.; Colaco, A.; Paz Montoya, J.; Ferguson, C.; Damme, M.; Moniatte, M.; Parton, R.G.; et al. Drug-induced increase in lysobisphosphatidic acid reduces the cholesterol overload in Niemann-Pick type C cells and mice. EMBO Rep. 2019, 20, e47055. [Google Scholar] [CrossRef]
- Yasmin, N.; Ishitsuka, Y.; Fukaura, M.; Yamada, Y.; Nakahara, S.; Ishii, A.; Kondo, Y.; Takeo, T.; Nakagata, N.; Motoyama, K.; et al. In Vitro and In Vivo Evaluation of 6-O-α-Maltosyl-β-Cyclodextrin as a Potential Therapeutic Agent Against Niemann-Pick Disease Type C. Int. J. Mol. Sci. 2019, 20, 1152. [Google Scholar] [CrossRef] [Green Version]
- Park, M.H.; Choi, B.J.; Jeong, M.S.; Lee, J.Y.; Jung, I.K.; Park, K.H.; Lee, H.W.; Yamaguchi, T.; Marti, H.H.; Lee, B.H.; et al. Characterization of the Subventricular-Thalamo-Cortical Circuit in the NP-C Mouse Brain, and New Insights Regarding Treatment. Mol. Ther. 2019, 27, 1507–1526. [Google Scholar] [CrossRef] [PubMed]
- Mitroi, D.N.; Pereyra-Gómez, G.; Soto-Huelin, B.; Senovilla, F.; Kobayashi, T.; Esteban, J.A.; Ledesma, M.D. NPC1 enables cholesterol mobilization during long-term potentiation that can be restored in Niemann-Pick disease type C by CYP46A1 activation. EMBO Rep. 2019, 20, e48143. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.L.; Fawaz, M.V.; Azaria, R.D.; Hollon, T.C.; Liu, E.A.; Kunkel, T.J.; Halseth, T.A.; Krus, K.L.; Ming, R.; Morin, E.E.; et al. Synthetic high-density lipoprotein nanoparticles for the treatment of Niemann-Pick diseases. BMC Med. 2019, 17, 200. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.; Yeh, W.H.; Pendse, N.; Davis, J.R.; Hennessey, E.; Butcher, R.; Koblan, L.W.; Comander, J.; Liu, Q.; Liu, D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020, 4, 97–110. [Google Scholar] [CrossRef]
- Hung, Y.H.; Lotan, A.; Yeshurun, S.; Schroeder, A.; Bush, A.I. Iron chelation by deferiprone does not rescue the Niemann-Pick Disease Type C1 mouse model. Biometals 2020, 33, 87–95. [Google Scholar] [CrossRef]
- Jiang, D.; Lee, H.; Pardridge, W.M. Plasmid DNA gene therapy of the Niemann-Pick C1 mouse with transferrin receptor-targeted Trojan horse liposomes. Sci. Rep. 2020, 10, 13334. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Marçais, C.; Rafi, M.A.; Yamamoto, T.; Morris, J.A.; Pentchev, P.G.; Ohno, K.; Wenger, D.A.; Vanier, M.T. Niemann-Pick C1 disease: The I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am. J. Hum. Genet. 1999, 65, 1321–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, W.D.; O’Brien, J.F.; Lundquist, P.A.; Kraft, D.L.; Vockley, C.W.; Karnes, P.S.; Patterson, M.C.; Snow, K. Identification of 58 novel mutations in Niemann-Pick disease type C: Correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum. Mutat. 2003, 22, 313–325. [Google Scholar] [CrossRef]
- Millat, G.; Marçais, C.; Tomasetto, C.; Chikh, K.; Fensom, A.H.; Harzer, K.; Wenger, D.A.; Ohno, K.; Vanier, M.T. Niemann-Pick C1 disease: Correlations between NPC1 mutations, levels of NPC1 protein, and phenotypes emphasize the functional significance of the putative sterol-sensing domain and of the cysteine-rich luminal loop. Am. J. Hum. Genet. 2001, 68, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Benussi, A.; Alberici, A.; Premi, E.; Bertasi, V.; Cotelli, M.S.; Turla, M.; Dardis, A.; Zampieri, S.; Marchina, E.; Paghera, B.; et al. Phenotypic heterogeneity of Niemann-Pick disease type C in monozygotic twins. J. Neurol. 2015, 262, 642–647. [Google Scholar] [CrossRef]
- Miyawaki, S.; Yoshida, H.; Mitsuoka, S.; Enomoto, H.; Ikehara, S. A mouse model for Niemann-Pick disease. Influence of genetic background on disease expression in spm/spm mice. J. Hered 1986, 77, 379–384. [Google Scholar] [CrossRef]
- Zhang, J.; Erickson, R.P. A modifier of Niemann Pick C 1 maps to mouse chromosome 19. Mamm. Genome 2000, 11, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Parra, J.; Klein, A.D.; Castro, J.; Morales, M.G.; Mosqueira, M.; Valencia, I.; Cortés, V.; Rigotti, A.; Zanlungo, S. Npc1 deficiency in the C57BL/6J genetic background enhances Niemann-Pick disease type C spleen pathology. Biochem. Biophys. Res. Commun. 2011, 413, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.A.; Watkins-Chow, D.E.; Palladino, G.; Deutsch, G.; Chandran, K.; Pavan, W.J.; Erickson, R.P. In Niemann-Pick C1 mouse models, glial-only expression of the normal gene extends survival much further than do changes in genetic background or treatment with hydroxypropyl-beta-cyclodextrin. Gene 2018, 643, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Turley, S.D.; Pentchev, P.G.; Dietschy, J.M. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am. J. Physiol 1999, 276, E336–E344. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Lund, E.G.; Turley, S.D.; Russell, D.W.; Dietschy, J.M. Quantitation of two pathways for cholesterol excretion from the brain in normal mice and mice with neurodegeneration. J. Lipid Res. 2003, 44, 1780–1789. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Turley, S.D.; Liu, B.; Repa, J.J.; Dietschy, J.M. GM2/GD2 and GM3 gangliosides have no effect on cellular cholesterol pools or turnover in normal or NPC1 mice. J. Lipid Res. 2008, 49, 1816–1828. [Google Scholar] [CrossRef] [Green Version]
- Kaptzan, T.; West, S.A.; Holicky, E.L.; Wheatley, C.L.; Marks, D.L.; Wang, T.; Peake, K.B.; Vance, J.; Walkley, S.U.; Pagano, R.E. Development of a Rab9 transgenic mouse and its ability to increase the lifespan of a murine model of Niemann-Pick type C disease. Am. J. Pathol. 2009, 174, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Maulik, M.; Ghoshal, B.; Kim, J.; Wang, Y.; Yang, J.; Westaway, D.; Kar, S. Mutant human APP exacerbates pathology in a mouse model of NPC and its reversal by a beta-cyclodextrin. Hum. Mol. Genet. 2012, 21, 4857–4875. [Google Scholar] [CrossRef]
- Jelinek, D.; Castillo, J.J.; Garver, W.S. The C57BL/6J Niemann-Pick C1 mouse model with decreased gene dosage has impaired glucose tolerance independent of body weight. Gene 2013, 527, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Maulik, M.; Thinakaran, G.; Kar, S. Alterations in gene expression in mutant amyloid precursor protein transgenic mice lacking Niemann-Pick type C1 protein. PLoS ONE 2013, 8, e54605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Lee, J.K.; Bae, Y.C.; Yang, S.H.; Okino, N.; Schuchman, E.H.; Yamashita, T.; Bae, J.S.; Jin, H.K. Inhibition of GM3 synthase attenuates neuropathology of Niemann-Pick disease Type C. by affecting sphingolipid metabolism. Mol. Cells 2014, 37, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.; Elrick, M.J.; Dell’Orco, J.M.; Qin, Z.S.; Kalyana-Sundaram, S.; Chinnaiyan, A.M.; Shakkottai, V.G.; Lieberman, A.P. Heat Shock Protein Beta-1 Modifies Anterior to Posterior Purkinje Cell Vulnerability in a Mouse Model of Niemann-Pick Type C Disease. PLoS Genet. 2016, 12, e1006042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, A.M.; Jones, R.D.; Repa, J.J.; Turley, S.D. Niemann-Pick C1-deficient mice lacking sterol O-acyltransferase 2 have less hepatic cholesterol entrapment and improved liver function. Am. J. Physiol. Liver Physiol. 2018, 315, G454–G463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.D.; Shin, A.; Mayagoitia, K.; Siebold, L.; Rubini, M.; Wilson, C.G.; Bellinger, D.L.; Soriano, S. Loss of amyloid precursor protein exacerbates early inflammation in Niemann-Pick disease type C. J. Neuroinflamm. 2019, 16, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cawley, N.X.; Lyons, A.T.; Abebe, D.; Wassif, C.A.; Porter, F.D. Evaluation of the Potential Role of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Niemann–Pick Disease, Type C1. Int. J. Mol. Sci. 2020, 21, 2430. [Google Scholar] [CrossRef] [Green Version]
- Cougnoux, A.; Yerger, J.C.; Fellmeth, M.; Serra-Vinardell, J.; Martin, K.; Navid, F.; Iben, J.R.; Wassif, C.A.; Cawley, N.X.; Porter, F.D. Single Cell Transcriptome Analysis of Niemann–Pick Disease, Type C1 Cerebella. Int. J. Mol. Sci. 2020, 21, 5368. [Google Scholar] [CrossRef]
- Klein, A.D.; De La Vega, J.G.; Zanlungo, S. Complement Component C3 Participates in Early Stages of Niemann–Pick C Mouse Liver Damage. Int. J. Mol. Sci. 2020, 21, 2127. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.A.; Schultz, M.L.; Mochida, C.; Chung, C.; Paulson, H.L.; Lieberman, A.P. Fbxo2 mediates clearance of damaged lysosomes and modifies neurodegeneration in the Niemann-Pick C brain. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Ramirez, C.M.; Taylor, A.M.; Lopez, A.M.; Repa, J.J.; Turley, S.D. Delineation of metabolic responses of Npc1(-/-nih) mice lacking the cholesterol-esterifying enzyme SOAT2 to acute treatment with 2-hydroxypropyl-β-cyclodextrin. Steroids 2020, 164, 108725. [Google Scholar] [CrossRef]
- Smith, A.F.; Vanderah, T.W.; Erickson, R.P. Haploinsufficiency of tau decreases survival of the mouse model of Niemann–Pick disease type C1 but does not alter tau phosphorylation. J. Appl. Genet. 2020, 61, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Võikar, V.; Rauvala, H.; Ikonen, E. Cognitive deficit and development of motor impairment in a mouse model of Niemann-Pick type C disease. Behav. Brain Res. 2002, 132, 1–10. [Google Scholar] [CrossRef]
- Cougnoux, A.; Fellmeth, M.; Gu, T.; Davidson, C.D.; Gibson, A.L.; Pavan, W.J.; Porter, F.D. Maternal immune activation modifies the course of Niemann-pick disease, type C1 in a gender specific manner. Mol. Genet. Metab. 2020, 129, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Houben, T.; Bitorina, A.V.; Oligschlaeger, Y.; Jeurissen, M.L.; Rensen, S.; Köhler, S.E.; Westerterp, M.; Lütjohann, D.; Theys, J.; Romano, A.; et al. Sex-opposed inflammatory effects of 27-hydroxycholesterol are mediated via differences in estrogen signaling. J. Pathol. 2020, 251, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Tchernof, A.; Després, J.P. Pathophysiology of human visceral obesity: An update. Physiol. Rev. 2013, 93, 359–404. [Google Scholar] [CrossRef] [PubMed]
- Regitz-Zagrosek, V.; Kararigas, G. Mechanistic Pathways of Sex Differences in Cardiovascular Disease. Physiol. Rev. 2017, 97, 1–37. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Bairey Merz, N.; Barnes, P.J.; Brinton, R.D.; Carrero, J.J.; DeMeo, D.L.; De Vries, G.J.; Epperson, C.N.; Govindan, R.; Klein, S.L.; et al. Sex and gender: Modifiers of health, disease, and medicine. Lancet 2020, 396, 565–582. [Google Scholar] [CrossRef]
- Segatto, M.; Di Giovanni, A.; Marino, M.; Pallottini, V. Analysis of the protein network of cholesterol homeostasis in different brain regions: An age and sex dependent perspective. J. Cell Physiol. 2013, 228, 1561–1567. [Google Scholar] [CrossRef]
- Tonini, C.; Segatto, M.; Pallottini, V. Impact of Sex and Age on the Mevalonate Pathway in the Brain: A Focus on Effects Induced by Maternal Exposure to Exogenous Compounds. Metab. 2020, 10, 304. [Google Scholar] [CrossRef]
- Yu, T.; Shakkottai, V.G.; Chung, C.; Lieberman, A.P. Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum. Mol. Genet. 2011, 20, 4440–4451. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Lieberman, A.P. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 2013, 9, e1003462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borbon, I.; Totenhagen, J.; Fiorenza, M.T.; Canterini, S.; Ke, W.; Trouard, T.; Erickson, R.P. Niemann-Pick C1 mice, a model of “juvenile Alzheimer’s disease”, with normal gene expression in neurons and fibrillary astrocytes show long term survival and delayed neurodegeneration. J. Alzheimers Dis 2012, 30, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.D.; Oyarzún, J.E.; Cortez, C.; Zanlungo, S. Gadolinium Chloride Rescues Niemann–Pick Type C Liver Damage. Int. J. Mol. Sci. 2018, 19, 3599. [Google Scholar] [CrossRef] [Green Version]
- Slezak, M.; Goritz, C.; Niemiec, A.; Frisen, J.; Chambon, P.; Metzger, D.; Pfrieger, F.W. Transgenic mice for conditional gene manipulation in astroglial cells. GLIA 2007, 55, 1565–1576. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Slezak, M. Genetic approaches to study glial cells in the rodent brain. GLIA 2012, 60, 681–701. [Google Scholar] [CrossRef]
- German, D.C.; Liang, C.L.; Song, T.; Yazdani, U.; Xie, C.; Dietschy, J.M. Neurodegeneration in the Niemann-Pick C mouse: Glial involvement. Neuroscience 2002, 109, 437–450. [Google Scholar] [CrossRef]
- Baudry, M.; Yao, Y.; Simmons, D.; Liu, J.; Bi, X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: Immunohistochemical observations of microglia and astroglia. Exp. Neurol. 2003, 184, 887–903. [Google Scholar] [CrossRef]
- Kavetsky, L.; Green, K.K.; Boyle, B.R.; Yousufzai, F.A.K.; Padron, Z.M.; Melli, S.E.; Kuhnel, V.L.; Jackson, H.M.; Blanco, R.E.; Howell, G.R.; et al. Increased interactions and engulfment of dendrites by microglia precede Purkinje cell degeneration in a mouse model of Niemann Pick Type-C. Sci. Rep. 2019, 9, 14722. [Google Scholar] [CrossRef] [Green Version]
- Walkley, S.U. Pyramidal neurons with ectopic dendrites in storage diseases exhibit increased GM2 ganglioside immunoreactivity. Neuroscience 1995, 68, 1027–1035. [Google Scholar] [CrossRef]
- March, P.A.; Thrall, M.A.; Brown, D.E.; Mitchell, T.W.; Lowenthal, A.C.; Walkley, S.U. GABAergic neuroaxonal dystrophy and other cytopathological alterations in feline Niemann-Pick disease type C. Acta Neuropathol. 1997, 94, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Vite, C.H.; Ding, W.; Bryan, C.; O’Donnell, P.; Cullen, K.; Aleman, D.; Haskins, M.E.; Winkle, T.V. Clinical, Electrophysiological, and Serum Biochemical Measures of Progressive Neurological and Hepatic Dysfunction in Feline Niemann-Pick Type C Disease. Pediatraic Res. 2008, 64, 544–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagel, J.H.; Sikora, T.U.; Prociuk, M.; Pesayco, J.P.; Mizisin, A.P.; Shelton, G.D.; Vite, C.H. Electrodiagnostic testing and histopathologic changes confirm peripheral nervous system myelin abnormalities in the feline model of niemann-pick disease type C. J. Neuropathol. Exp. Neurol. 2013, 72, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Roszell, B.R.; Tao, J.-Q.; Yu, K.J.; Gao, L.; Huang, S.; Ning, Y.; Feinstein, S.I.; Vite, C.H.; Bates, S.R. Pulmonary Abnormalities in Animal Models Due to Niemann-Pick Type C1 (NPC1) or C2 (NPC2) Disease. PLoS ONE 2013, 8, e67084. [Google Scholar] [CrossRef] [Green Version]
- Mauler, D.A.; Gandolfi, B.; Reinero, C.R.; O’Brien, D.P.; Spooner, J.L.; Lyons, L.A. Precision Medicine in Cats: Novel Niemann-Pick Type C1 Diagnosed by Whole-Genome Sequencing. J. Vet. Intern. Med. 2017, 31, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, S.; Bianchi, E.; Cantile, C.; Saleri, R.; Bembi, B.; Dardis, A. Characterization of a Spontaneous Novel Mutation in the NPC2 Gene in a Cat Affected by Niemann Pick Type C Disease. PLoS ONE 2014, 9, e112503. [Google Scholar] [CrossRef] [Green Version]
- Sloan, H.R.; Uhlendorf, B.W.; Kanfer, J.N.; Brady, R.O.; Fredrickson, D.S. Deficiency of sphingomyelin-cleaving enzyme activity in tissue cultures derived from patients with Niemann-Pick disease. Biochem. Biophys. Res. Commun. 1969, 34, 582–588. [Google Scholar] [CrossRef]
- Pentchev, P.G.; Boothe, A.D.; Kruth, H.S.; Weintroub, H.; Stivers, J.; Brady, R.O. A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. J. Biol. Chem. 1984, 259, 5784–5791. [Google Scholar] [PubMed]
- Liscum, L.; Faust, J.R. Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J. Biol. Chem. 1987, 262, 17002–17008. [Google Scholar]
- Mazière, C.; Mazière, J.C.; Mora, L.; Lageron, A.; Polonovski, C.; Polonovski, J. Alterations in cholesterol metabolism in cultured fibroblasts from patients with Niemann-Pick disease type C. J. Inherit. Metab. Dis. 1987, 10, 339–346. [Google Scholar] [CrossRef]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 Disease Gene: Homology to Mediators of Cholesterol Homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelsthorpe, M.E.; Baumann, N.; Millard, E.; Gale, S.E.; Langmade, S.J.; Schaffer, J.E.; Ory, D.S. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 2008, 283, 8229–8236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liscum, L.; Faust, J.R. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J. Biol. Chem. 1989, 264, 11796–11806. [Google Scholar] [PubMed]
- Rodriguez-Lafrasse, C.; Rousson, R.; Bonnet, J.; Pentchev, P.G.; Louisot, P.; Vanier, M.T. Abnormal cholesterol metabolism in imipramine-treated fibroblast cultures. Similarities with Niemann-Pick type C disease. Biochim. Biophys. Acta 1990, 1043, 123–128. [Google Scholar] [CrossRef]
- Roff, C.F.; Goldin, E.; Comly, M.E.; Cooney, A.; Brown, A.; Vanier, M.T.; Miller, S.P.; Brady, R.O.; Pentchev, P.G. Type C Niemann-Pick disease: Use of hydrophobic amines to study defective cholesterol transport. Dev. Neurosci. 1991, 13, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.H.; Cheung, N.S. Cellular mechanism of U18666A-mediated apoptosis in cultured murine cortical neurons: Bridging Niemann-Pick disease type C and Alzheimer’s disease. Cell Signal. 2006, 18, 1844–1853. [Google Scholar] [CrossRef]
- Cenedella, R.J. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids 2009, 44, 477–487. [Google Scholar] [CrossRef]
- Lu, F.; Liang, Q.; Abi-Mosleh, L.; Das, A.; De Brabander, J.K.; Goldstein, J.L.; Brown, M.S. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. eLife 2015, 4, e12177. [Google Scholar] [CrossRef]
- Levade, T.; Salvayre, R.; Lenoir, G.; Douste-Blazy, L. Sphingomyelinase and nonspecific phosphodiesterase activities in Epstein-Barr virus-transformed lymphoid cell lines from Niemann-Pick disease A, B and C. Biochim. Biophys. Acta 1984, 793, 321–324. [Google Scholar] [CrossRef]
- Ohno, K.; Nanba, E.; Miyawaki, S.; Sakiyama, T.; Kitagawa, T.; Takeshita, K. A cell line derived from sphingomyelinosis mouse shows alterations in intracellular cholesterol metabolism similar to those in type C Niemann-Pick disease. Cell Struct. Funct. 1992, 17, 229–235. [Google Scholar] [CrossRef] [Green Version]
- Byers, D.M.; Douglas, J.A.; Cook, H.W.; Palmer, F.B.; Ridgway, N.D. Regulation of intracellular cholesterol metabolism is defective in lymphoblasts from Niemann-Pick type C and type D patients. Biochim. Biophys. Acta 1994, 1226, 173–180. [Google Scholar] [CrossRef]
- Brown, A.; Patel, S.; Ward, C.; Lorenz, A.; Ortiz, M.; DuRoss, A.; Wieghardt, F.; Esch, A.; Otten, E.G.; Heiser, L.M.; et al. PEG-lipid micelles enable cholesterol efflux in Niemann-Pick Type C1 disease-based lysosomal storage disorder. Sci. Rep. 2016, 6, 31750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Pike, D.; Sleat, D.E.; Nanda, V.; Lobel, P. Potential pitfalls and solutions for use of fluorescent fusion proteins to study the lysosome. PLoS ONE 2014, 9, e88893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, E.M.; Walker, T.N.; Hildreth, J.E. Loss of Niemann Pick type C proteins 1 and 2 greatly enhances HIV infectivity and is associated with accumulation of HIV Gag and cholesterol in late endosomes/lysosomes. Virol. J. 2012, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadigan, K.M.; Spillane, D.M.; Chang, T.Y. Isolation and characterization of Chinese hamster ovary cell mutants defective in intracellular low density lipoprotein-cholesterol trafficking. J. Cell Biol. 1990, 110, 295–308. [Google Scholar] [CrossRef]
- Dahl, N.K.; Reed, K.L.; Daunais, M.A.; Faust, J.R.; Liscum, L. Isolation and characterization of Chinese hamster ovary cells defective in the intracellular metabolism of low density lipoprotein-derived cholesterol. J. Biol. Chem. 1992, 267, 4889–4896. [Google Scholar]
- Higaki, K.; Ninomiya, H.; Sugimoto, Y.; Suzuki, T.; Taniguchi, M.; Niwa, H.; Pentchev, P.G.; Vanier, M.T.; Ohno, K. Isolation of NPC1-deficient Chinese hamster ovary cell mutants by gene trap mutagenesis. J. Biochem. 2001, 129, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Cruz, J.C.; Sugii, S.; Yu, C.; Chang, T.Y. Role of Niemann-Pick type C1 protein in intracellular trafficking of low density lipoprotein-derived cholesterol. J. Biol. Chem. 2000, 275, 4013–4021. [Google Scholar] [CrossRef] [Green Version]
- Millard, E.E.; Srivastava, K.; Traub, L.M.; Schaffer, J.E.; Ory, D.S. Niemann-pick type C1 (NPC1) overexpression alters cellular cholesterol homeostasis. J. Biol. Chem. 2000, 275, 38445–38451. [Google Scholar] [CrossRef] [Green Version]
- Ko, D.C.; Binkley, J.; Sidow, A.; Scott, M.P. The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc. Natl. Acad. Sci. USA 2003, 100, 2518–2525. [Google Scholar] [CrossRef] [Green Version]
- Ko, D.C.; Gordon, M.D.; Jin, J.Y.; Scott, M.P. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol. Biol. Cell 2001, 12, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Dwyer, N.K.; Love, D.C.; Cooney, A.; Comly, M.; Neufeld, E.; Pentchev, P.G.; Blanchette-Mackie, E.J.; Hanover, J.A. Cessation of rapid late endosomal tubulovesicular trafficking in Niemann-Pick type C1 disease. Proc. Natl. Acad. Sci. USA 2001, 98, 4466–4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Zhang, Y.Y.; Luo, J.; Wang, J.Q.; Zhou, Y.X.; Miao, H.H.; Shi, X.J.; Qu, Y.X.; Xu, J.; Li, B.L.; et al. The GARP Complex Is Involved in Intracellular Cholesterol Transport via Targeting NPC2 to Lysosomes. Cell Rep. 2017, 19, 2823–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimi, V.; Hashemi, A. Challenges of in vitro genome editing with CRISPR/Cas9 and possible solutions: A review. Gene 2020, 753, 144813. [Google Scholar] [CrossRef]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab. 2009, 10, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Lukmantara, I.; Yang, H. CRISPR/Cas9-Mediated Generation of Niemann-Pick C1 Knockout Cell Line. Methods Mol. Biol. 2017, 1583, 73–83. [Google Scholar]
- Tharkeshwar, A.K.; Trekker, J.; Vermeire, W.; Pauwels, J.; Sannerud, R.; Priestman, D.A.; Te Vruchte, D.; Vints, K.; Baatsen, P.; Decuypere, J.P.; et al. A novel approach to analyze lysosomal dysfunctions through subcellular proteomics and lipidomics: The case of NPC1 deficiency. Sci. Rep. 2017, 7, 41408. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Ridgway, N.D. Oxysterol-Binding Protein-Related Protein 1L Regulates Cholesterol Egress from the Endo-Lysosomal System. Cell Rep. 2017, 19, 1807–1818. [Google Scholar] [CrossRef] [Green Version]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Pascau, L.; Coll, M.J.; Casas, J.; Vilageliu, L.; Grinberg, D. Generation of a human neuronal stable cell model for niemann-pick C disease by RNA interference. JIMD Rep. 2012, 4, 29–37. [Google Scholar]
- Ulatowski, L.; Parker, R.; Davidson, C.; Yanjanin, N.; Kelley, T.J.; Corey, D.; Atkinson, J.; Porter, F.; Arai, H.; Walkley, S.U.; et al. Altered vitamin E status in Niemann-Pick type C disease. J. Lipid Res. 2011, 52, 1400–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twu, Y.C.; Lee, T.S.; Lin, Y.L.; Hsu, S.M.; Wang, Y.H.; Liao, C.Y.; Wang, C.K.; Liang, Y.C.; Liao, Y.J. Niemann-Pick Type C2 Protein Mediates Hepatic Stellate Cells Activation by Regulating Free Cholesterol Accumulation. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.P.; Chen, F.W.; Ioannou, Y.A. Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science 2000, 290, 2295–2298. [Google Scholar] [CrossRef] [PubMed]
- Maulik, M.; Peake, K.; Chung, J.; Wang, Y.; Vance, J.E.; Kar, S. APP overexpression in the absence of NPC1 exacerbates metabolism of amyloidogenic proteins of Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 7132–7150. [Google Scholar] [CrossRef] [Green Version]
- Watabe, K.; Ida, H.; Uehara, K.; Oyanagi, K.; Sakamoto, T.; Tanaka, J.; Garver, W.S.; Miyawaki, S.; Ohno, K.; Eto, Y. Establishment and characterization of immortalized Schwann cells from murine model of Niemann-Pick disease type C (spm/spm). J. Peripher. Nerv. Syst. 2001, 6, 85–94. [Google Scholar] [CrossRef]
- Erwood, S.; Brewer, R.A.; Bily, T.M.I.; Maino, E.; Zhou, L.; Cohn, R.D.; Ivakine, E.A. Modeling Niemann-Pick disease type C in a human haploid cell line allows for patient variant characterization and clinical interpretation. Genome Res. 2019, 29, 2010–2019. [Google Scholar] [CrossRef]
- Wang, M.L.; Motamed, M.; Infante, R.E.; Abi-Mosleh, L.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 2010, 12, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Vanharanta, L.; Peränen, J.; Pfisterer, S.G.; Enkavi, G.; Vattulainen, I.; Ikonen, E. High-content imaging and structure-based predictions reveal functional differences between Niemann-Pick C1 variants. Traffic 2020, 21, 386–397. [Google Scholar] [CrossRef] [Green Version]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Stoeck, I.K.; Lee, J.Y.; Tabata, K.; Romero-Brey, I.; Paul, D.; Schult, P.; Lohmann, V.; Kaderali, L.; Bartenschlager, R. Hepatitis C Virus Replication Depends on Endosomal Cholesterol Homeostasis. J. Virol. 2018, 92, e01196-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foo, L.C.; Allen, N.J.; Bushong, E.A.; Ventura, P.B.; Chung, W.S.; Zhou, L.; Cahoy, J.D.; Daneman, R.; Zong, H.; Ellisman, M.H.; et al. Development of a method for the purification and culture of rodent astrocytes. Neuron 2011, 71, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohl, S.G.; Reh, T.A. The microRNA expression profile of mouse Müller glia in vivo and in vitro. Sci. Rep. 2016, 6, 35423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohlen, C.J.; Bennett, F.C.; Tucker, A.F.; Collins, H.Y.; Mulinyawe, S.B.; Barres, B.A. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 2017, 94, 759–773.e758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.A.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Paşca, S.P. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 2017, 95, 779–790.e776. [Google Scholar] [CrossRef]
- Falk, T.; Garver, W.S.; Erickson, R.P.; Wilson, J.M.; Yool, A.J. Expression of Niemann-Pick type C transcript in rodent cerebellum in vivo and in vitro. Brain Res. 1999, 839, 49–57. [Google Scholar] [CrossRef]
- Henderson, L.P.; Lin, L.; Prasad, A.; Paul, C.A.; Chang, T.Y.; Maue, R.A. Embryonic striatal neurons from niemann-pick type C mice exhibit defects in cholesterol metabolism and neurotrophin responsiveness. J. Biol. Chem. 2000, 275, 20179–20187. [Google Scholar] [CrossRef] [Green Version]
- Karten, B.; Vance, D.E.; Campenot, R.B.; Vance, J.E. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J. Neurochem. 2002, 83, 1154–1163. [Google Scholar] [CrossRef]
- Jin, L.W.; Shie, F.S.; Maezawa, I.; Vincent, I.; Bird, T. Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am. J. Pathol. 2004, 164, 975–985. [Google Scholar] [CrossRef]
- Tashiro, Y.; Yamazaki, T.; Shimada, Y.; Ohno-Iwashita, Y.; Okamoto, K. Axon-dominant localization of cell-surface cholesterol in cultured hippocampal neurons and its disappearance in Niemann-Pick type C model cells. Eur. J. Neurosci. 2004, 20, 2015–2021. [Google Scholar] [CrossRef] [PubMed]
- Wasser, C.R.; Ertunc, M.; Liu, X.; Kavalali, E.T. Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. J.Physiol 2007, 579, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Demais, V.; Barthélémy, A.; Perraut, M.; Ungerer, N.; Keime, C.; Reibel, S.; Pfrieger, F.W. Reversal of Pathologic Lipid Accumulation in NPC1-Deficient Neurons by Drug-Promoted Release of LAMP1-Coated Lamellar Inclusions. J. Neurosci. 2016, 36, 8012–8025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buard, I.; Pfrieger, F.W. Relevance of neuronal and glial NPC1 for synaptic input to cerebellar Purkinje cells. Mol. Cell Neurosci. 2014, 61, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hawes, C.M.; Wiemer, H.; Krueger, S.R.; Karten, B. Pre-synaptic defects of NPC1-deficient hippocampal neurons are not directly related to plasma membrane cholesterol. J. Neurochem. 2010, 114, 311–322. [Google Scholar] [CrossRef]
- Suresh, S.; Yan, Z.; Patel, R.C.; Patel, Y.C.; Patel, S.C. Cellular cholesterol storage in the Niemann-Pick disease type C mouse is associated with increased expression and defective processing of apolipoprotein D. J. Neurochem. 1998, 70, 242–251. [Google Scholar] [CrossRef]
- Strauss, K.; Goebel, C.; Runz, H.; Mobius, W.; Weiss, S.; Feussner, I.; Simons, M.; Schneider, A. Exosome secretion ameliorates lysosomal storage of cholesterol in Niemann-Pick type C disease. J. Biol. Chem. 2010, 285, 26279–26288. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Feng, X.; Rolfs, A.; Luo, J. Lovastatin promotes myelin formation in NPC1 mutant oligodendrocytes. J. Neurol. Sci. 2018, 386, 56–63. [Google Scholar] [CrossRef]
- De Nuccio, C.; Bernardo, A.; Ferrante, A.; Pepponi, R.; Martire, A.; Falchi, M.; Visentin, S.; Popoli, P.; Minghetti, L. Adenosine A(2A) receptor stimulation restores cell functions and differentiation in Niemann-Pick type C-like oligodendrocytes. Sci. Rep. 2019, 9, 9782. [Google Scholar] [CrossRef]
- Peake, K.B.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Niemann-Pick Type C1 deficiency in microglia does not cause neuron death in vitro. Biochim. Biophys. Acta 2011, 1812, 1121–1129. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.; Kim, H.S.; Kang, I.; Choi, S.W.; Shin, T.H.; Shin, J.H.; Lee, B.C.; Lee, J.Y.; Kim, J.J.; Kook, M.G.; et al. Cathepsin S contributes to microglia-mediated olfactory dysfunction through the regulation of Cx3cl1-Cx3cr1 axis in a Niemann-Pick disease type C1 model. Glia 2016, 64, 2291–2305. [Google Scholar] [CrossRef] [PubMed]
- Walterfang, M.; Di Biase, M.A.; Cropley, V.L.; Scott, A.M.; O’Keefe, G.; Velakoulis, D.; Pathmaraj, K.; Ackermann, U.; Pantelis, C. Imaging of neuroinflammation in adult Niemann-Pick type C disease: A cross-sectional study. Neurology 2020, 94, e1716–e1725. [Google Scholar] [CrossRef] [PubMed]
- Marschalek, N.; Albert, F.; Meske, V.; Ohm, T.G. The natural history of cerebellar degeneration of Niemann-Pick C mice monitored in vitro. Neuropathol. Appl. Neurobiol. 2014, 40, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Reid, P.C.; Sugii, S.; Chang, T.Y. Trafficking defects in endogenously synthesized cholesterol in fibroblasts, macrophages, hepatocytes, and glial cells from Niemann-Pick type C1 mice. J. Lipid Res. 2003, 44, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Vainio, S.; Bykov, I.; Hermansson, M.; Jokitalo, E.; Somerharju, P.; Ikonen, E. Defective insulin receptor activation and altered lipid rafts in Niemann-Pick type C disease hepatocytes. Biochem. J. 2005, 391, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164.e110. [Google Scholar] [CrossRef]
- Te Vruchte, D.; Jeans, A.; Platt, F.M.; Sillence, D.J. Glycosphingolipid storage leads to the enhanced degradation of the B cell receptor in Sandhoff disease mice. J. Inherit. Metab. Dis. 2010, 33, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.R.; Coleman, T.; Langmade, S.J.; Scherrer, D.E.; Lane, L.; Lanier, M.H.; Feng, C.; Sands, M.S.; Schaffer, J.E.; Semenkovich, C.F.; et al. Niemann-Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J. Clin. Investig. 2008, 118, 2281–2290. [Google Scholar] [CrossRef] [Green Version]
- Roszell, B.R.; Tao, J.Q.; Yu, K.J.; Huang, S.; Bates, S.R. Characterization of the Niemann-Pick C pathway in alveolar type II cells and lamellar bodies of the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L919–L932. [Google Scholar] [CrossRef] [Green Version]
- Cougnoux, A.; Movassaghi, M.; Picache, J.A.; Iben, J.R.; Navid, F.; Salman, A.; Martin, K.; Farhat, N.Y.; Cluzeau, C.; Tseng, W.C.; et al. Gastrointestinal Tract Pathology in a BALB/c Niemann-Pick Disease Type C1 Null Mouse Model. Dig. Dis. Sci. 2018, 63, 870–880. [Google Scholar] [CrossRef]
- Speak, A.O.; Platt, N.; Salio, M.; te Vruchte, D.; Smith, D.A.; Shepherd, D.; Veerapen, N.; Besra, G.S.; Yanjanin, N.M.; Simmons, L.; et al. Invariant natural killer T cells are not affected by lysosomal storage in patients with Niemann-Pick disease type C. Eur. J. Immunol. 2012, 42, 1886–1892. [Google Scholar] [CrossRef]
- Pereira, C.S.; Pérez-Cabezas, B.; Ribeiro, H.; Maia, M.L.; Cardoso, M.T.; Dias, A.F.; Azevedo, O.; Ferreira, M.F.; Garcia, P.; Rodrigues, E.; et al. Lipid Antigen Presentation by CD1b and CD1d in Lysosomal Storage Disease Patients. Front. Immunol. 2019, 10, 1264. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, Y.; Hudspeth, K.; Mattner, J.; Schrantz, N.; Stern, R.K.; Zhou, D.; Savage, P.B.; Teyton, L.; Bendelac, A. Cutting edge: Impaired glycosphingolipid trafficking and NKT cell development in mice lacking Niemann-Pick type C1 protein. J. Immunol. 2006, 177, 26–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, N.; Speak, A.O.; Colaco, A.; Gray, J.; Smith, D.A.; Williams, I.M.; Wallom, K.L.; Platt, F.M. Immune dysfunction in Niemann-Pick disease type C. J. Neurochem. 2016, 136, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Csepeggi, C.; Jiang, M.; Frolov, A. Somatic cell plasticity and Niemann-pick type C2 protein: Adipocyte differentiation and function. J. Biol. Chem. 2010, 285, 30347–30354. [Google Scholar] [CrossRef] [Green Version]
- Busso, D.; Oñate-Alvarado, M.J.; Balboa, E.; Castro, J.; Lizama, C.; Morales, G.; Vargas, S.; Härtel, S.; Moreno, R.D.; Zanlungo, S. Spermatozoa from mice deficient in Niemann-Pick disease type C2 (NPC2) protein have defective cholesterol content and reduced in vitro fertilising ability. Reprod. Fertil. Dev. 2014, 26, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.R.; Kim, S.J.; Byun, K.H.; Hutchinson, B.; Lee, B.H.; Michikawa, M.; Lee, Y.S.; Kang, K.S. NPC1 gene deficiency leads to lack of neural stem cell self-renewal and abnormal differentiation through activation of p38 mitogen-activated protein kinase signaling. Stem. Cells 2006, 24, 292–298. [Google Scholar] [CrossRef]
- Ordonez, M.P.; Roberts, E.A.; Kidwell, C.U.; Yuan, S.H.; Plaisted, W.C.; Goldstein, L.S.B. Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum. Mol. Genet. 2012, 21, 2651–2662. [Google Scholar] [CrossRef]
- Bergamin, N.; Dardis, A.; Beltrami, A.; Cesselli, D.; Rigo, S.; Zampieri, S.; Domenis, R.; Bembi, B.; Beltrami, C.A. A human neuronal model of Niemann Pick C disease developed from stem cells isolated from patient’s skin. Orphanet. J. Rare Dis. 2013, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Trilck, M.; Hübner, R.; Seibler, P.; Klein, C.; Rolfs, A.; Frech, M.J. Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J. Rare Dis. 2013, 8, 144. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Lee, J.K.; Park, M.H.; Hong, Y.R.; Marti, H.H.; Kim, H.; Okada, Y.; Otsu, M.; Seo, E.J.; Park, J.H.; et al. Pathological roles of the VEGF/SphK pathway in Niemann-Pick type C neurons. Nat. Commun. 2014, 5, 5514. [Google Scholar] [CrossRef] [Green Version]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and Chemical Correction of Cholesterol Accumulation and Impaired Autophagy in Hepatic and Neural Cells Derived from Niemann-Pick Type C Patient-Specific iPS Cells. Stem. Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Swaroop, M.; Wang, M.; Baxa, U.; Yang, R.; Yan, Y.; Coksaygan, T.; DeTolla, L.; Marugan, J.J.; Austin, C.P.; et al. Niemann-Pick Disease Type C: Induced Pluripotent Stem Cell-Derived Neuronal Cells for Modeling Neural Disease and Evaluating Drug Efficacy. J. Biomol. Screen 2014, 19, 1164–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efthymiou, A.G.; Steiner, J.; Pavan, W.J.; Wincovitch, S.; Larson, D.M.; Porter, F.D.; Rao, M.S.; Malik, N. Rescue of an in vitro neuron phenotype identified in Niemann-Pick disease, type C1 induced pluripotent stem cell-derived neurons by modulating the WNT pathway and calcium signaling. Stem. Cells Transl. Med. 2015, 4, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Völkner, C.; Peter, F.; Liedtke, M.; Krohn, S.; Lindner, I.; Murua Escobar, H.; Cimmaruta, C.; Lukas, J.; Hermann, A.; Frech, M.J. Generation of the Niemann–Pick type C2 patient-derived iPSC line AKOSi001-A. Stem. Cell Res. 2019, 41, 101606. [Google Scholar] [CrossRef]
- Kuo, S.Y.; Castoreno, A.B.; Aldrich, L.N.; Lassen, K.G.; Goel, G.; Dančík, V.; Kuballa, P.; Latorre, I.; Conway, K.L.; Sarkar, S.; et al. Small-molecule enhancers of autophagy modulate cellular disease phenotypes suggested by human genetics. Proc. Natl. Acad. Sci. USA 2015, 112, E4281–E4287. [Google Scholar] [CrossRef] [Green Version]
- Gläser, A.; Hammerl, F.; Gräler, M.H.; Coldewey, S.M.; Völkner, C.; Frech, M.J.; Yang, F.; Luo, J.; Tönnies, E.; von Bohlen Und Halbach, O.; et al. Identification of Brain-Specific Treatment Effects in NPC1 Disease by Focusing on Cellular and Molecular Changes of Sphingosine-1-Phosphate Metabolism. Int. J. Mol. Sci. 2020, 21, 4520. [Google Scholar] [CrossRef]
- Liscum, L.; Arnio, E.; Anthony, M.; Howley, A.; Sturley, S.L.; Agler, M. Identification of a pharmaceutical compound that partially corrects the Niemann-Pick C phenotype in cultured cells. J. Lipid Res. 2002, 43, 1708–1717. [Google Scholar] [CrossRef] [Green Version]
- Swaroop, M.; Thorne, N.; Rao, M.S.; Austin, C.P.; McKew, J.C.; Zheng, W. Evaluation of cholesterol reduction activity of methyl-beta-cyclodextrin using differentiated human neurons and astrocytes. J. Biomol. Screen. 2012, 17, 1243–1251. [Google Scholar] [CrossRef] [Green Version]
- Sung, E.-A.; Yu, K.-R.; Shin, J.-H.; Seo, Y.; Kim, H.-S.; Koog, M.G.; Kang, I.; Kim, J.-J.; Lee, B.-C.; Shin, T.-H.; et al. Generation of patient specific human neural stem cells from Niemann-Pick disease type C patient-derived fibroblasts. Oncotarget 2017, 8, 85428–85441. [Google Scholar] [CrossRef]
- Pipalia, N.H.; Huang, A.; Ralph, H.; Rujoi, M.; Maxfield, F.R. Automated microscopy screening for compounds that partially revert cholesterol accumulation in Niemann-Pick C cells. J. Lipid Res. 2006, 47, 284–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Liu, K.; Swaroop, M.; Porter, F.D.; Sidhu, R.; Firnkes, S.; Ory, D.S.; Marugan, J.J.; Xiao, J.; Southall, N.; et al. δ-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders. J. Biol. Chem. 2012, 287, 39349–39360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugach, E.K.; Feltes, M.; Kaufman, R.J.; Ory, D.S.; Bang, A.G. High-content screen for modifiers of Niemann-Pick type C disease in patient cells. Hum. Mol. Genet. 2018, 27, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Neises, G.R.; Dwek, R.A.; Butters, T.D. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J. Biol. Chem. 1994, 269, 8362–8365. [Google Scholar]
- Cruz, J.C.; Chang, T.Y. Fate of endogenously synthesized cholesterol in Niemann-Pick type C1 cells. J. Biol. Chem. 2000, 275, 41309–41316. [Google Scholar] [CrossRef] [Green Version]
- Zervas, M.; Somers, K.L.; Thrall, M.A.; Walkley, S.U. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 2001, 11, 1283–1287. [Google Scholar] [CrossRef] [Green Version]
- Crini, G. Review: A history of cyclodextrins. Chem Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef]
- Camargo, F.; Erickson, R.P.; Garver, W.S.; Hossain, G.S.; Carbone, P.N.; Heidenreich, R.A.; Blanchard, J. Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 2001, 70, 131–142. [Google Scholar] [CrossRef]
- Davidson, J.; Molitor, E.; Moores, S.; Gale, S.E.; Subramanian, K.; Jiang, X.; Sidhu, R.; Kell, P.; Zhang, J.; Fujiwara, H.; et al. 2-Hydroxypropyl-β-cyclodextrin is the active component in a triple combination formulation for treatment of Niemann-Pick C1 disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1545–1561. [Google Scholar] [CrossRef]
- Kao, M.L.; Stellar, S.; Solon, E.; Lordi, A.; Kasica, N.; Swain, G.; Bagel, J.H.; Gurda, B.L.; Vite, C.H. Pharmacokinetics and distribution of 2-hydroxypropyl-β-cyclodextrin following a single intrathecal dose to cats. J. Inherit. Metab. Dis. 2020, 43, 618–634. [Google Scholar] [CrossRef]
- Pontikis, C.C.; Davidson, C.D.; Walkley, S.U.; Platt, F.M.; Begley, D.J. Cyclodextrin alleviates neuronal storage of cholesterol in Niemann-Pick C disease without evidence of detectable blood-brain barrier permeability. J. Inherit. Metab. Dis. 2013, 36, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, C.D.; Fishman, Y.I.; Puskas, I.; Szeman, J.; Sohajda, T.; McCauliff, L.A.; Sikora, J.; Storch, J.; Vanier, M.T.; Szente, L.; et al. Efficacy and ototoxicity of different cyclodextrins in Niemann-Pick C disease. Ann. Clin. Transl. Neurol. 2016, 3, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Homma, K.; Zhou, Y.; Nishimura, S.; Duan, C.; Chen, J.; Ahmad, A.; Cheatham, M.A.; Zheng, J. Susceptibility of outer hair cells to cholesterol chelator 2-hydroxypropyl-β-cyclodextrine is prestin-dependent. Sci Rep. 2016, 6, 21973. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Drummond, R.A.; Collar, A.L.; Iben, J.R.; Salman, A.; Westgarth, H.; Wassif, C.A.; Cawley, N.X.; Farhat, N.Y.; Ozato, K.; et al. Microglia activation in Niemann-Pick disease, type C1 is amendable to therapeutic intervention. Hum. Mol. Genet. 2018, 27, 2076–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebner, L.; Gläser, A.; Bräuer, A.; Witt, M.; Wree, A.; Rolfs, A.; Frank, M.; Vollmar, B.; Kuhla, A. Evaluation of Two Liver Treatment Strategies in a Mouse Model of Niemann-Pick-Disease Type C1. Int. J. Mol. Sci. 2018, 19, 972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacca, F.; Vossio, S.; Mercier, V.; Moreau, D.; Johnson, S.; Scott, C.C.; Montoya, J.P.; Moniatte, M.; Gruenberg, J. Cyclodextrin triggers MCOLN1-dependent endo-lysosome secretion in Niemann-Pick type C cells. J. Lipid Res. 2019, 60, 832–843. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.W.; Li, C.; Ioannou, Y.A. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS ONE 2010, 5, e15054. [Google Scholar] [CrossRef] [Green Version]
- Abi-Mosleh, L.; Infante, R.E.; Radhakrishnan, A.; Goldstein, J.L.; Brown, M.S. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19316–19321. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, A.I.; Zhang, G.; Warren, J.D.; Maxfield, F.R. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5477–5482. [Google Scholar] [CrossRef] [Green Version]
- Wehrmann, Z.T.; Hulett, T.W.; Huegel, K.L.; Vaughan, K.T.; Wiest, O.; Helquist, P.; Goodson, H. Quantitative comparison of the efficacy of various compounds in lowering intracellular cholesterol levels in niemann-pick type C fibroblasts. PLoS.ONE 2012, 7, e48561. [Google Scholar] [CrossRef] [Green Version]
- Peake, K.B.; Vance, J.E. Normalization of cholesterol homeostasis by 2-hydroxypropyl-beta-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J. Biol. Chem. 2012, 287, 9290–9298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meske, V.; Erz, J.; Priesnitz, T.; Ohm, T.G. The autophagic defect in Niemann-Pick disease type C neurons differs from somatic cells and reduces neuronal viability. Neurobiol. Dis. 2014, 64, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Carroll, B.; Buganim, Y.; Maetzel, D.; Ng, A.H.; Cassady, J.P.; Cohen, M.A.; Chakraborty, S.; Wang, H.; Spooner, E.; et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013, 5, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Ory, D.S.; Ottinger, E.A.; Farhat, N.Y.; King, K.A.; Jiang, X.; Weissfeld, L.; Berry-Kravis, E.; Davidson, C.D.; Bianconi, S.; Keener, L.A.; et al. Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: A non-randomised, open-label, phase 1-2 trial. Lancet 2017, 390, 1758–1768. [Google Scholar] [CrossRef] [Green Version]
- Berry-Kravis, E.; Chin, J.; Hoffmann, A.; Winston, A.; Stoner, R.; LaGorio, L.; Friedmann, K.; Hernandez, M.; Ory, D.S.; Porter, F.D.; et al. Long-Term Treatment of Niemann-Pick Type C1 Disease With Intrathecal 2-Hydroxypropyl-β-Cyclodextrin. Pediatr. Neurol. 2018, 80, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Lee, B.H.; Lee, Y.S.; Kang, K.S. Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 2007, 360, 593–599. [Google Scholar] [CrossRef]
- Pipalia, N.H.; Cosner, C.C.; Huang, A.; Chatterjee, A.; Bourbon, P.; Farley, N.; Helquist, P.; Wiest, O.; Maxfield, F.R. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. USA 2011, 108, 5620–5625. [Google Scholar] [CrossRef] [Green Version]
- Nunes, M.J.; Moutinho, M.; Gama, M.J.; Rodrigues, C.M.; Rodrigues, E. Histone deacetylase inhibition decreases cholesterol levels in neuronal cells by modulating key genes in cholesterol synthesis, uptake and efflux. PLoS ONE 2013, 8, e53394. [Google Scholar] [CrossRef] [Green Version]
- Newton, J.; Hait, N.C.; Maceyka, M.; Colaco, A.; Maczis, M.; Wassif, C.A.; Cougnoux, A.; Porter, F.D.; Milstien, S.; Platt, N.; et al. FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibroblasts. FASEB J. 2017, 31, 1719–1730. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, K.; Hutt, D.M.; Scott, S.M.; Gupta, V.; Mao, S.; Balch, W.E. Correction of Niemann-Pick type C1 trafficking and activity with the histone deacetylase inhibitor valproic acid. J. Biol. Chem. 2020, 295, 8017–8035. [Google Scholar] [CrossRef]
- Subramanian, K.; Rauniyar, N.; Lavalleé-Adam, M.; Yates, J.R., 3rd; Balch, W.E. Quantitative Analysis of the Proteome Response to the Histone Deacetylase Inhibitor (HDACi) Vorinostat in Niemann-Pick Type C1 disease. Mol. Cell Proteom. 2017, 16, 1938–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, P.S.; Gonzalez-Zuñiga, M.; González-Hódar, L.; Yáñez, M.J.; Dulcey, A.; Marugan, J.; Seto, E.; Alvarez, A.R.; Zanlungo, S. Neuronal gene repression in Niemann-Pick type C models is mediated by the c-Abl/HDAC2 signaling pathway. Biochim. Biophys. Acta 2016, 1859, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.S.; Getz, M.; Haldar, K. Chronic administration of an HDAC inhibitor treats both neurological and systemic Niemann-Pick type C disease in a mouse model. Sci. Transl. Med. 2016, 8, 326ra323. [Google Scholar] [CrossRef] [PubMed]
- Nakasone, N.; Nakamura, Y.S.; Higaki, K.; Oumi, N.; Ohno, K.; Ninomiya, H. Endoplasmic reticulum-associated degradation of Niemann-Pick C1: Evidence for the role of heat shock proteins and identification of lysine residues that accept ubiquitin. J. Biol. Chem. 2014, 289, 19714–19725. [Google Scholar] [CrossRef] [Green Version]
- Paul, C.A.; Reid, P.C.; Boegle, A.K.; Karten, B.; Zhang, M.; Jiang, Z.G.; Franz, D.; Lin, L.; Chang, T.Y.; Vance, J.E.; et al. Adenovirus expressing an NPC1-GFP fusion gene corrects neuronal and nonneuronal defects associated with Niemann pick type C disease. J. Neurosci. Res. 2005, 81, 706–719. [Google Scholar] [CrossRef]
- Loftus, S.K.; Erickson, R.P.; Walkley, S.U.; Bryant, M.A.; Incao, A.; Heidenreich, R.A.; Pavan, W.J. Rescue of neurodegeneration in Niemann-Pick C mice by a prion-promoter-driven Npc1 cDNA transgene. Hum. Mol. Genet. 2002, 11, 3107–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.G.; Pfrieger, F.; Dotti, C.G. Cholesterol in brain disease: Sometimes determinant and frequently implicated. EMBO Rep. 2014, 15, 1036–1052. [Google Scholar] [CrossRef] [Green Version]
- Arenas, F.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Intracellular Cholesterol Trafficking and Impact in Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Lamri, A.; Pigeyre, M.; Garver, W.S.; Meyre, D. The Extending Spectrum of NPC1-Related Human Disorders: From Niemann-Pick C1 Disease to Obesity. Endocr. Rev. 2018, 39, 192–220. [Google Scholar] [CrossRef] [PubMed]
- Trezza, V.; Campolongo, P.; Vanderschuren, L.J. Evaluating the rewarding nature of social interactions in laboratory animals. Dev. Cogn. Neurosci. 2011, 1, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, J.; Barbaric, I.; Andrews, P.W. Acquired genetic changes in human pluripotent stem cells: Origins and consequences. Nat. Rev. Mol. Cell Biol. 2020, 21, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Aulner, N.; Bickle, M.; Davies, A.M.; Nery, E.D.; Ebner, D.; Montoya, M.C.; Östling, P.; Pietiäinen, V.; Price, L.S.; et al. Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 2016, 15, 751–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; State, M.W. Psychiatric disorders: Diagnosis to therapy. Cell 2014, 157, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, R.E.; Seeman, M.V.; Greig, N.H.; Lahiri, D.K. What can triumphs and tribulations from drug research in Alzheimer’s disease tell us about the development of psychotropic drugs in general? Lancet Psychiatry 2015, 2, 756–764. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Dowle, M. Data.Table: Extension of ‘Data.Frame’. Available online: https://CRAN.R-project.org/package=data.table (accessed on 25 November 2020).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Wickham, H. Readr: Read Rectangular Text Data. Available online: https://cran.r-project.org/web/packages/readr/index.html (accessed on 25 November 2020).
Figure 1.
Growth of the NPC research field. Cumulative counts of publications obtained by Boolean queries in PubMed using the keywords “Niemann–Pick type c OR Niemann–Pick type c1 OR Niemann-Pick type c2 OR npc1 OR npc2” (Appendix A). Black and orange lines indicate original articles “(...) NOT review [pt]” and reviews [pt] “(...) AND review [pt]”, respectively. To retrieve publications more specifically related to NPC, we restricted the query to titles [ti] or abstracts [ab] by adding the corresponding field tags to each keyword “Niemann–Pick type c [tiab] OR Niemann–Pick type c1 [tiab] OR Niemann–Pick type c2 [tiab] OR npc1 [tiab] OR npc2 [tiab]”. Sky blue and green lines indicate original articles and reviews of this subset, respectively.
Figure 1.
Growth of the NPC research field. Cumulative counts of publications obtained by Boolean queries in PubMed using the keywords “Niemann–Pick type c OR Niemann–Pick type c1 OR Niemann-Pick type c2 OR npc1 OR npc2” (Appendix A). Black and orange lines indicate original articles “(...) NOT review [pt]” and reviews [pt] “(...) AND review [pt]”, respectively. To retrieve publications more specifically related to NPC, we restricted the query to titles [ti] or abstracts [ab] by adding the corresponding field tags to each keyword “Niemann–Pick type c [tiab] OR Niemann–Pick type c1 [tiab] OR Niemann–Pick type c2 [tiab] OR npc1 [tiab] OR npc2 [tiab]”. Sky blue and green lines indicate original articles and reviews of this subset, respectively.

Figure 2.
Use of experimental models in NPC research. Cumulative counts (log10 values) of publications obtained by respective Boolean queries in PubMed [e.g., for mouse: (Niemann–Pick type c [tiab] OR Niemann–Pick type c1 [tiab] OR Niemann–Pick type c2 [tiab] OR npc1 [tiab] OR npc2 [tiab]) AND (mice [tiab] OR mouse [tiab] OR mus musculus [tiab]) NOT review]. Inset, the histogram shows that most publications relate to one animal model and that only a small fraction of articles contributes to multiple cumulative counts.
Figure 2.
Use of experimental models in NPC research. Cumulative counts (log10 values) of publications obtained by respective Boolean queries in PubMed [e.g., for mouse: (Niemann–Pick type c [tiab] OR Niemann–Pick type c1 [tiab] OR Niemann–Pick type c2 [tiab] OR npc1 [tiab] OR npc2 [tiab]) AND (mice [tiab] OR mouse [tiab] OR mus musculus [tiab]) NOT review]. Inset, the histogram shows that most publications relate to one animal model and that only a small fraction of articles contributes to multiple cumulative counts.

Figure 3.
NPC research on specific types of cells. Cumulative counts of publications obtained by respective Boolean queries in PubMed [e.g., for fibroblasts: (Niemann–Pick type c[tiab] OR Niemann–Pick type c1[tiab] OR Niemann–Pick type c2[tiab] OR npc1[tiab] OR npc2[tiab]) AND (fibroblast[tiab] OR fibroblasts[tiab]) NOT review]. Inset, the histogram shows that most publications relate to one cell model and that only a small fraction of articles contributes to multiple cumulative counts.
Figure 3.
NPC research on specific types of cells. Cumulative counts of publications obtained by respective Boolean queries in PubMed [e.g., for fibroblasts: (Niemann–Pick type c[tiab] OR Niemann–Pick type c1[tiab] OR Niemann–Pick type c2[tiab] OR npc1[tiab] OR npc2[tiab]) AND (fibroblast[tiab] OR fibroblasts[tiab]) NOT review]. Inset, the histogram shows that most publications relate to one cell model and that only a small fraction of articles contributes to multiple cumulative counts.

Figure 4.
Models to study the impact of NPC1 deficiency on selected neurons in the retina. (a) Fluorescence micrographs of retinal neurons from one-week-old wild-type (WT) and NPC1-deficient (NPC) mice in vivo. NPC1 deficiency causes an intracellular accumulation of unesterified cholesterol in neurons of the ganglion cell layer in vivo (arrowheads). (b) Phase-contrast (left) and fluorescence micrographs (middle, right) of retinal neurons acutely isolated from one-week-old wild-type (WT: left, middle) and NPC1-deficient mice (NPC). In this ex vivo model, NPC1-deficient neurons maintain the increased levels of cholesterol as shown by filipin staining. (c) Phase-contrast (left) and fluorescence micrographs (middle, right) of neurons purified from the retina of one-week-old rats, cultured for 48 h and stained with filipin. Treatment with the NPC1-inhibiting drug U18666A induced an accumulation of unesterified cholesterol. Scale bars: 20 µm. In (a–c), the distribution of unesterified cholesterol was shown by the staining of chemically fixed material with filipin (a,b): Barthélémy, Pfrieger, unpublished; (c): modified from [317].
Figure 4.
Models to study the impact of NPC1 deficiency on selected neurons in the retina. (a) Fluorescence micrographs of retinal neurons from one-week-old wild-type (WT) and NPC1-deficient (NPC) mice in vivo. NPC1 deficiency causes an intracellular accumulation of unesterified cholesterol in neurons of the ganglion cell layer in vivo (arrowheads). (b) Phase-contrast (left) and fluorescence micrographs (middle, right) of retinal neurons acutely isolated from one-week-old wild-type (WT: left, middle) and NPC1-deficient mice (NPC). In this ex vivo model, NPC1-deficient neurons maintain the increased levels of cholesterol as shown by filipin staining. (c) Phase-contrast (left) and fluorescence micrographs (middle, right) of neurons purified from the retina of one-week-old rats, cultured for 48 h and stained with filipin. Treatment with the NPC1-inhibiting drug U18666A induced an accumulation of unesterified cholesterol. Scale bars: 20 µm. In (a–c), the distribution of unesterified cholesterol was shown by the staining of chemically fixed material with filipin (a,b): Barthélémy, Pfrieger, unpublished; (c): modified from [317].


{kind=link}
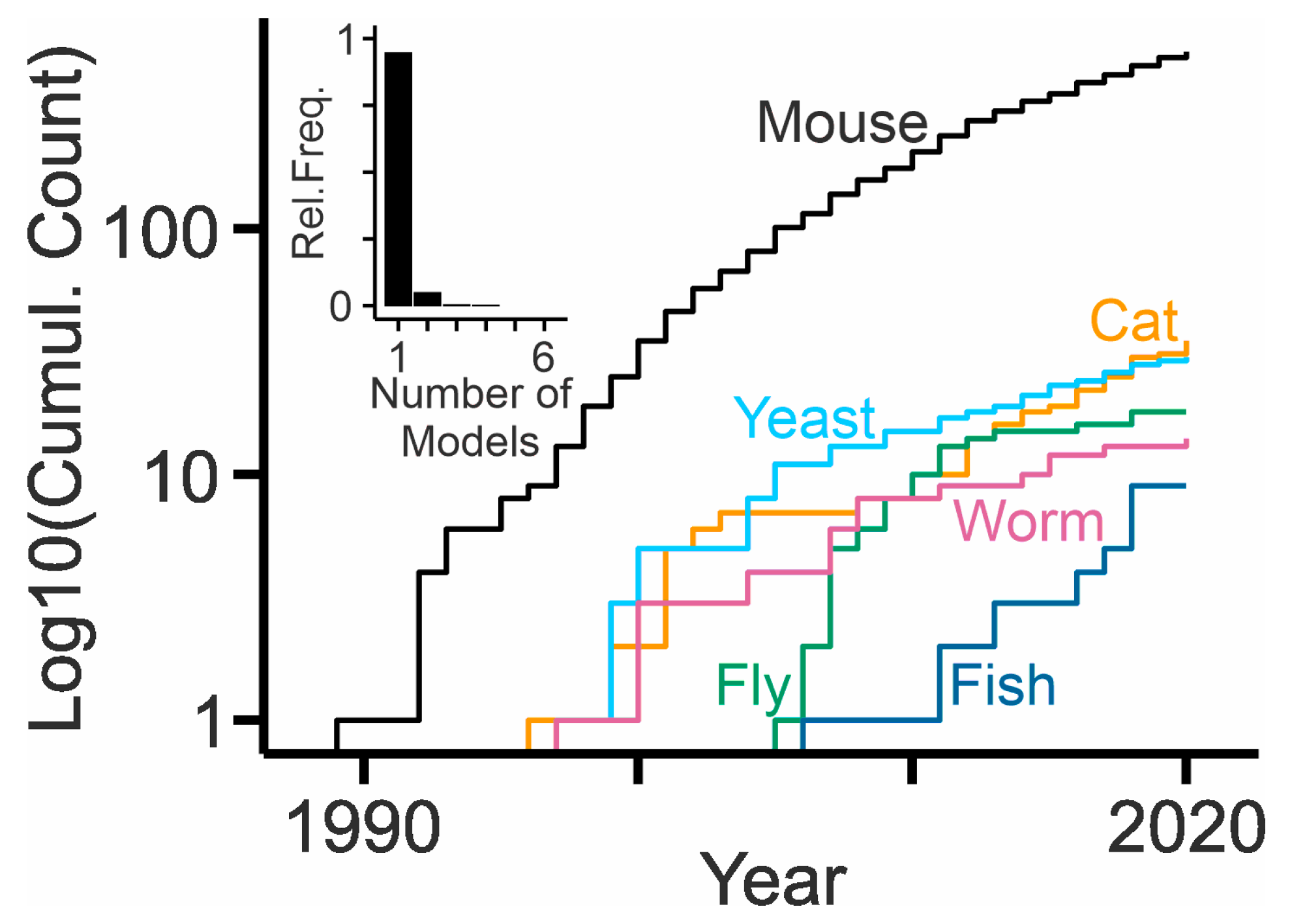{kind=link}
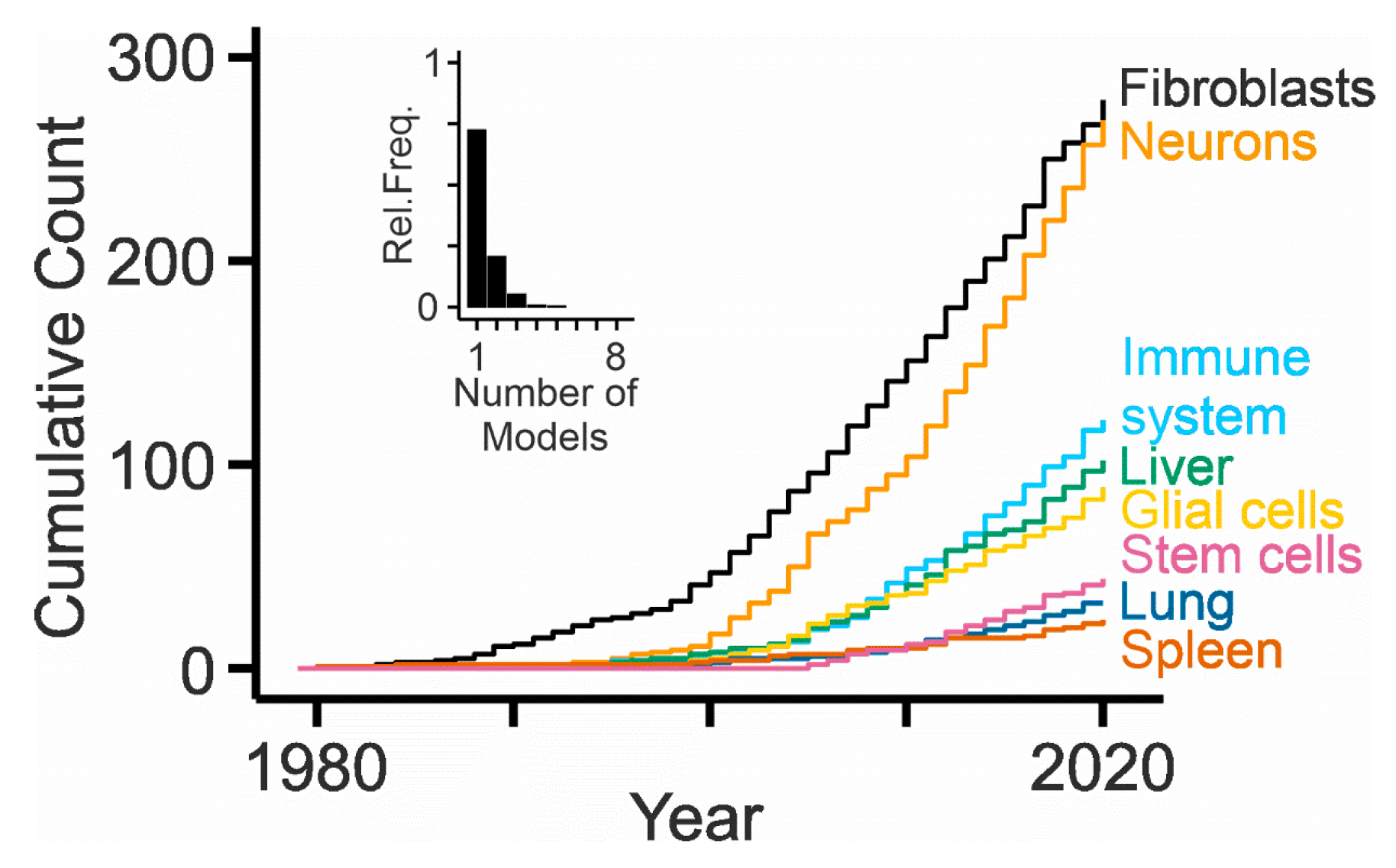{kind=link}
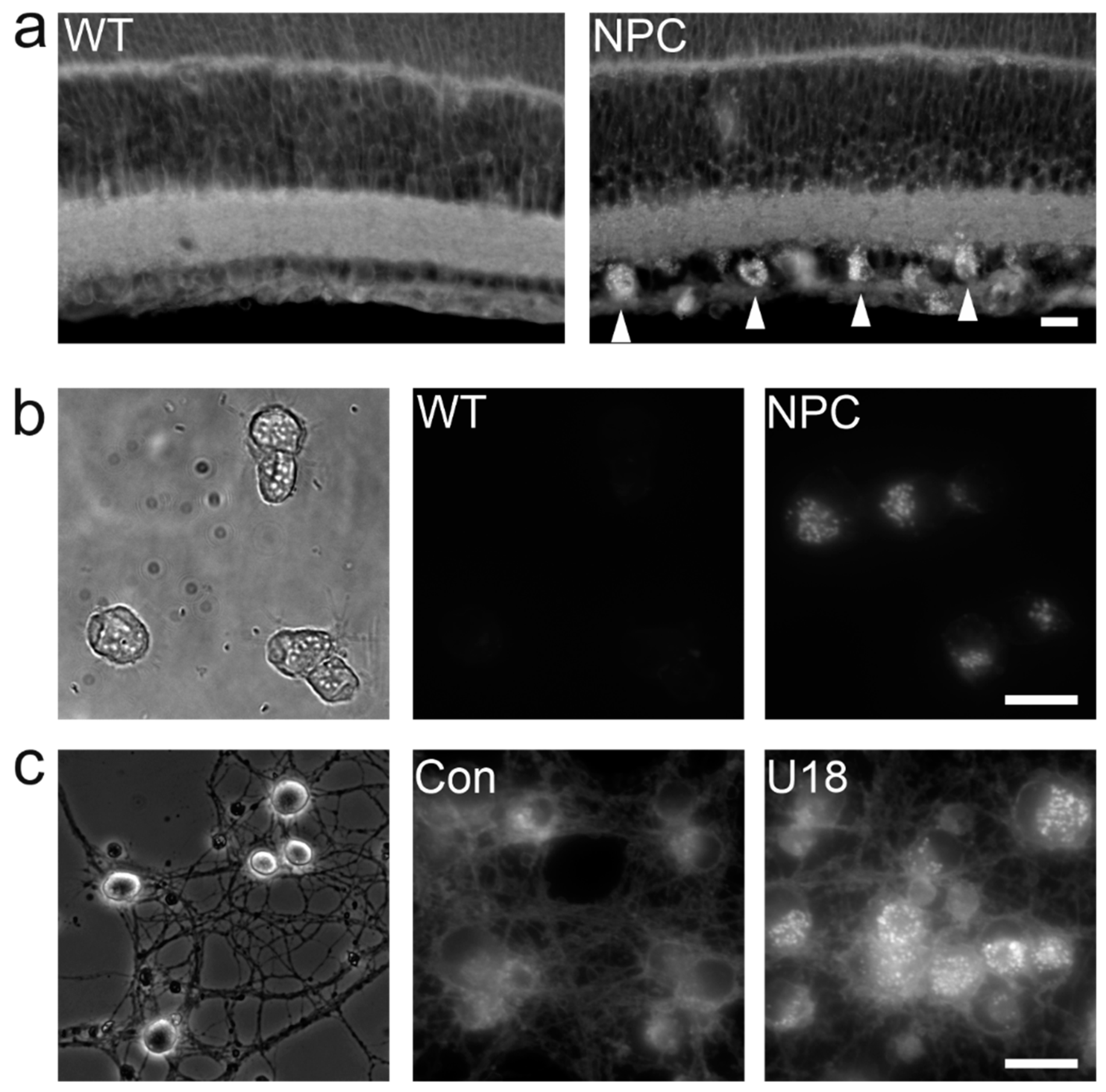{kind=link}
Table 1.
Animal models available to study NPC disease.
| Species, Gene, Animal model | Symptom Onset | Life Span | Visceral Symptoms | Neurologic Symptoms | Lipid Accumulation in Tissues | References |
|---|---|---|---|---|---|---|
| Caenorhabditis elegans ncr-1 ncr-1(nr2022) ncr-2 ncr-2(nr2023) | ND | Dauer formation | ND | Modified trafficking or release of synaptic vesicles | Nerve ring, spermatheca and oocytes: DHE accumulation | [113,114,115] |
| Drosophila melanogaster Npc1a Npc1a57A | ND | Larval lethality | More efficient sterol absorption than wild-type animals | ND | Malpighian tubules and midgut: Sterol accumulation. Brain and retina: Chol aggregates | [119,120] |
| Drosophila melanogaster Npc1b Npc1bR9-28 | ND | Larval lethality | Defects in sterol absorption; similar to NPC1L1 | ND | No Chol accumulation | [121] |
| Drosophila melanogaster Npc2a Npc2a376 Npc2b Npc2b19 | ND | Larval lethality | Apoptotic cell death in the nervous system | No sterol distribution abnormality | [122,123] | |
| Danio rerio npc1 npc1ihb334 npc1ihb335 npc1hg37 npc1y535 | Larval stage | 99% animals die within the first MPF; 1% die before 8 months of age | Hepatomegaly, splenomegaly | Disturbed balance and motor control, loss of Purkinje cells | Liver: accumulation of Chol, CER, DG, LPA, PA, PC, PE, PS, TG, SL | [125,126,127,128] |
| Mus musculus Npc1 BALB/cNctr-Npc1m1n/J Npc1nih | 6 wks (N) | 9–11 wks | Hepatomegaly, splenomegaly, decreased weight gain, increased lung mass | Disturbed motor coordination, tremor, ataxia, loss of Purkinje cells | Spleen, liver, lung, lymph nodes, thymus, bone marrow, brain: accumulation of FA, CER, Chol, SL | [18,129] |
| Mus musculus C57BLKS/J-Npc1spm/J Npc1spm | 4 wks (V) | 11–15 wks | Hepatomegaly, splenomegaly, decreased weight gain | Disturbed motor coordination, tremor, ataxia, loss of Purkinje cells | Liver: accumulation of FA, CER, Chol, SL. Brain: Chol accumulation | [130] |
| Mus musculus Npc1tm1Mbjg Npc1pf | 7 wks (N) | 12 wks | Hepatomegaly, splenomegaly, decreased weight gain | Tremor, ataxia, loss of Purkinje cells | Brain, kidney, liver, lung, and spleen: Chol accumulation. Brain and liver: GM accumulation. | [131] |
| Mus musculus Npc1nmf164/J Npc1nmf164 | 4 wks (N) | 16 wks | Hepatomegaly, splenomegaly, decreased weight gain, foamy pulmonary macrophages | Loss of Purkinje cells, abnormal acoustic startle response, decreased strength and motor capabilities | Brain: Chol and GM accumulation. Liver: accumulation of CER, Chol, SL, GM. | [132] |
| Mus musculus Npc1tm(I1061T)Dso Npc1I1061T | 8 wks (N) | 17–18 wks | ND Decreased weight gain | Decreased motor coordination, tremor, loss of Purkinje cells | Liver and brain: Chol accumulation | [133] |
| Mus musculus Npc1tm1Tacf/J Npc1Imagine/Imagine | 7 wks (N) | 9 wks | ND | Decreased motor coordination, tremor, ataxia, age-dependent hyperactivity, reduced anxiety, cortico-hippocampal defects, higher pain threshold | ND | [134] |
| Mus musculus Npc1tm2Tacf/J Npc1Pioneer/Pioneer | ND | Only 2% live births | ND | ND | ND | [134] |
| Mus musculus Npc1Imagine/Pioneer | 7 wks (N) | 9 wks | ND | Decreased motor coordination, tremor, ataxia, age-dependent hyperactivity, reduced anxiety, higher pain threshold | Liver: Chol and CER accumulation Brain: Chol, CER, GM accumulation | [134] |
| Mus musculus Npc1em1Pav | 4 wks (V,N) | 10–12 wks, strain-dependent | ND | Loss of Purkinje cells, decreased motor coordination | Liver, brain, spleen: GM accumulation | [135] |
| Mus musculus Npc1tm1.1Apl Cell-specific knock-out based on Cre/loxP | Depends on target cells/tissues | [136] | ||||
| Mus musculus Npc1(ASO) Knock-down of NPC1 in liver and lung by antisense oligonucleotides (ASOs) | Hepatomegaly; foamy, vacuolated macrophages and increased apoptosis/proliferation in liver | No neurologic symptoms | Liver: Chol accumulation | [137] | ||
| Mus musculus Tg(Gfap-Npc1) Rescue of Npc1 expression in Gfap-expressing cells | Delayed onset with respect to NPC1-/- (N) | 24 wks | Weight gain with respect to NPC1-/- | Reduced numbers of axonal spheroids and reactive astrocytes, restoration of myelin, loss of Purkinje cells, decreased neurodegeneration with respect to NPC1-/- | Reduced Chol accumulation in some brain areas with respect to NPC1-/- | [138] |
| Mus musculus Tg(tetO-Npc1/YFP)1Mps Cell-specific over-expression based on Tet-On/Off | Depends on target cells/tissues | [139] | ||||
| Mus musculus Npc1fl/fl Cell-specific reversal of Npc1 knock-out based on Cre/loxP | Depends on target cells/tissue | [140] | ||||
| Felis catus NPC1 | 6 wks (N) | 20 wks | Hepatomegaly, spleen and lung with multifocal histiocytosis | Tremor, ataxia, loss of Purkinje cells, astroglyosis, myelin abnormalities in peripheral nervous system | Pyramidal neurons: GM2 accumulation | [141,142,143] |
| Bos taurus NPC1 | 3 months (N) | before 8 months (N = 1) | Marked hypertrophy of Purkinje cells in heart, foamy macrophages in lymph nodes | Limb weakness, dysmetria, incoordination, a wide based stance, walking sideways or falling over and recumbency, vacuolation of Purkinje cells, astrocytosis, microgliosis | Fibroblasts: Chol, GM, SL accumulation | [144] |
| Mus musculus Npc2tm1Plob | 4 wks (V) | 18 wks | Decreased weight gain | Tremor, motor defects, ataxia, loss of Purkinje cells | Liver: Chol accumulation, neocortex, dentate gyrus, hippocampus, and cerebellum: Chol accumulation | [145] |
| Mus musculus Npc2Gt(LST105)Byg | 8 wks (N) | ND | Decreased weight gain | Tremor, ataxia, loss of Purkinje cell, astrocytosis | Liver, spleen, kidney, lung: Chol accumulation. | [146,147] |
| Mus musculus Tg(Apoe-Npc2) Over-expression of NPC2 in liver | ND | ND | [148] |
Abbreviations: not determined (ND); weeks (wks); neurological symptoms (N); visceral symptoms (V); cholesterol (Chol); ceramide (CER); dehydroergosterol (DHE); diacylglycerol (DG); gangliosides (GM); lysophosphatidic acid (LPA); months post fertilization (MPF); phosphatidic acid (PA); phosphatidyl–choline (PC); phosphatidyl–ethanolamine (PE); phosphatidyl–serine (PS); sphingolipids (SL); triglycerides (TG)
Table 2.
Summary of therapeutic approaches for NPC explored with animal models.
| Treatment | Model | Effect | Reference |
|---|---|---|---|
| Cholesterol lowering drugs | Npc1nih | No | [162] |
| Apoptosis, inhibition | Npc1nih | No | [163] |
| Mitogen-activated protein kinase, inhibition | Npc1nih | No | [164] |
| Dietary restriction | Cat | No | [165] |
| Implantation of neural stem cells | Npc1nih | No | [166] |
| Transplantation of mesenchymal stem cell | Npc1nih | Small | [167,168,169] |
| Vitamin C | Npc1nih | No | [170] |
| Vitamin E | Npc1nih | Yes | [171,172] |
| Liver X receptor, activation | Npc1nih | Yes | [173] |
| Pregnane X receptor, activation | Npc1nih | Yes | [174] |
| Estradiol | Npc1nih | Small | [175] |
| C-Abl inhibition (Imatinib) | Npc1nih | Yes | [176] |
| 2-hydroxypropyl-beta-cyclodextrin | Npc1nih, cat | Yes | [177,178,179,180,181] |
| Cyclin-dependent kinase-5, inhibition | Npc1nih | Small | [182] |
| Non-steroidal anti-inflammatory drugs | Npc1nih | Yes | [170] |
| Protein replacement, NPC2 | 129P2/OlaHsd-Npc2Gt(LST105)BygNya | Small | [147] |
| Curcumin | Npc1nih | No | [183] |
| Glucosylceramide synthase, inhibition | Npc1nih, cat | Yes | [184,185] |
| N-acetylcysteine | Npc1nih, Npc1(ASO) | Small | [186] |
| Copper chelation | Npc1nih | Yes, not CNS | [187] |
| Acetylcholinesterase, inhibition | Npc1nih | Small | [188] |
| Combination miglustat, curcumin, ibuprofen | Npc1nih | Yes | [189] |
| Glucocerebrosidase, inhibition | Npc1nih | Yes | [190] |
| Necroptosis, inhibition | Npc1nih | Yes | [191,192] |
| Heat shock protein, activation (Arimoclomol) | Npc1nih | Yes | [193] |
| Histone deacetylases, inhibition (Vorinostat) | Npc1nmf164, Npc1nih | Yes, not CNS | [194] |
| Gene therapy, AAV9-NPC1 | Npc1nih | Yes | [195,196,197] |
| Gene therapy, AAV rh.10-NPC2 | Npc2tm1Plob | Yes | [198] |
| Glutathion | Npc1nih | Yes | [199] |
| Adenosine A2A receptor, activation | Npc1nih | Yes | [200] |
| Polymeric beta-cyclodextrin | Npc1nmf164 | Small | [201] |
| Pneumococcal immunization | Npc1nih | Yes | [202] |
| Histamine H3 receptor, activation | Npc1nih | No | [203] |
| 6-O-alpha-maltosyl-beta-cyclodextrin | Npc1nih | Yes | [204] |
| Implantation of VEGF-overexpressing neural stem cells | Npc1nih | Yes | [205] |
| CYP46A1, activation | Npc1nmf164 | Yes | [206] |
| High-density lipoprotein nanoparticles | Npc1I1061T | Small | [207] |
| Gene therapy, AAV-mediated base editing | Npc1I1061T | small | [208] |
| Iron chelation | Npc1nih | No | [209] |
| Gene therapy, Trojan horse liposomes | Npc1nih | No | [210] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pallottini, V.; Pfrieger, F.W. Understanding and Treating Niemann–Pick Type C Disease: Models Matter. Int. J. Mol. Sci. 2020, 21, 8979. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238979
AMA Style
Pallottini V, Pfrieger FW. Understanding and Treating Niemann–Pick Type C Disease: Models Matter. International Journal of Molecular Sciences. 2020; 21(23):8979. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238979
Chicago/Turabian StylePallottini, Valentina, and Frank W. Pfrieger. 2020. "Understanding and Treating Niemann–Pick Type C Disease: Models Matter" International Journal of Molecular Sciences 21, no. 23: 8979. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238979
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.