Length Polymorphism and Methylation Status of UPS29 Minisatellite of the ACAP3 Gene as Molecular Biomarker of Epilepsy. Sex Differences in Seizure Types and Symptoms


Abstract
:1. Introduction
2. Results
2.1. Association of UPS29 Length Polymorphism with Symptomatic and Cryptogenic Epilepsy (Epilepsy at the Onset, Etiology, Clinical Course)
2.2. Association of UPS29 Length Polymorphism with Clinical Features of Seizure
2.3. Association of UPS29 Methylation Status with Epilepsy Type and Clinical Features of the Seizure
3. Discussion
4. Materials and Methods
4.1. Participant Recruitment (Patients and Healthy Volunteers)
4.2. Genome DNA Isolation and Genotyping
4.3. Analysis of UPS29 DNA Methylation Status
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| nt | nucleotide |
| bp | base pairs |
| VNTR | variable number tandem repeat |
| SNP | single nucleotide polymorphisms |
| RR | Risk Ratio |
| OR | Odds Ratio |
| CI | Confidence Interval |
| phi | Coefficient of association |
| PAAG | polyacrylamide gel |
References
- Engel, J., Jr. Report of the ILAE classification core group. Epilepsia 2006, 47, 1558–1568. [Google Scholar] [CrossRef]
- Berg, A.T.; Cross, J.H. Classification of epilepsies and seizures: Historical perspective and future directions. Handb. Clin. Neurol. 2012, 107, 99–111. [Google Scholar]
- Legnani, M.; Bertin, A.; Decima, R.; Demicheli, E.; Higgie, J.R.; Preve, F.; Braga, P.; Bogacz, A.; Scaramelli, A. Applicability, and contribution of the new ILAE 2017 classification of epileptic seizures and epilepsies. Epilept. Disord. 2019, 21, 549–554. [Google Scholar]
- Chen, T.; Giri, M.; Xia, Z.; Subedi, Y.N.; Li, Y. Genetic and epigenetic mechanisms of epilepsy: A review. Neuropsychiatr. Dis. Treat. 2017, 13, 1841–1859. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.Y.; Wang, H.Y.; Wang, Y. Recent progress in identifying genetic and epigenetic contributions to epilepsy. Reprod. Dev. Med. 2017, 1, 239–249. [Google Scholar]
- Nam, S.M.; Cho, K.-O. The role of epigenetics in the pathophysiology of epilepsy. In Neuropsychiatric Disorders and Epigenetics; Yasui, D.H., Peedicayil, J., Grayson, D.R., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Chapter 12; pp. 233–260. [Google Scholar]
- Holmes, G.L.; Noebels, J.L. The epilepsy spectrum: Targeting future research challenges. Cold Spring Harb. Perspect. Med. 2016, 6, a028043. [Google Scholar] [CrossRef]
- Weber, Y.G.; Biskup, S.; Helbig, K.L.; Spiczak, S.V.; Lerche, H. The role of genetic testing in epilepsy diagnosis and management. Expert. Rev. Mol. Diagn. 2017, 17, 739–750. [Google Scholar] [CrossRef]
- Sterlini, B.; Fruscione, F.; Baldassari, S.; Benfenati, F.; Zara, F.; Corradi, A. Progress of induced pluripotent stem cell technologies to understand genetic epilepsy. Int. J. Mol. Sci. 2020, 21, 482. [Google Scholar] [CrossRef] [Green Version]
- Pulido Fontes, L.; Quesada Jimenez, P.; Mendioroz Iriarte, M. Epigenetics and epilepsy. Neurologia 2015, 30, 111–118. [Google Scholar] [CrossRef]
- Hauser, R.M.; Henshall, D.C.; Lubin, F.D. The epigenetics of epilepsy and its progression. Neuroscientist 2018, 24, 186–200. [Google Scholar] [CrossRef]
- Kobow, K.; Blümcke, I. Epigenetics in epilepsy. Neurosc. Lett. 2018, 667, 40–46. [Google Scholar] [CrossRef]
- Ozdemir, O.; Egemen, E.; Ugur Iseri, S.A.; Sezerman, O.U.; Bebek, N.; Baykan, B.; Ozbek, U. Identification of epilepsy-related pathways using genome-wide DNA methylation measures: A trio-based approach. PLoS ONE 2019, 14, e0211917. [Google Scholar] [CrossRef]
- Caramaschi, D.; Hatcher, C.; Mulder, R.H.; Felix, J.F.; Cecil, C.A.M.; Relton, C.L.; Walton, E. Epigenome-wide association study of seizures in childhood and adolescence. Clin. Epigenet. 2020, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Rawat, C.; Kushwaha, S.; Srivastava, A.K.; Kukreti, R. Peripheral blood gene expression signatures associated with epilepsy and its etiologic classification. Genomics 2020, 112, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Virtaneva, K.; D’Amato, E.; Miao, J.; Koskiniemi, M.; Norio, R.; Avanzini, G.; Franceschetti, S.; Michelucci, R.; Tassinari, C.A.; Omer, S.; et al. Unstable minisatellite expansion causing recessively inherited myoclonus epilepsy, EPM1. Nat. Genet. 1997, 15, 393–396. [Google Scholar] [CrossRef]
- Alakurtti, K.; Virtaneva, K.; Joensuu, T.; Palvimo, J.J.; Lehesjoki, A.E. Characterization of the cystatin B gene promoter harboring the dodecamer repeat expanded in progressive myoclonus epilepsy, EPM1. Gene 2000, 242, 65–73. [Google Scholar] [CrossRef]
- Horiuchi, H.; Osawa, M.; Furutani, R.; Morita, M.; Tian, W.; Awatsu, Y.; Shimazaki, H.; Umetsu, K. Polymerase chain reaction-based analysis using deaminated DNA of dodecamer expansions in CSTB, associated with Unverricht-Lundborg myoclonus epilepsy. Genet. Test. 2005, 9, 328–333. [Google Scholar] [CrossRef]
- Manna, I.; Labate, A.; Gambardella, A.; Forabosco, P.; La Russa, A.; Le Piane, E.; Aguglia, U.; Quattrone, A. Serotonin transporter gene (5-Htt): Association analysis with temporal lobe epilepsy. Neurosci. Lett. 2007, 421, 52–56. [Google Scholar] [CrossRef]
- Hecimovic, H.; Stefulj, J.; Cicin-Sain, L.; Demarin, V.; Jernej, B. Association of serotonin transporter promoter (5-HTTLPR) and intron 2 (VNTR-2) polymorphisms with treatment response in temporal lobe epilepsy. Epilepsy Res. 2010, 91, 35–38. [Google Scholar]
- Vincentiis, S.; Alcantara, J.; Rzezak, P.; Kerr, D.; Dos Santos, B.; Alessi, R.; van der Linden, H.; Arruda, F.; Chaim-Avancini, T.; Serpa, M.; et al. Higher transcription alleles of the MAOA-uVNTR polymorphism are associated with higher seizure frequency in temporal lobe epilepsy. Epilepsy Res. 2019, 149, 26–29. [Google Scholar] [CrossRef]
- Lei, X.X.; Liu, Q.; Huang, Y.; Zhou, X.Q.; Sun, H.Y.; Wu, L.W.; Cui, L.Y.; Zhang, X. TTTCA repeat expansion causes familial cortical myoclonic tremor with epilepsy. Eur. J. Neurol. 2019, 26, 513–518. [Google Scholar] [CrossRef]
- Ali, F.R.; Vasiliou, S.A.; Haddley, K.; Paredes, U.M.; Roberts, J.C.; Miyajima, F.; Klenova, E.; Bubb, V.J.; Quinn, J.R. Combinatorial interaction between two human serotonin transporter gene variable number tandem repeats and their regulation by CTCF. J. Neurochem. 2010, 112, 296–306. [Google Scholar] [CrossRef]
- Breen, G.; Collier, D.; Craig, J.; Quinn, J. Variable number tandem repeats as agents of functional regulation in the genome. IEEE Eng. Med. Biol. Mag. 2008, 27, 103–108. [Google Scholar] [CrossRef]
- MacKenzie, A.; John Quinn, J. A serotonin transporter gene intron 2 polymorphic region, correlated with affective disorders, has allele-dependent differential enhancer-like properties in the mouse embryo. Proc. Natl. Acad. Sci. USA 1999, 96, 15251–15255. [Google Scholar] [CrossRef] [Green Version]
- Vergnaud, G.; Denoeud, F. Minisatellites: Mutability and genome architecture. Genome Res. 2000, 10, 899–907. [Google Scholar] [CrossRef] [Green Version]
- Lauthier, V.; Mariat, D.; Vergnaud, G. CEB 15 detects a VNTR locus (Het: 92%) on chromosome 1p. Hum. Mol. Genet. 1992, 1, 63. [Google Scholar] [CrossRef]
- Myers, S.; Freeman, C.; Auton, A.; Donnelly, P.; McVean, G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008, 40, 1124–1129. [Google Scholar] [CrossRef]
- Vergnaud, G.; Institute of Genetics and Microbiology, University of Paris, Paris, France; Patkin, E.L.; Department of Molecular Genetics, Institute of Experimental Medicine, St. Petersburg, Russia. Personal communication, 1999.
- NCBI. Genome. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/genome/gdv/browser/gene/?id=4524 (accessed on 4 June 2020).
- Suchkova, I.O.; Shubina, D.M.; Sasina, L.K.; Slominskaya, N.A.; Vasilyev, V.B.; Alenina, N.V.; Bader, M.; Patkin, E.L. Molecular-genetic characteristics of the non-hypervariable GC-rich human minisatellite UPS29 of CENTB5 gene. Ecol. Genet. 2007, 5, 35–45. (In Russian) [Google Scholar] [CrossRef] [Green Version]
- Ensembl. Blast/Blat Search. Available online: https://asia.ensembl.org/Homo_sapiens/Tools/Blast/Blat%20search (accessed on 17 May 2020).
- Smith, G.R.; Kunes, S.M.; Schultz, D.W.; Taylor, A.; Triman, K.L. Structure of chi hotspots of generalized recombination. Cell 1981, 24, 429–436. [Google Scholar] [CrossRef]
- Weinreb, A.; Katzenberg, D.R.; Gilmore, G.L.; Birshtein, B.K. The site of unequal sister chromatid exchange contains a potential Z-DNA-forming tract. Proc. Natl. Acad. Sci. USA 1988, 85, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Spitzner, J.R.; Muller, M.T. A consensus sequence for cleavage by vertebrate DNA topoisomerase II. Nucl. Acids Res. 1988, 16, 5533–5556. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Son, H.; Lee, S.-W.; Hwang, W.; Jung, S.-R.; Byl, J.A.W.; Osheroff, N.; Le, S. Selection of DNA cleavage sites by topoisomerase II results from enzyme-induced flexibility of DNA. Cell Chem. Biol. 2019, 26, 502–511.e3. [Google Scholar] [CrossRef]
- Mar-Wiz (Detection of Putative Matrix Attachment Regions in Eukaryotic DNA Sequences). Available online: https://www.futuresoft.org/MAR-Wiz/ (accessed on 17 March 2003).
- Denoeud, F.; Vergnaud, G.; Benson, G. Predicting Human Minisatellite Polymorphism. Genome Res. 2003, 13, 856–867. [Google Scholar] [CrossRef] [Green Version]
- Suchkova, I.O.; Tran, V.T.; Sasina, L.K.; Baranova, T.V.; Mustafina, O.E.; Khusnutdinova, E.K.; Wo, V.C.; Patkin, E.L. Population differences in the length polymorphism UPS29 of the ACAP3 gene in cohorts belonging to different ethnic groups. in preparation.
- Tynan, K.M.; Field, L.L.; Hoar, D.I. Ethnic-specific allelic variation within the D1Z2 locus. Hum. Hered. 1990, 40, 1–14. [Google Scholar] [CrossRef]
- Chevalier, D.; Allorge, D.; Lo-Guidice, J.-M.; Cauffiez, C.; Lepetit, C.; Migot-Nabias, F.; Kenani, A.; Lhermitte, M.; Broly, F. Sequence analysis, frequency and ethnic distribution of VNTR polymorphism in the 5′-untranslated region of the human prostacyclin synthase gene (CYP8A1). Prostaglandins Other Lipid Mediat. 2002, 70, 31–37. [Google Scholar] [CrossRef]
- Mansoor, A.; Mazhar, K.; Qamar, R. VNTR polymorphism of the DRD4 locus in different Pakistani ethnic groups. Genet Test. 2008, 12, 299–304. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, Y.; Xiong, N.; Qing, H.; Wang, T.; Lin, Z. Presence of recombination hotspots throughout SLC6A3. PLoS ONE 2019, 14, e0218129. [Google Scholar] [CrossRef]
- Nagase, T.; Kikuno, R.; Hattori, A.; Kondo, Y.; Okumura, K.; Ohara, O. Prediction of the coding sequences of unidentified human genes. XIX. The complete sequences of 100 new cDNA clones from the brain which code for large proteins in vitro. DNA Res. 2000, 7, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Ensembl. Gene Expression. Available online: http://www.ensembl.org/Homo_sapiens/Gene/ExpressionAtlas?db=core;g=ENSG00000131584;r=1:1292390-1309609;t=ENST00000354700 (accessed on 21 May 2020).
- The Human Protein Atlas. ACAP3. Available online: https://www.proteinatlas.org/ENSG00000131584-ACAP3 (accessed on 21 May 2020).
- Miura, Y.; Kanaho, Y. ACAP3, the GTPase-activating protein specific to the small GTPase Arf6, regulates neuronal migration in the developing cerebral cortex. Biochem. Biophys. Res. Commun. 2017, 493, 1089–1094. [Google Scholar] [CrossRef]
- UniProtKB. Q96P50. Available online: http://www.uniprot.org/uniprot/Q96P50 (accessed on 21 May 2020).
- Randazzo, P.A.; Miura, K.; Jackson, T.R. Assay and purification of phosphoinositide-dependent ADP-Ribosylation Factor (ARF) GTPase activating protein. Methods Enzymol. 2001, 329, 343–355. [Google Scholar]
- Moore, C.D.; Thacker, E.E.; Larimore, J.; Gaston, D.; Underwood, A.; Kearns, B.; Patterson, S.I.; Jackson, T.; Chapleau, C.; Pozzo-Miller, L.; et al. The neuronal Arf GAP centaurin α1 modulates dendritic differentiation. J. Cell Sci. 2007, 120, 2683–2693. [Google Scholar] [CrossRef] [Green Version]
- Kahn, R.A.; Bruford, E.; Inoue, H.; Logsdon, J.M.; Nie, Z.; Premont, R.T.; Randazzo, P.A.; Satake, M.; Theibert, A.B.; Zapp, M.L.; et al. Consensus nomenclature for the human ArfGAP domain-containing proteins. J. Cell Biol. 2008, 182, 1039–1044. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Jackson, C.L. ARF family G proteins and their regulators: Roles in membrane transport, development, and disease. Nat. Rev. Mol. Cell Biol. 2011, 12, 362–375. [Google Scholar] [CrossRef]
- Bento, C.F.; Puri, C.; Moreau, K.; Rubinsztain, D.C. The role of membrane-trafficking small GTPases in the regulation of autophagy. J. Cell Sci. 2013, 126, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Miura, Y.; Hongu, T.; Yamauchi, Y.; Funakoshi, Y.; Katagiri, N.; Ohbayashi, N.; Kanaho, Y. ACAP3 regulates neurite outgrowth through its GAP activity specific to Arf6 in mouse hippocampal neurons. Biochem. J. 2016, 473, 2591–2602. [Google Scholar] [CrossRef]
- Hongu, T.; Yamauchi, Y.; Funakoshi, Y.; Katagiri, N.; Ohbayashi, N.; Kanaho, Y. Pathological functions of the small GTPase Arf6 in cancer progression: Tumor angiogenesis and metastasis. Small GTPases 2016, 7, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Szatmari, E.M.; Oliveira, A.F.; Sumner, E.J.; Yasuda, R. Centaurin-α1-Ras-Elk-1 signaling at mitochondria mediates β-amyloid-induced synaptic dysfunction. J. Neurosci. 2013, 33, 5367–5374. [Google Scholar] [CrossRef] [Green Version]
- Sticker, R.; Reiser, G. Functions of the neuron-specific protein ADAP1 (centaurin-α1) in neuronal differentiation and neurodegenerative diseases, with an overview of structural and biochemical properties of ADAP1. Biol. Chem. 2014, 395, 1321–1340. [Google Scholar] [CrossRef]
- Falace, A.; Filipello, F.; Padula, V.L.; Vanni, N.; Madia, F.; Pietri, T.D.D.; de Falco, F.A.; Striano, P.; Bricarelli, F.D.; Minetti, C.; et al. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am. J. Hum. Genet. 2010, 87, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Suchkova, I.O.; Shubina, D.M.; Yakimovskii, A.F.; Borisova, E.V.; Eliseeva, N.G.; Sasina, L.K.; Baranova, T.V.; Baranov, V.S.; Patkin, E.L. Analysis of the Association of Minisatellite UPS29 of CENTB5 gene with Parkinson’s Disease. Russ. J. Genet. Appl. Res. 2011, 1, 128–137. [Google Scholar] [CrossRef]
- Atlas of Genetics and Cytogenetics in Oncology and Haematology, Chromosome 1 Genes. Available online: http://atlasgeneticsoncology.org/Indexbychrom/idxg_1.html (accessed on 22 May 2020).
- Nakamura, Y.; Koyama, K.; Matsushima, M. VNTR (variable number of tandem repeat) sequences as transcriptional, translational, or functional regulators. J. Hum. Genet. 1998, 43, 149–152. [Google Scholar] [CrossRef]
- Rose, A.B. Introns as gene regulators: A brick on the accelerator. Front. Genet. 2019, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.Y.; Shao, W.; Chang, L.; Yin, Y.; Li, T.; Zhang, H.; Hong, Y.; Perchade, M.; Guo, L.; Wu, Z.; et al. Genomic repeats categorize genes with distinct functions for orchestrated regulation. Cell Rep. 2020, 30, 3296–3311e5. [Google Scholar] [CrossRef] [Green Version]
- Shubina, D.M.; Suchkova, I.O.; Slominskaya, N.A.; Alenina, N.; Bader, M.; Patkin, E.L. CENTB5 gene expression in humans and mice. Mol. Biol. 2009, 43, 374–380. [Google Scholar] [CrossRef]
- Brykczynska, U.; Pecho-Vrieseling, E.; Thiemeyer, A.; Klein, J.; Fruh, I.; Doll, T.; Manneville, C.; Fuchs, S.; Iazeolla, M.; Beibel, M.; et al. CGG repeat-induced FMR1 silencing depends on the expansion size in human iPSCs and neurons carrying unmethylated full mutations. Stem Cell Rep. 2016, 7, 1059–1071. [Google Scholar] [CrossRef]
- Polak, U.; Li, Y.; Butler, J.S.; Napierala, M. Alleviating GAA repeat-induced transcriptional silencing of the Friedreich’s ataxia gene during somatic cell reprogramming. Stem Cells Dev. 2016, 25, 1788–1800. [Google Scholar] [CrossRef] [Green Version]
- Suchkova, I.O.; Borisova, E.V.; Borovkova, N.K.; Rogozina, A.A.; Milyukhina, I.V.; Belotserkovskaya, E.V.; Patkin, E.L. Minisatellite repeat polymorphism 5-HTTLPR, STin2, and B2-VNTR in patients with symptomatic epilepsy. In Materials of the International Virtual Internet Conference “Medicine in the XXI Century: Trends and Prospects”, April 19–20; Kazan-IP Sinyaev D.N.: Penza, Russia, 2013; Volume 2, pp. 106–109. [Google Scholar]
- Scorza, F.A. Parkinson’s disease, epileptic seizures, and sudden death: Three faces of the same coin. Epilepsy Behav. 2018, 83, 239–241. [Google Scholar] [CrossRef]
- Gruntz, K.; Bloechliger, M.; Becker, C.; Jick, S.; Peter Fuhr, P.; Meier, C.R.; Rüegg, S. Parkinson disease and the risk of epileptic seizures. Ann. Neurol. 2018, 83, 363–374. [Google Scholar] [CrossRef]
- Sasina, L.K.; Slominskaia, N.A.; Suchkova, I.O.; Pitsik, E.V.; Solovyov, K.V.; Grudinina, N.A.; Klinskaia, T.A.; Patkin, E.L. Human intra-intronic minisatellite UPS29 associated with neurological diseases regulate reporter gene EGFP expression depending on cell type. Tsytology 2010, 52, 715–723. (In Russian) [Google Scholar]
- Sasina, L.K.; Fedorova, E.M.; Grudinina, N.A.; Belotserkovskaya, E.V.; Solovyov, K.V.; Suchkova, I.O.; Patkin, E.L. Modulation of reporter EGFP gene expression by a disease-associated human intra-intronic minisatellite upon transient and stable transfection. Int. J. Agric. Biol. Eng. 2013, 3, 1–10. [Google Scholar]
- Belotserkovskaya, E.V.; Suchkova, I.O.; Borisova, E.V.; Borovkova, N.K.; Pavlinova, L.I.; Patkin, E.L. Concurrent changes of CSTB and ACAP3 genes expression in symptomatic epilepsy and Parkinson’s disease. Int. Res. J. 2017, 66, 96–103. [Google Scholar]
- Lalioti, M.D.; Antonarakis, S.E.; Scott, H.S. Epilepsy, the protease inhibitor, and the dodecamer: Progressive myoclonus epilepsy, cystatin B and a 12-mer repeat expansion. Cytogenet. Genome Res. 2003, 100, 213–223. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [Green Version]
- Scelfo, A.; Fachinetti, D. Keeping the centromere under control: A promising role for DNA methylation. Cells 2019, 8, 912. [Google Scholar] [CrossRef] [Green Version]
- Dion, V.; Lin, Y.; Price, B.A.; Fyffe, S.L.; Seluanov, A.; Gorbunova, V.; Wilson, J.H. Genome-wide demethylation promotes triplet repeat instability independently of homologous recombination. DNA Repair 2008, 7, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Imai, K. Microsatellite instability: An update. Arch. Toxicol. 2015, 89, 899–921. [Google Scholar] [CrossRef]
- Mitsuda, S.-H.; Shimizu, N. Epigenetic repeat-induced gene silencing in the chromosomal and extrachromosomal contexts in human cells. PLoS ONE 2016, 11, e0161288. [Google Scholar] [CrossRef] [Green Version]
- Tsegay, P.S.; Lai, Y.; Liu, Y. Replication stress and consequential instability of the genome and epigenome. Molecules 2019, 24, 3870. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.; Dugan, P.; Kirsch, H.E.; Friedman, D. Sex differences in seizure types and symptoms. Epilepsy Behav. 2014, 41, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Credendino, S.C.; Neumayer, C.; Cantone, I. Genetics and Epigenetics of Sex Bias: Insights from Human Cancer and Autoimmunity. Trends Genet. 2020, 36, 650–653. [Google Scholar] [CrossRef]
- Luedi, P.P.; Dietrich, F.S.; Weidman, J.R.; Bosko, J.M.; Jirtle, R.L.; Hartemink, A.J. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007, 17, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Brideau, C.M.; Eilertson, K.E.; Hagarman, J.A.; Bustamante, C.D.; Soloway, P.D. Successful computational prediction of novel imprinted genes from epigenomic features. Mol. Cell Biol. 2010, 30, 3357–3370. [Google Scholar] [CrossRef] [Green Version]
- Girardot, M.; Cavaille, J.; Feil, R. Small regulatory RNAs controlled by genomic imprinting and their contribution to human disease. Epigenetics 2012, 7, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Hutter, B.; Helms, V.; Paulsen, M. Tandem repeats in the CpG islands of imprinted genes. Genomics 2006, 88, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Khatib, H.; Zaitoun, I.; Kim, E.-S. Comparative analysis of Sequence characteristics of imprinted genes in humans, mice, and cattle. Mamm. Genome 2007, 18, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Hara, S.; Kato, T.; Tamano, M.; Muramatsu, A.; Asahara, H.; Takada, S. A tandem repeat array in IG-DMR is essential for imprinting of paternal allele at the Dlk1-Dio3 domain during embryonic development. Hum. Mol. Genet. 2018, 27, 3283–3292. [Google Scholar] [CrossRef]
- Corre, S.; Galibert, M.-D. USF is a key regulatory element of gene expression. Med. Sci. 2006, 22, 62–67. [Google Scholar]
- Skabkin, M.A.; Lyabin, D.N.; Ovchinnikov, L.P. Nonspecific and specific interactions of Y-box-binding protein 1 (YB-1) with mRNA and posttranscriptional regulation of protein synthesis in animal cells. Mol. Biol. 2006, 40, 551–563. [Google Scholar] [CrossRef]
- Chukanova, A.S.; Tushkanov, M.A.; Barsky, V.I.; Grazhdan, I.K.; Cricova, E.V.; Aksenova, M.G.; Burd, S.G.; Gusev, E.I. Analysis of the SERT and TPH2 Genes polymorphism relationship with side effect during topiramate therapy taking into account gender features. Epilepsy Paroxysmal Cond. 2011, 3, 45–54. (In Russian) [Google Scholar]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, R.; Champigny, G.; Bassilana, F.C.; Voilley, N.; Lazdunski, M. Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J. Biol. Chem. 1995, 270, 27411–27414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perosa, S.R.; Argañaraz, G.A.; Goto, E.M.; Costa, L.G.P.; Konno, A.C.; Varella, P.P.V.; Santiago, J.F.C.; Pesquero, J.B.; Canzian, M.; Amado, D.; et al. Kinin B1 and B2 receptors are overexpressed in the hippocampus of humans with temporal lobe epilepsy. Hippocampus 2007, 17, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Gibson, L.M.; Hanby, M.F.; Al-Bachari, S.M.; Parkes, L.M.; Allan, S.M.; Emsley, H.C.A. Late-onset epilepsy and occult cerebrovascular disease. J. Cereb. Blood Flow Metab. 2014, 34, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, R. Penfield, and the vascular hypothesis of focal epilepsy. J. Neurosurg. 2019, 131, 1947–1953. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Bragatti, J.A.; Becker, J.A.; Torres, C.M.; Martin, K.C.; de Souza, A.C.; Manfro, G.G.; Leistner-Segal, S.; Bianchin, M.M. Serotonin gene polymorphisms and psychiatry comorbidities in temporal lobe epilepsy. Epilepsy Res. 2012, 99, 260–266. [Google Scholar] [CrossRef] [Green Version]
- Politsky, J.M. Brain tumor-related epilepsy: A current review of the etiologic basis and diagnostic and treatment approaches. Curr. Neurol. Neurosci. Rep. 2017, 17, 70. [Google Scholar] [CrossRef]
- Chen, D.Y.; Chen, C.C.; Crawford, J.R.; Wang, S.G. Tumor-related epilepsy: Epidemiology, pathogenesis, and management. J. Neurooncol. 2018, 139, 13–21. [Google Scholar] [CrossRef]
- Hanukoglu, I.; Hanukoglu, A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene 2016, 579, 95–132. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Forty years of sodium channels: Structure, function, pharmacology, and epilepsy. Neurochem. Res. 2017, 42, 2495–2504. [Google Scholar] [CrossRef]
- Nikolić, I.; Ristić, A.; Vojvodić, N.; Baščarević, V.; Ilanković, A.; Berisavac, I.; Đukić, T.; Sokić, D. The association of arachnoid cysts and focal epilepsy: Hospital-based case-control study. Clin. Neurol. Neurosurg. 2017, 159, 39–41. [Google Scholar] [CrossRef]
- Meyer, M.Z.; Déliot, N.; Chasserot-Golaz, S.; Premont, R.T.; Bader, M.-F. Vitale N regulation of neuroendocrine exocytosis by the ARF6 GTPase-activating protein GIT1. J. Biol. Chem. 2006, 281, 7919–7926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba, Y.; Randazzo, P.A. ArfGAP1 function in COPI mediated membrane traffic: Currently debated models and comparison to other coat-binding ArfGAPs. Histol. Histopathol. 2012, 27, 1143–1153. [Google Scholar]
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989, 30, 389–399. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waalwijk, C.; Flavell, R.A. MspI an isoschizomer of HpaII which cleaves both unmethylated and methylated HpaII sites. Nucleic Acids Res. 1978, 5, 3231–3236. [Google Scholar] [CrossRef] [Green Version]
- Henninger, N.; Mayasi, Y. Nucleic Acid Therapies for Ischemic Stroke. Neurotherapeutics 2019, 16, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Zain, R.; Smith, E. Targeted Oligonucleotides for Treating Neurodegenerative Tandem Repeat Diseases. Neurotherapeutics 2019, 16, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.K.; Brown, R.H.; Khvorova, A. Nucleic Acid Therapeutics for Neurological Diseases. Neurotherapeutics 2019, 16, 245–247. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
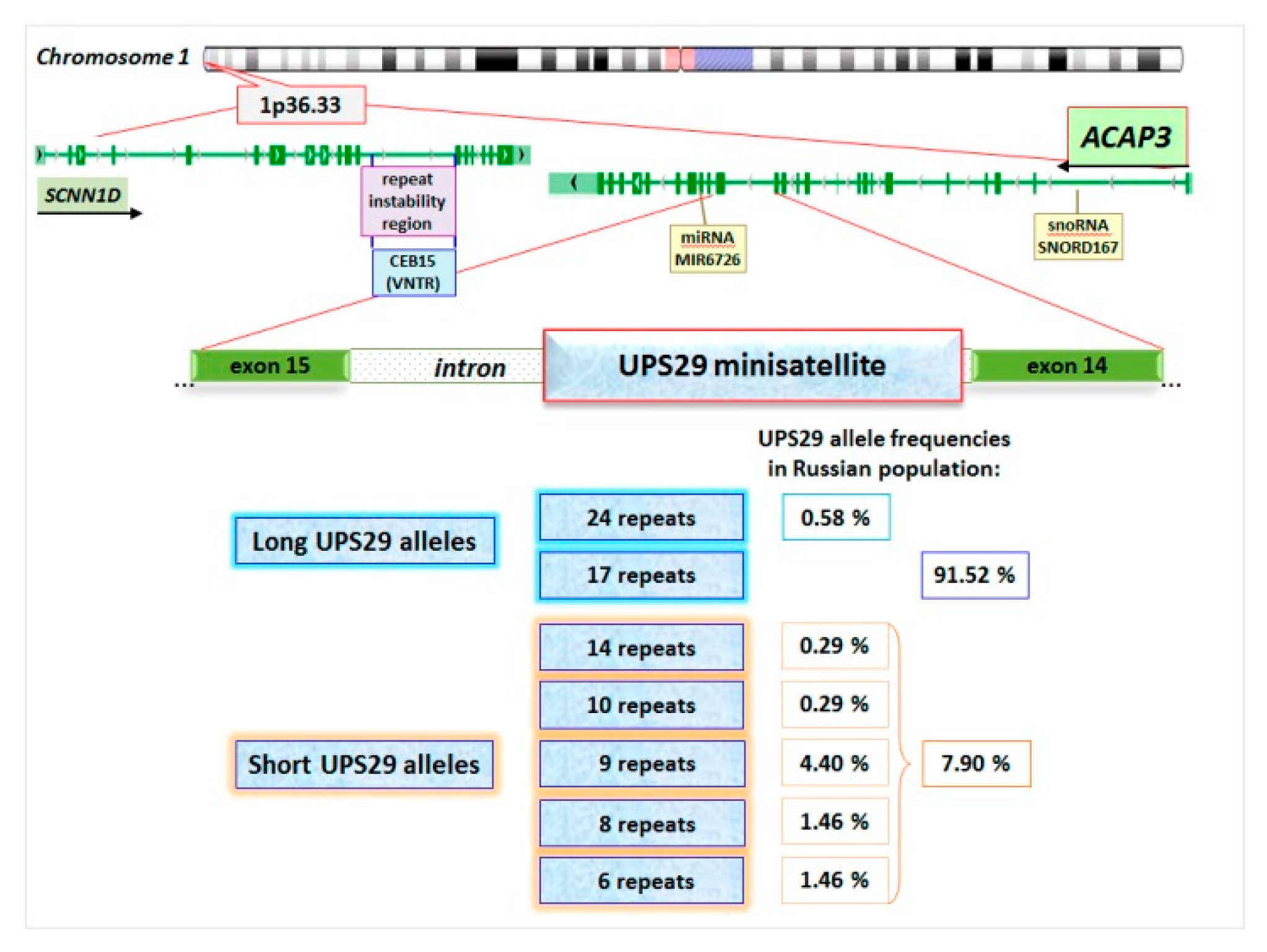
| UPS29 Minisatellite | Symptomatic Epilepsy | Cryptogenic Epilepsy | Control | |||
|---|---|---|---|---|---|---|
| N | Frequency, % (95% CI) | N | Frequency, % (95% CI) | N | Frequency, % (95% CI) | |
| Allele | ||||||
| 17 repeats | 119 | 87.5 (80.9–92.1) | 24 | 85.7 (68.5–94.3) | 170 | 92.4 (87.6–95.4) |
| 9 repeats | 10 | 7.4 (4.0–13.0) | 1 | 3.6 (0.6–17.7) | 8 | 4.4 (2.2–8.4) |
| 8 repeats | 5 | 3.7 (1.6–8.3) | 2 | 7.1 (2.0–22.6) | 3 | 1.6 (0.6–4.7) |
| 6 repeats | 2 | 1.5 (0.4–5.2) | 1 | 3.6 (0.6–17.7) | 3 | 1.6 (0.6–4.7) |
| Short alleles (sum) | 17 | 12.5 (8.0–19.1) | 4 | 14.3 (5.7–31.5) | 14 | 7.6 (4.6–12.4) |
| Total alleles | 136 | 28 | 184 | |||
| p-value | 0.181 | 0.269 | ||||
| Genotype | ||||||
| 17/17 | 52 | 76.5 (65.1–85.0) | 10 | 71.4 (45.4–88.3) | 80 | 87.0 (78.6–92.4) |
| 17/9 | 9 | 13.2 (7.1–23.3) | 1 | 7.1 (1.3–31.5) | 4 | 4.4 (1.7–10.7) |
| 17/8 | 4 | 5.9 (2.3–14.2) | 2 | 14.3 (4.0–40.0) | 3 | 3.3 (1.1–9.1) |
| 17/6 | 2 | 2.9 (0.8–10.1) | 1 | 7.14 (1.3–31.5) | 3 | 3.3 (1.1–9.1) |
| 9/8 | 1 | 1.5 (0.3–7.9) | 0 | 0 (0–21.5) | 0 | 0 (0–4.0) |
| 9/9 | 0 | 0 (0–5.4) | 0 | 0 (0–21.5) | 2 | 2.2 (0.6–7.6) |
| Short alleles carriers (sum) | 16 | 23.5 (15.0–34.8) | 4 | 28.57 (11.7–54.7) | 12 | 13. 0 (7.6–21.4) |
| Sample size | 68 | 14 | 92 | |||
| p-value | 0.096 | 0.220 | ||||
| UPS29 Minisatellite | Symptomatic Epilepsy | Cryptogenic Epilepsy | Control | |||
|---|---|---|---|---|---|---|
| N | Frequency, % (95% CI) | N | Frequency, % (95% CI) | N | Frequency, % (95% CI) | |
| Allele | ||||||
| 17 repeats | 174 | 83.7 (78.0–88.1) | 36 | 78.3 (64.4–87.7) | 250 | 93.3 (89.6–95.7) |
| 9 repeats | 20 | 9.6 (6.3–14.4) | 6 | 13.0 (6.1–25.7) | 7 | 2.6 (1.3–5.3) |
| 8 repeats | 7 | 3.4 (1.6–6.8) | 1 | 2.2 (0.4–11.3) | 6 | 2.2 (1.0–4.8) |
| 6 repeats | 7 | 3.4 (1.6–6.8) | 3 | 6.5 (2.2–17.5) | 5 | 1.9 (0.8–4.3) |
| Short alleles (sum) | 34 | 16.4 (11.9–22.0) | 10 | 21.7 (12.3–35.6) | 18 | 6.7 (4.3–10.4) |
| Total alleles | 208 | 46 | 268 | |||
| p-value | <0.001 | 0.003 | ||||
| Genotype | ||||||
| 17/17 | 73 | 70.2 (60.8–78.1) | 13 | 56.5 (36.8–74.4) | 117 | 87.3 (80.6–91.9) |
| 17/9 | 16 | 15.4 (9.7–23.5) | 6 | 26.1 (12.6–46.5) | 7 | 5.2 (2.6–10.4) |
| 17/8 | 5 | 4.8 (2.1–10.8) | 1 | 4.4 (0.8–21.0) | 4 | 3.0 (1.2–7.4) |
| 17/6 | 7 | 6.7 (3.3–13.2) | 3 | 13.0 (4.5–32.1) | 5 | 3.7 (1.6–8.4) |
| 9/8 | 2 | 1.9 (0.5–6.7) | 0 | 0 (0–14.3) | 0 | 0 (0–2.8) |
| 9/9 | 1 | 1.0 (0.2–5.3) | 0 | 0 (0–14.3) | 0 | 0 (0–2.8) |
| 8/8 | 0 | 0 (0–3.6) | 0 | 0 (0–14.3) | 1 | 0.8 (0.1–4.1) |
| Short alleles carriers (sum) | 31 | 29.8 (21.9–39.2) | 10 | 43.5 (25.6–63.2) | 17 | 12.7 (8.1–19.4) |
| Sample size | 104 | 23 | 134 | |||
| p-value | 0.001 | 0.001 | ||||
| Coefficient of association, phi | +0.21 | +0.29 | ||||
| RR (95% CI) | 2.5 (1.4–4.0) | 3.4 (1.9–6.5) | ||||
| OR (95% CI) | 2.9 (1.2–5.7) | 5.3 (2.0–14.0) | ||||
| Epilepsy Type | Age at Onset of a Seizure, Years (M ± SD) | |||||
|---|---|---|---|---|---|---|
| Men | Women | |||||
| Excluding UPS29 Genotype | UPS29 Homozygotes 17/17 | Subjects with Short UPS29 Alleles | Excluding UPS29 Genotype | UPS29 Homozygotes 17/17 | Subjects with Short UPS29 Alleles | |
| Symptomatic | 19.8 ± 16.3 | 17.5 ± 14.5 | 25.5 ± 19.5 | 20.4 ± 17.2 | 21.1 ± 16.6 | 18.4 ± 19.5 |
| Cryptogenic | 11.9 ± 5.7 | 11.9 ± 6.6 | 11.8 ± 2.3 | 14.0 ± 6.3 | 11.6 ± 5.4 | 17.3 ± 6.3 |
| Kruskal–Wallis chi-squared statistic | 3.625 | 2.499 | ||||
| p-value | 0.305 | 0.475 | ||||
| Epilepsy Type | Seizure Types (at Onset) | Total Cases, N | Frequency of UPS29 Genotype, % (95% CI) | p-Value * | phi | RR (95% CI), OR (95% CI) | |
|---|---|---|---|---|---|---|---|
| Homozygotes 17/17 | Subjects with Short UPS29 Alleles | ||||||
| Men | |||||||
| Symptomatic | Generalized | 47 | 83.0 (69.9–91.1) n = 39 | 17.0 (8.9–30.1) n = 8 | 0.611 | - | - |
| Focal (partial) | 21 | 62.0 (40.9–79.3) n = 13 | 38.1 (20.8–59.1) n = 8 | 0.012 | +0.26 | 2.9 (1.4–6.2) 4.1 (1.4–12.0) | |
| Cryptogenic | Generalized | 11 | 72.7 (43.4–90.3) n = 8 | 27.3 (9.7–56.6) n = 3 | 0.359 | - | - |
| Focal (partial) | 3 | 66.7 (20.8–93.9) n = 2 | 33.3 (6.2–79.2) n = 1 | 0.361 | - | - | |
| Total | Generalized | 58 | 81.0 (69.1–89.1) n = 47 | 19.0 (10.9–30.9) n = 11 | 0.358 | - | - |
| Focal (partial) | 24 | 62.5 (42.7–78.8) n = 15 | 37.5 (21.2–57.3) n = 9 | 0.009 | +0.26 | 2.9 (1.4–6.0) 4.0 (1.4–11.2) | |
| Women | |||||||
| Symptomatic | Generalized | 73 | 65.8 (54.3–75.6) n = 48 | 34.3 (24.4–45.7) n = 25 | <0.001 | + 0.26 | 2.7 (1.6–4.7) 3.6 (1.8–7.2) |
| Focal (partial) | 31 | 80.7 (63.7–90.8) n = 25 | 19.4 (9.18–36.3) n = 6 | 0.387 | - | - | |
| Cryptogenic | Generalized | 15 | 53.3 (30.1–75.2) n = 8 | 46.7 (24.8–69.9) n = 7 | 0.003 | + 0.28 | 3.7 (1.8–7.4) 6.0 (1.9–18.7) |
| Focal (partial) | 8 | 62.5 (30.6–86.3) n = 5 | 37.5 (13.7–69.4) n = 3 | 0.085 | - | - | |
| Total | Generalized | 88 | 63.6 (53.2–72.9) n = 56 | 36.4 (27.1–46.8) n = 32 | <0.001 | + 0.28 | 2.9 (1.7–4.8) 3.9 (2.0–7.7) |
| Focal (partial) | 39 | 76.9 (61.7–87.4) n = 30 | 23.1 (12.7–38.3) n = 9 | 0.128 | - | - | |
| Epilepsy Type | Seizure Types (at Onset) | Men | Women | ||
|---|---|---|---|---|---|
| N | Frequency, % (95% CI) | N | Frequency, % (95% CI) | ||
| Symptomatic | Generalized | 47 | 69.1 (57.4–78.8) | 73 | 70.2 (60.8–78.1) |
| Focal (partial) | 21 | 30.9 (21.2–42.6) | 31 | 29.8 (21.9–39.2) | |
| Cryptogenic | Generalized | 11 | 78.6 (52.4–92.4) | 15 | 65.2 (44.9–81.2) |
| Focal (partial) | 3 | 21.4 (7.6–47.6) | 8 | 34.8 (18.8–55.1) | |
| Total | Generalized | 58 | 70.7 (60.1–79.5) | 88 | 69.3 (60.8–76.7) |
| Focal (partial) | 24 | 29.3 (20.5–39.9) | 39 | 30.7 (23.4–39.2) | |
| Sample | Frequency, % (95% CI) | |
|---|---|---|
| Men | Women | |
| Etiological factors | ||
| Perinatal pathology | 41.2 (30.3–53.0) | 55.8 (46.2–65.0) |
| Traumatic brain injuries | 35.3 (25.0–47.2) | 24.0 (16.9–33.1) |
| Cerebrovascular pathologies | 10.3 (5.1–19.8) | 10.6 (6.0–18.0) |
| Neuroinfection and intoxication | 11.8 (6.1–21.5) | 6.7 (3.3–13.2) |
| Brain tumors | 1.5 (0.3–7.9) | 2.9 (1.0–8.1) |
| Total, N | 68 | 104 |
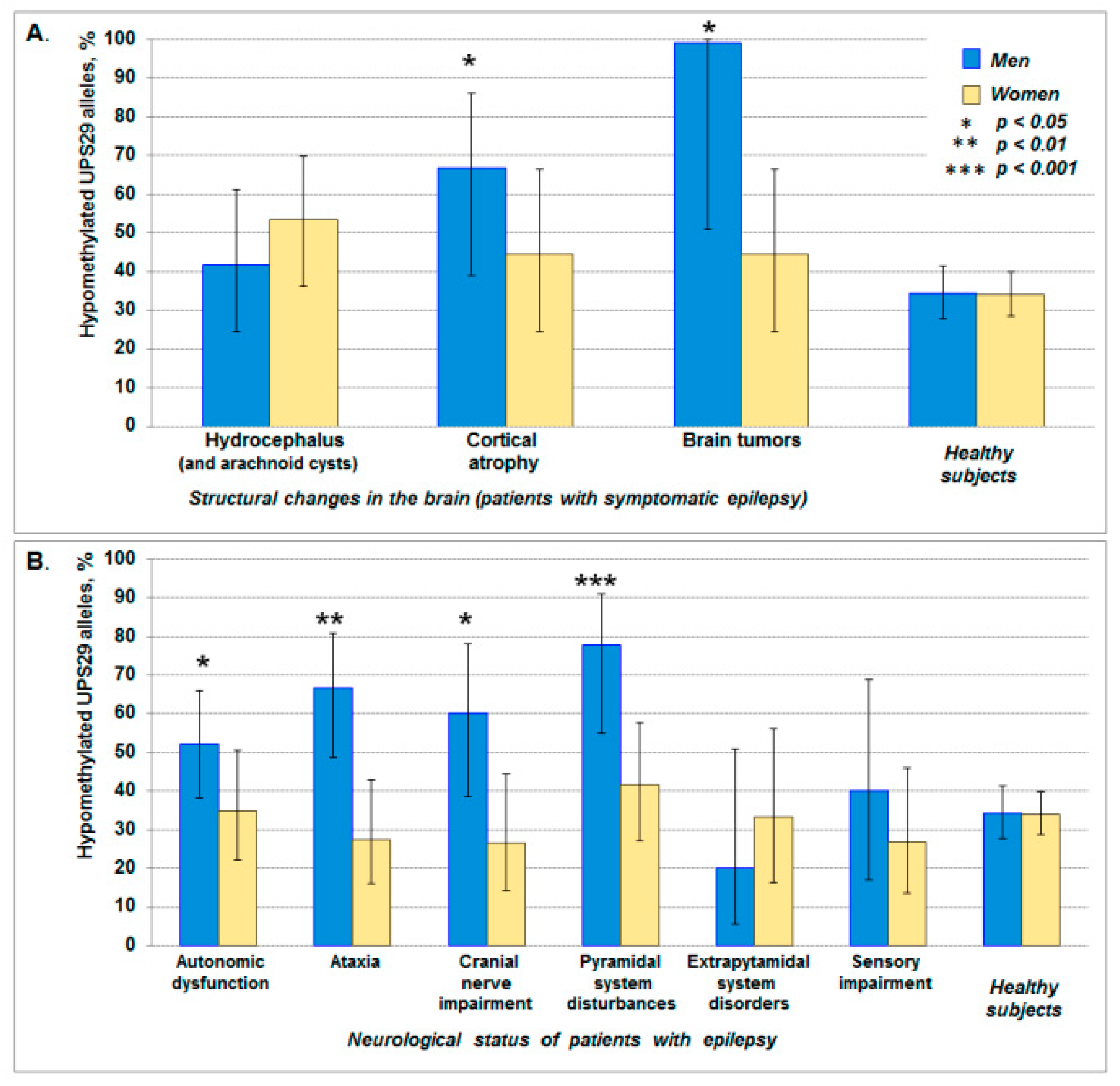
| Structural changes in the brain * | ||
| Hydrocephalus (and arachnoid cysts) | 57.4 (45.5–68.4) | 47.1 (37.8–56.6) |
| Cortical atrophy | 32.4 (22.4–44.2) | 26.0 (18.5–35.1) |
| Brain tumors | 10.3 (5.1–19.8) | 26.9 (19.3–36.2) |
| Total, N | 68 | 104 |
| Neurological status | ||
| Autonomic dysfunction | 34.3 (24.1–46.3) | 21.1 (14.1–30.3) |
| Ataxia | 22.4 (14.1–33.7) | 21.1 (14.1–30.3) |
| Cranial nerve impairment | 14.9 (8.3–25.4) | 15.8 (9.8–24.4) |
| Pyramidal system disturbances | 13.4 (7.2–23.6) | 19.0 (7.2–23.6) |
| Extrapyramidal system disorders | 7.5 (3.2–16.3) | 9.5 (5.1–17.0) |
| Sensory impairment | 7.5 (3.2–16.3) | 13.7 (8.2–22.0) |
| Total, N | 67 | 95 |
| Sample | Frequency, % (95% CI) | |
|---|---|---|
| Men | Women | |
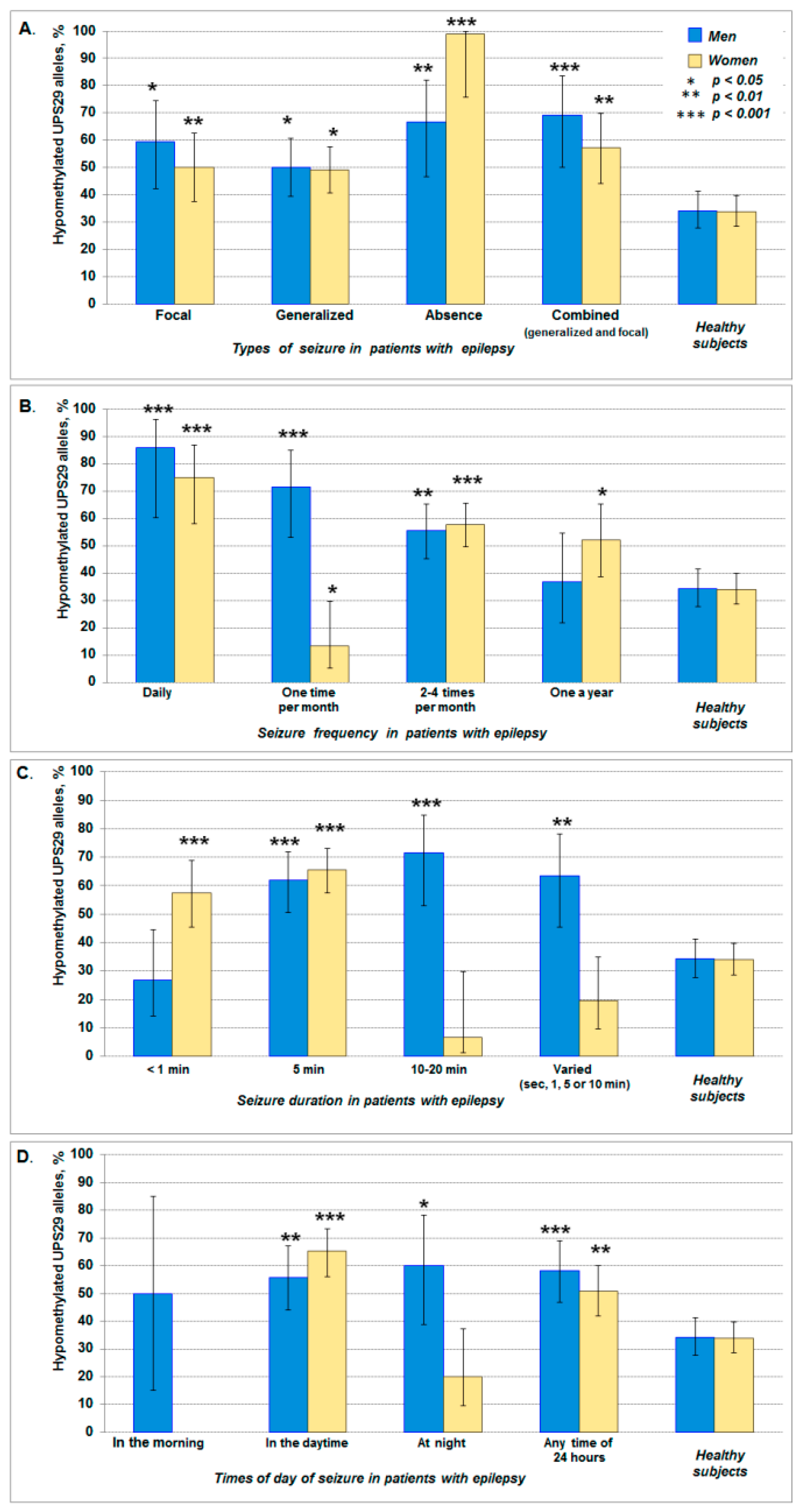
| Seizure types | ||
| Focal (partial) | 19.5 (12.4–29.4) | 22.8 (16.4–30.9) |
| Generalized | 50.0 (39.4–60.6) | 51.2 (42.6–59.7) |
| Absence | 14.6 (8.6–23.9) | 4.7 (2.2–9.9) |
| Combined | 15.9 (9.5–25.3) | 21.3 (15.0–29.2) |
| Seizure frequency | ||
| Daily | 8.5 (4.0–16.6) | 12.6 (7.9–19.5) |
| One time per month | 17.1 (10.5–26.6) | 11.8 (7.3–18.6) |
| 2–4 times per month | 56.1 (45.3–66.3) | 55.9 (47.2–64.3) |
| Once a year | 18.3 (11.4–28.0) | 19.7 (13.7–27.5) |
| Seizure duration * | ||
| <1 min | 18.3 (11.4–28.0) | 26.0 (19.1–34.2) |
| 5 min | 46.3 (36.0–57.1) | 54.3 (45.7–62.7) |
| 10–20 min | 17.1 (10.5–26.6) | 5.5 (2.7–10.9) |
| Varied (sec, 1, 5 or 10 min) | 18.3 (11.4–28.0) | 14.2 (9.2–21.3) |
| Times of day for seizure onset | ||
| In the morning | 2.4 (0.7–8.5) | 0 (0.0–2.9) |
| In the daytime | 41.5 (31.4–52.3) | 44.1 (35.8–52.8) |
| At night | 12.2 (6.8–21.0) | 11.8 (7.3–18.6) |
| Any time of 24 h | 43.9 (33.7–54.7) | 44.1 (35.8–52.8) |
| Total, N | 82 | 127 |
| UPS29 Epialleles | Symptomatic Epilepsy | Cryptogenic Epilepsy | Control | |||||
|---|---|---|---|---|---|---|---|---|
| N | Frequency, % (95% CI) | p-Value | N | Frequency, % (95% CI) | p-Value | N | Frequency, % (95% CI) | |
| Men | ||||||||
| Lmeth+ | 49 | 36.0 (28.5–44.4) | <0.001 | 12 | 42.9 (26.5–60.9) | 0.170 | 113 | 61.4 (54.2–68.1) |
| Lmeth− | 70 | 51.5 (43.2–59.7) | 12 | 42.9 (26.5–60.9) | 57 | 31.0 (24.7–38.0) | ||
| Shmeth+ | 7 | 5.2 (2.5–10.3) | 0.479 | 2 | 7.1 (2.0–22.6) | 1.000 | 8 | 4.4 (2.2–8.4) |
| Shmeth− | 10 | 7.4 (4.0–13.0) | 2 | 7.1 (2.0–22.6) | 6 | 3.3 (1.5–6.9) | ||
| Methylated alleles (sum) | 56 | 41.2 (33.3–49.6) | <0.001 | 14 | 50.0 (32.6–67.4) | 0.139 | 121 | 65.8 (58.6–72.2) |
| Hypomethylated alleles (sum) | 80 | 58.8 (50.4–66.7) | 14 | 50.0 (32.6–67.4) | 63 | 34.2 (27.8–41.4) | ||
| Total | 136 | 28 | 184 | |||||
| Coefficient of association, phi | +0.24 | - | ||||||
| RR (95% CI) | 1.6 (1.3–2.0) | - | ||||||
| OR (95% CI) | 2.7 (1.7–4.3) | - | ||||||
| Women | ||||||||
| Lmeth+ | 85 | 40.9 (34.4–47.7) | <0.001 | 14 | 30.4 (19.1–44.8) | 0.002 | 165 | 61.5 (55.6–67.29) |
| Lmeth− | 89 | 42.8 (36.3–49.6) | 22 | 47.8 (34.1–61.9) | 85 | 31.7 (26.4–37.5) | ||
| Shmeth+ | 14 | 6.7 (4.1–11.0) | 0.144 | 5 | 10.9 (4.7–23.0) | 0.444 | 12 | 4.5 (2.68–7.7) |
| Shmeth− | 20 | 9.6 (6.3–14.4) | 5 | 10.9 (4.7–23.0) | 6 | 2.3 (1.0–4.8) | ||
| Methylated alleles (sum) | 99 | 47.6 (40.9–54.4) | <0.001 | 19 | 41.3 (28.3–55.7) | 0.002 | 177 | 66.0 (60.2–71.5) |
| Hypomethylated alleles (sum) | 109 | 52.4 (45.6–59.1) | 27 | 58.7 (44.3–71.7) | 91 | 34.0 (28.6–39.8) | ||
| Total | 208 | 46 | 268 | |||||
| Coefficient of association, phi | +0.19 | +0.18 | ||||||
| RR (95% CI) | 1.4 (1.2–1.6) | 1.6 (1.1–2.3) | ||||||
| OR (95% CI) | 2.1 (1.5–3.1) | 2.8 (1.5–5.2) | ||||||
| UPS29 Epigenotypes | Symptomatic Epilepsy | Cryptogenic Epilepsy | Control | |||
|---|---|---|---|---|---|---|
| N | Frequency, % (95% CI) | N | Frequency, % (95% CI) | N | Frequency, % (95% CI) | |
| Men | ||||||
| Lmeth+/Lmeth+ | 23 | 33.8 (23.7–45.7) | 5 | 35.7 (16.3–61.2) | 54 | 58.7 (48.5–68.2) |
| Lmeth−/Lmeth− | 29 | 42.7 (31.6–54.5) | 5 | 35.7 (16.3–61.2) | 26 | 28.3 (20.1–38.2) |
| Lmeth+/Shmeth+ | 3 | 4.4 (1.5–12.2) | 2 | 14.3 (4.0–40.0) | 5 | 5.4 (2.3–12.1) |
| Lmeth+/Shmeth− | 0 | 0 (0–3.4) | 0 | 0 (0–21.5) | 0 | 0 (0–4.0) |
| Lmeth−/Shmeth+ | 2 | 2.9 (0.8–10.1) | 0 | 0 (0–21.5) | 1 | 1.1 (0.2–5.9) |
| Lmeth−/Shmeth− | 10 | 14.7 (8.2–25.0) | 2 | 14.3 (4.0–40.0) | 4 | 4.3 (1.7–10.7) |
| Shmeth+/Shmeth+ | 1 | 1.5 (0.3–7.9) | 0 | 0 (0–21.5) | 1 | 1.1 (0.2–5.9) |
| Shmeth+/Shmeth− | 0 | 0 (0–3.4) | 0 | 0 (0–21.5) | 0 | 0 (0–4.0) |
| Shmeth−/Shmeth− | 0 | 0 (0–3.4) | 0 | 0 (0–21.5) | 1 | 1.1 (0.2–5.9) |
| Subjects with methylated UPS29 (sum) | 29 | 42.6 (31.6–54.5) | 7 | 50.0 (26.8–73.2) | 61 | 66.3 (56.2–75.1) |
| Subjects with hypomethylated UPS29 (sum) | 39 | 57.4 (45.5–68.4) | 7 | 50.0 (26.8–73.2) | 31 | 33.7 (24.9–43.8) |
| Sample size | 68 | 14 | 92 | |||
| p-value | 0.004 | 0.370 | ||||
| Coefficient of association, phi | +0.24 | - | ||||
| RR (95% CI) | 1.6 (1.1–2.1) | - | ||||
| OR (95% CI) | 2.7 (1.4–5.1) | - | ||||
| Women | ||||||
| Lmeth+/Lmeth+ | 38 | 36.5 (27.9–46.1) | 5 | 21.7 (9.7–41.9) | 80 | 59.7 (51.2–67.6) |
| Lmeth−/Lmeth− | 35 | 33.7 (25.3–43.2) | 8 | 34.8 (18.8–55.1) | 37 | 27.6 (20.7–35.7) |
| Lmeth+/Shmeth+ | 7 | 6.7 (3.3–13.2) | 3 | 13.0 (4.5–32.1) | 4 | 3.0 (1.2–7.4) |
| Lmeth+/Shmeth− | 2 | 1.9 (0.5–6.7) | 1 | 4.3 (0.8–21.0) | 1 | 0.7 (0.1–4.1) |
| Lmeth−/Shmeth+ | 2 | 1.9 (0.5–6.7) | 2 | 8.7 (2.4–26.8) | 6 | 4.5 (2.1–9.4) |
| Lmeth−/Shmeth− | 17 | 16.3 (10.5–24.6) | 4 | 17.4 (7.0–37.1) | 5 | 3.7 (1.6–8.4) |
| Shmeth+/Shmeth+ | 2 | 1.9 (0.5–6.7) | 0 | 0 (0–14.3) | 1 | 0.7 (0.1–4.1) |
| Shmeth+/Shmeth− | 1 | 1.0 (0.2–5.2) | 0 | 0 (0–14.3) | 0 | 0 (0–2.8) |
| Shmeth−/Shmeth− | 0 | 0 (0–3.6) | 0 | 0 (0–14.3) | 0 | 0 (0–2.8) |
| Subjects with methylated UPS29 (sum) | 52 | 50.0 (40.6–59.4) | 11 | 47.8 (29.2–67.0) | 92 | 68.7 (60.4–75.9) |
| Subjects with hypomethylated UPS29 (sum) | 52 | 50.0 (40.6–59.4) | 12 | 52.2 (33.0–70.8) | 42 | 31.3 (24.1–39.6) |
| Sample size | 104 | 23 | 134 | |||
| p-value | 0.005 | 0.060 | ||||
| Coefficient of association, phi | +0.19 | - | ||||
| RR (95% CI) | 1.4 (1.1–1.7) | - | ||||
| OR (95% CI) | 2.2 (1.3–3.7) | - | ||||
| Sample | Men | Women | ||||||
|---|---|---|---|---|---|---|---|---|
| p-Value | RR (95% CI) | OR (95% CI) | phi | p-Value | RR (95% CI) | OR (95% CI) | phi | |
| Seizure types | ||||||||
| Focal (partial) | 0.020 | 1.5 (1.1–2.0) | 1.9 (1.1–3.3) | +0.15 | 0.004 | 1.5 (1.1–1.9) | 1.9 (1.2–2.9) | +0.15 |
| Generalized | 0.010 | 1.7 (1.2–2.5) | 2.8 (1.3–6.1) | +0.18 | 0.025 | 1.5 (1.1–2.0) | 2.0 (1.1–3.5) | +0.13 |
| Absence | 0.003 | 2.0 (1.4–2.8) | 3.8 (1.6–9.5) | +0.21 | <0.001 | 3.0 (2.9–3.5) | 23.3 (3.0–18.2) | +0.28 |
| Combined | 0.001 | 2.0 (1.5–2.8) | 4.3 (1.8–10.5) | +0.24 | 0.001 | 1.7 (1.3–2.3) | 2.6 (1.5–4.8) | +0.18 |
| Seizure frequency | ||||||||
| Daily | < 0.001 | 2.5 (1.9–3.4) | 11.5 (2.5–53.1) | +0.27 | <0.001 | 2.2 (1.7–2.9) | 5.8 (2.5–13.5) | +0.26 |
| One time per month | < 0.001 | 2.1 (1.5–2.8) | 4.8 (2.0–11.5) | +0.26 | 0.023 | 2.6 (1.0–6.4) | 3.3 (1.1–9.9) | −0.13 |
| 2–4 times per month | 0.001 | 1.6 (1.2–2.1) | 2.4 (1.4–4.0) | +0.20 | <0.001 | 1.7 (1.4–2.1) | 2.7 (1.8–4.0) | +0.23 |
| Once a year | 0.837 | - | - | - | 0.017 | 1.5 (1.1–2.1) | 2.1 (1.2–3.9) | +0.14 |
| Seizure duration | ||||||||
| < 1 min | 0.532 | - | - | - | 0.001 | 1.7 (1.3–2.2) | 2.6 (1.5–4.6) | +0.19 |
| 5 min | < 0.001 | 1.8 (1.4–2.4) | 3.1 (1.8–5.4) | +0.25 | <0.001 | 2.0 (1.6–2.4) | 3.7 (2.4–5.8) | +0.30 |
| 10–20 min | < 0.001 | 2.1 (1.5–2.8) | 4.8 (2.0–11.5) | +0.26 | 0.042 | - | - | - |
| Varied (sec, 1, 5 or 10 min) | 0.003 | 1.9 (1.3–2.6) | 3.3 (1.5–7.4) | +0.21 | 0.090 | - | - | - |
| Times of day for seizure onset | ||||||||
| In the morning | 0.610 | - | - | - | - | - | - | - |
| In the daytime | 0.002 | 1.6 (1.2–2.2) | 2.4 (1.4–4.3) | +0.20 | <0.001 | 1.9 (1.6–2.4) | 3.6 (2.3–5.8) | +0.29 |
| At night | 0.028 | 1.8 (1.2–2.6) | 2.9 (1.2–7.4) | +0.16 | 0.152 | - | - | - |
| Any time of 24 h | 0.001 | 1.7 (1.3–2.3) | 2.7 (1.5–4.7) | +0.22 | 0.003 | 1.5 (1.2–2.0) | 2.0 (1.3–3.2) | +0.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suchkova, I.O.; Borisova, E.V.; Patkin, E.L. Length Polymorphism and Methylation Status of UPS29 Minisatellite of the ACAP3 Gene as Molecular Biomarker of Epilepsy. Sex Differences in Seizure Types and Symptoms. Int. J. Mol. Sci. 2020, 21, 9206. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239206
Suchkova IO, Borisova EV, Patkin EL. Length Polymorphism and Methylation Status of UPS29 Minisatellite of the ACAP3 Gene as Molecular Biomarker of Epilepsy. Sex Differences in Seizure Types and Symptoms. International Journal of Molecular Sciences. 2020; 21(23):9206. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239206
Chicago/Turabian StyleSuchkova, Irina O., Elena V. Borisova, and Eugene L. Patkin. 2020. "Length Polymorphism and Methylation Status of UPS29 Minisatellite of the ACAP3 Gene as Molecular Biomarker of Epilepsy. Sex Differences in Seizure Types and Symptoms" International Journal of Molecular Sciences 21, no. 23: 9206. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239206