Sleep/Wake Behavior and EEG Signatures of the TgF344-AD Rat Model at the Prodromal Stage

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Demographics
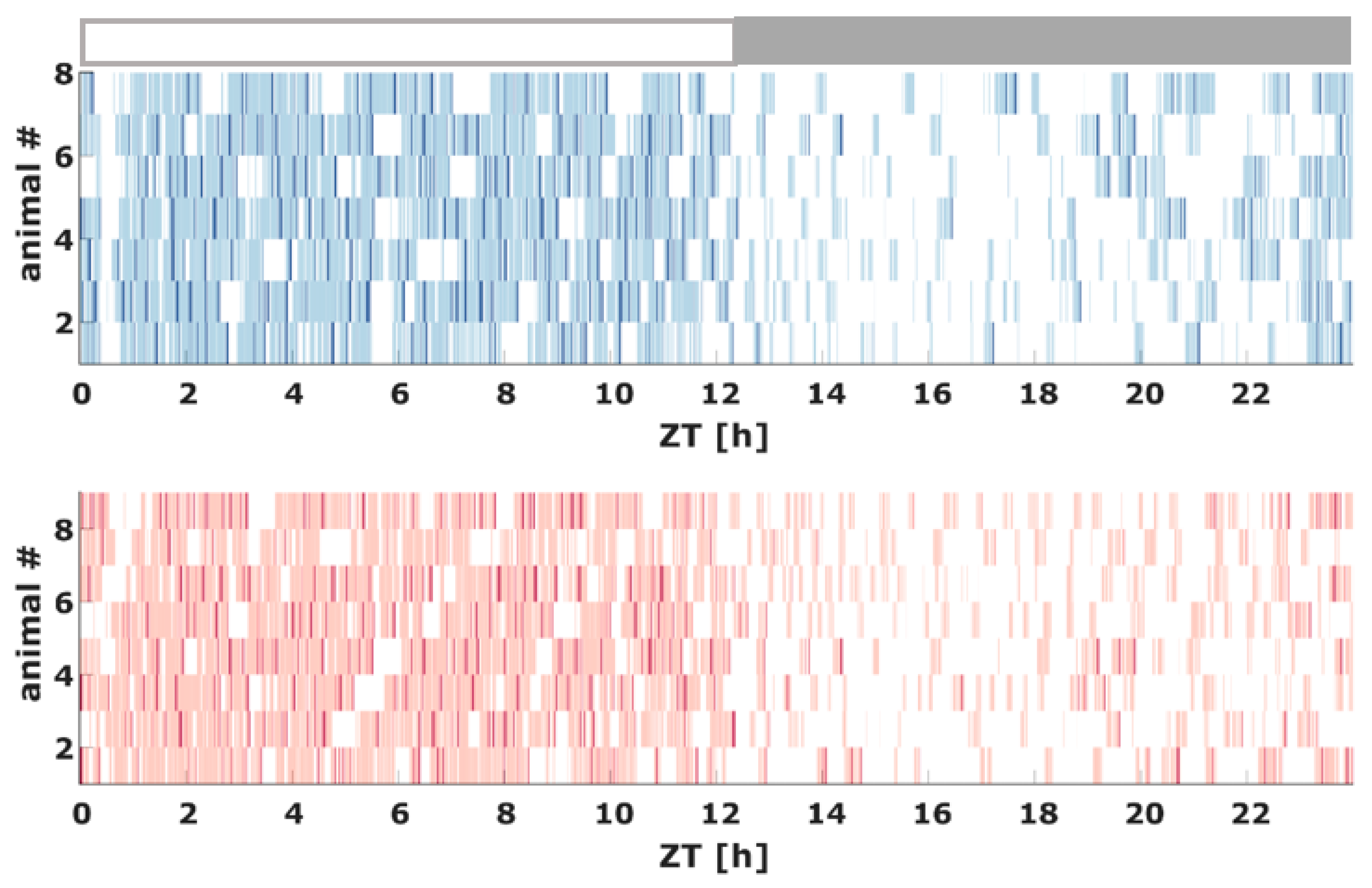
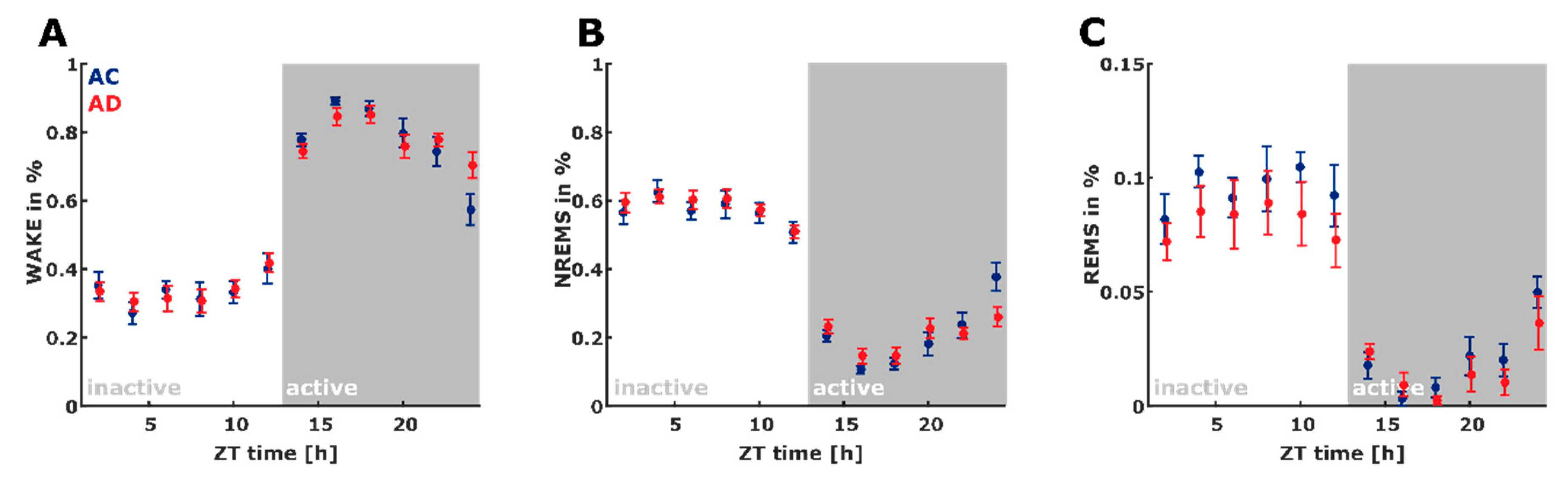
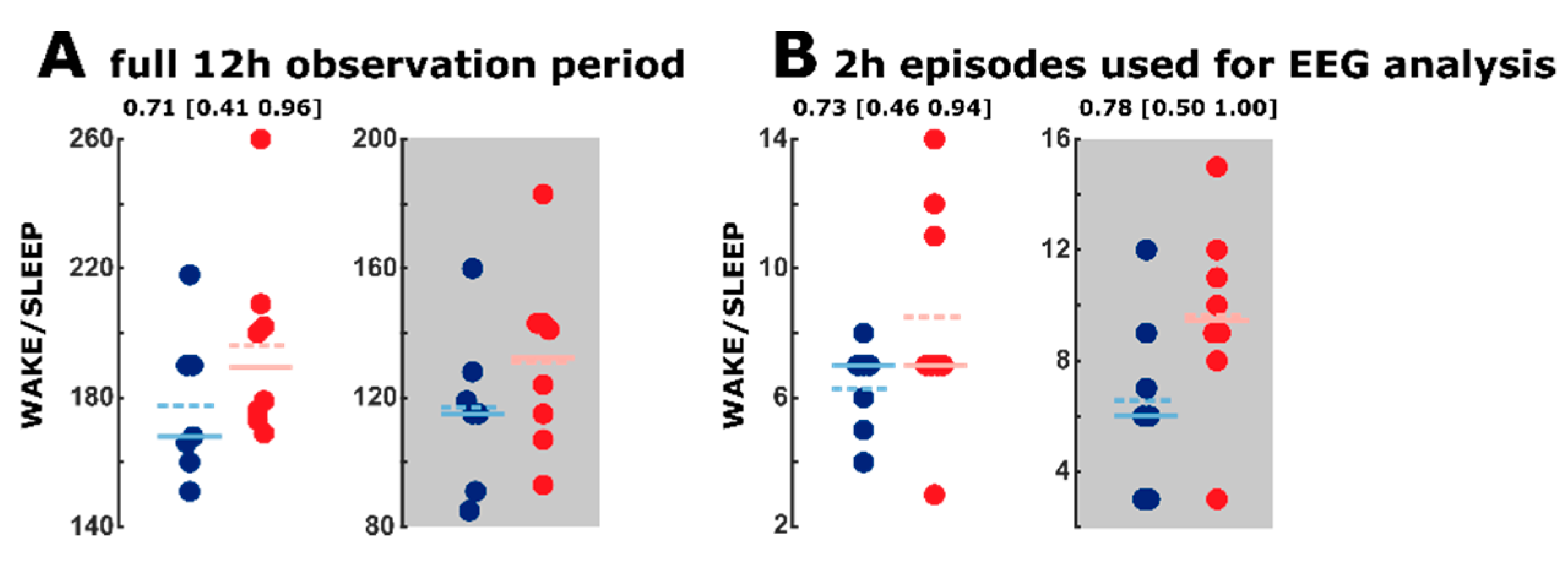
2.2. Sleep Macroarchitecture Based on the 10 s Sleep Scoring
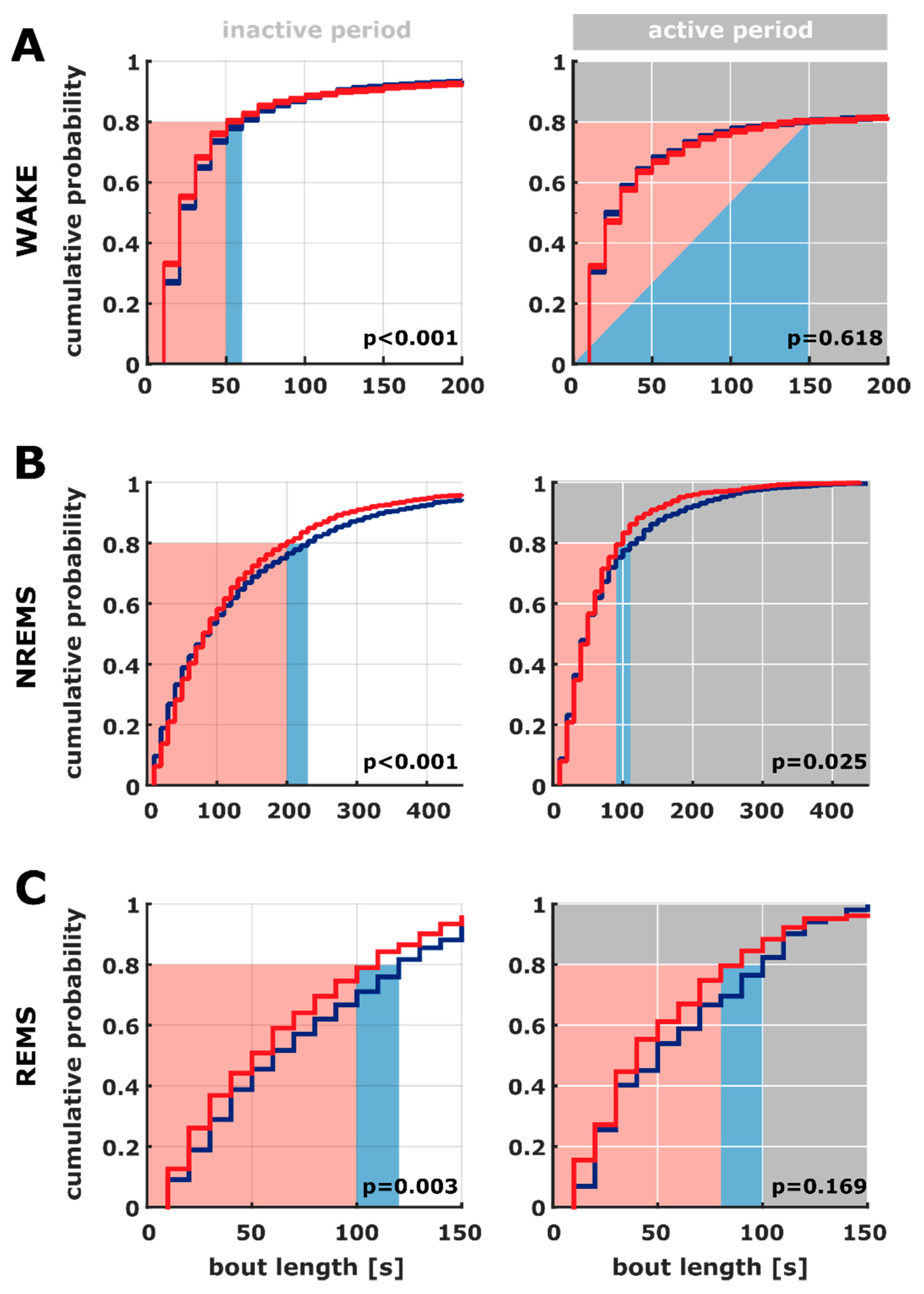
2.3. Bout Length Based on the 10 s Sleep Scoring
2.4. Transition between WAKE and Sleep Levels
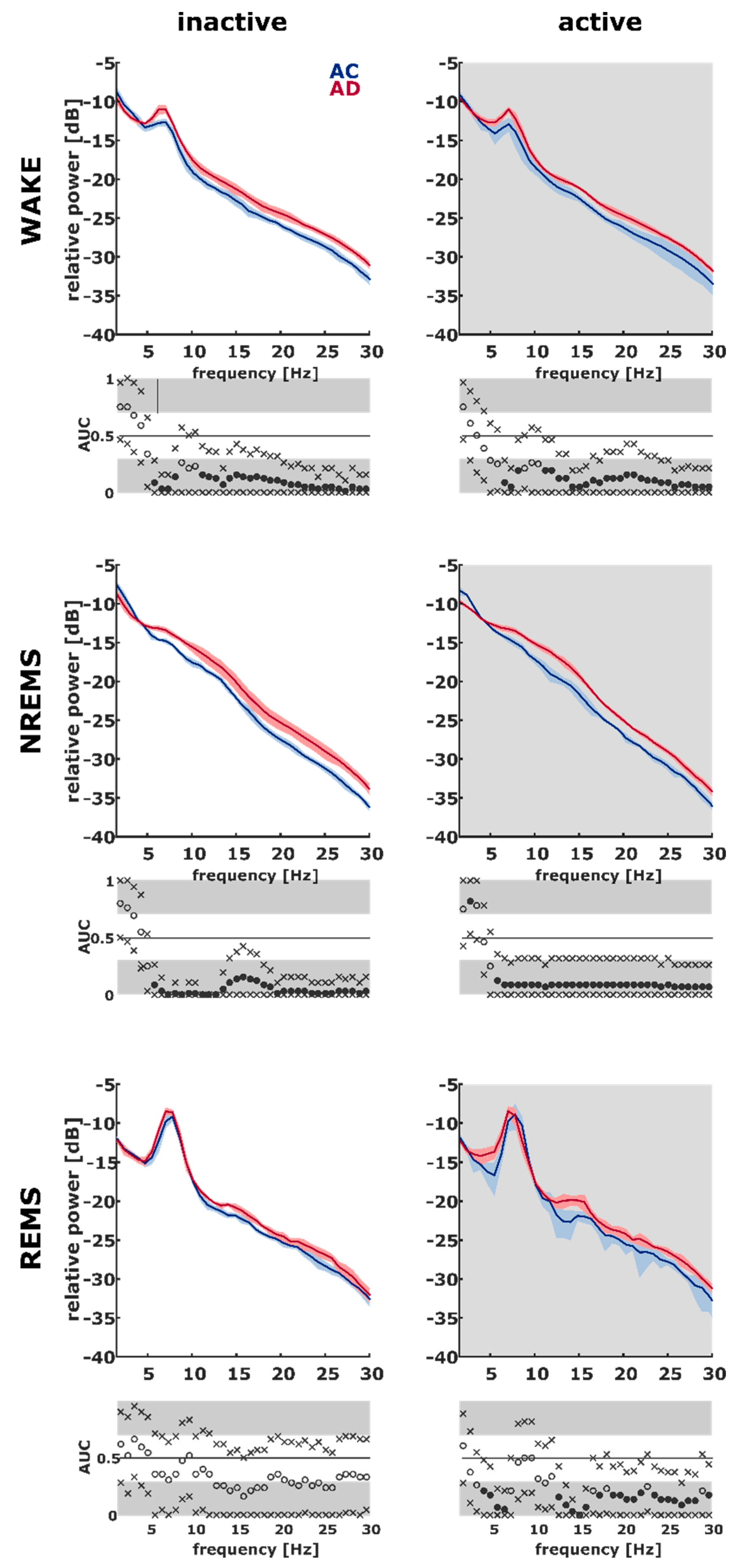
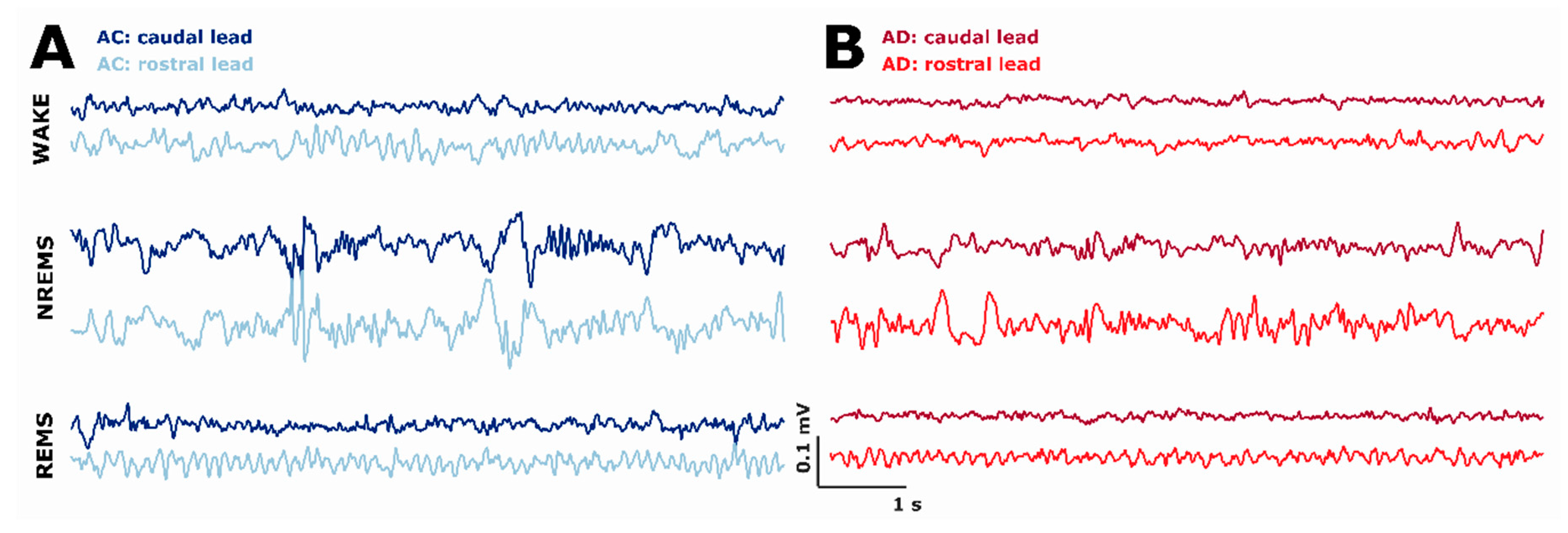
2.5. AD Rats Express Different Sleep Microarchitecture as Evaluated by Spectral EEG Properties
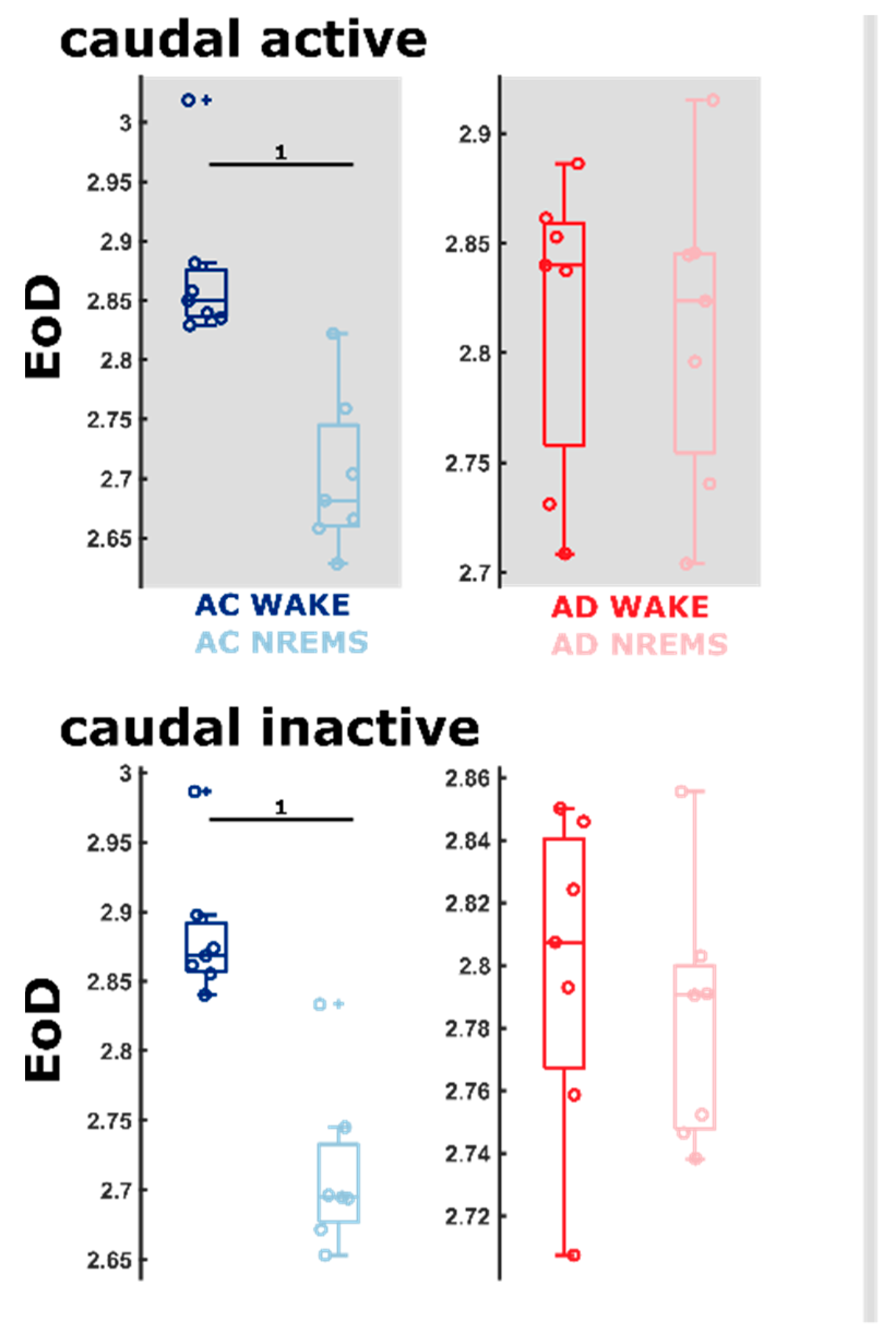
2.6. AD Rats Express Different Sleep Microarchitecture as Evaluated by Entropic EEG Properties
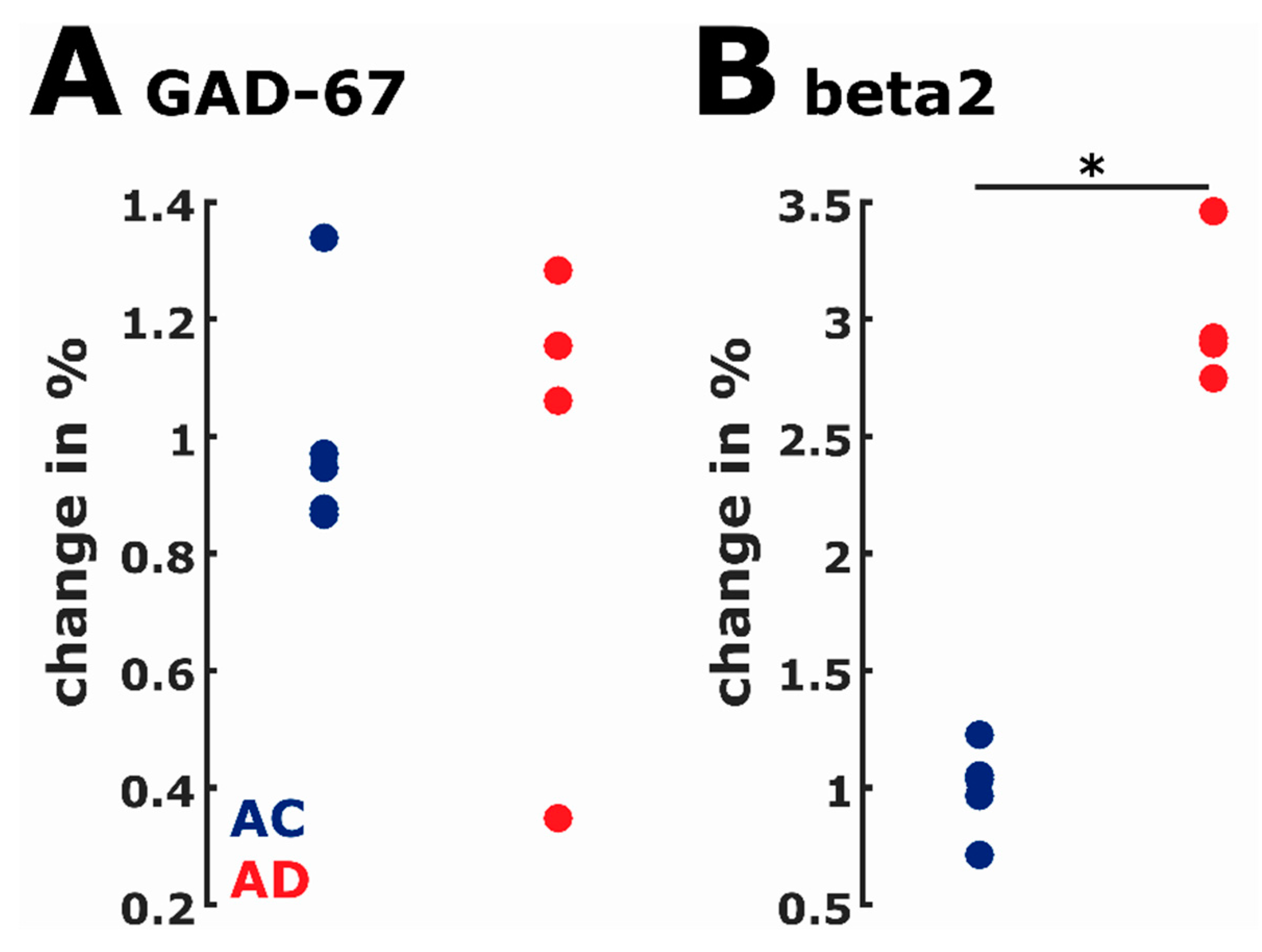
2.7. Protein Expression
3. Discussion
3.1. Early-Stage AD Influences Sleep Macro-Architecture
3.2. Differences in Sleep Microarchitecture between AD and AC Rats
4. Materials and Methods
4.1. EEG Surgery
4.2. Data Collection—EEG and EMG
4.3. EEG Recording Procedure
4.4. Assessment of Vigilance States
4.5. Analysis of Sleep Architecture
4.6. Quantitative EEG Analysis of WAKE, NREM Sleep, and REM Sleep
4.7. SDS-PAGE/Western Blotting
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Aged controls |
| AD | Alzheimer’s disease |
| AUC | Area under the receiver operating curve |
| EEG | Electroencephalogram |
| EMG | Electromyogram |
| EoD | Entropy of difference |
| MCI | Mild cognitive impairment |
| NREM | Non-rapid eye movement |
| NREMS | Non-rapid eye movement sleep |
| PeEn | Permutation entropy |
| REM | Rapid eye movement |
| REMS | Rapid eye movement sleep |
References
- Evans, D.A.; Funkenstein, H.H.; Albert, M.S.; Scherr, P.A.; Cook, N.R.; Chown, M.J.; Hebert, L.E.; Hennekens, C.H.; Taylor, J.O. Prevalence of Alzheimer’s disease in a community population of older persons: Higher than previously reported. JAMA 1989, 262, 2551–2556. [Google Scholar] [CrossRef]
- Schaller, S.; Mauskopf, J.; Kriza, C.; Wahlster, P.; Kolominsky-Rabas, P.L. The main cost drivers in dementia: A systematic review. Int. J. Geriatr. Psychiatry 2015, 30, 111–129. [Google Scholar] [CrossRef]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef]
- Wulff, K.; Gatti, S.; Wettstein, J.G.; Foster, R.G. Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat. Rev. Neurosci. 2010, 11, 589–599. [Google Scholar] [CrossRef]
- Jeong, J. EEG dynamics in patients with Alzheimer’s disease. Clin. Neurophysiol. 2004, 115, 1490–1505. [Google Scholar] [CrossRef]
- Kang, J.-E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef] [Green Version]
- Dustman, R.; Shearer, D.; Emmerson, R. Life-span changes in EEG spectral amplitude, amplitude variability and mean frequency. Clin. Neurophysiol. 1999, 110, 1399–1409. [Google Scholar] [CrossRef]
- Polich, J. EEG and ERP assessment of normal aging. Electroencephalogr. Clin. Neurophysiol. Potentials Sect. 1997, 104, 244–256. [Google Scholar] [CrossRef]
- Carrier, J.; Land, S.; Buysse, D.J.; Kupfer, D.J.; Monk, T.H. The effects of age and gender on sleep EEG power spectral density in the middle years of life (ages 20–60 years old). Psychophysiology 2001, 38, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Grouven, U.; Zander, I.; Beger, F.A.; Siedenberg, M.; Schultz, B. Age-related effects in the EEG during propofol anaesthesia. Acta Anaesthesiol. Scand. 2004, 48, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Purdon, P.L.; Pavone, K.J.; Akeju, O.; Smith, A.C.; Sampson, A.L.; Lee, J.; Zhou, D.W.; Solt, K.; Brown, E.N. The Ageing Brain: Age-dependent changes in the electroencephalogram during propofol and sevoflurane general anaesthesia. Br. J. Anaesth. 2015, 115, i46–i57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuzer, M.; Stern, M.A.; Hight, D.; Berger, S.; Schneider, G.; Sleigh, J.W.; García, P.S. Spectral and Entropic Features Are Altered by Age in the Electroencephalogram in Patients under Sevoflurane Anesthesia. Anesthesiology 2020, 132, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Roubicek, J. The electroencephalogram in the middle-aged and the elderly. J. Am. Geriatr. Soc. 1977, 25, 145–152. [Google Scholar] [CrossRef]
- McGinnis, S.M.; Brickhouse, M.; Pascual, B.; Dickerson, B.C. Age-related changes in the thickness of cortical zones in humans. Brain Topogr. 2011, 24, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Brunovsky, M.; Matousek, M.; Edman, A.; Cervena, K.; Krajca, V. Objective assessment of the degree of dementia by means of EEG. Neuropsychobiology 2003, 48, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Chemali, J.; Kenny, J.; Olutola, O.; Taylor, N.; Kimchi, E.; Purdon, P.; Brown, E.; Solt, K. Ageing delays emergence from general anaesthesia in rats by increasing anaesthetic sensitivity in the brain. Br. J. Anaesth. 2015, 115, i58–i65. [Google Scholar] [CrossRef] [Green Version]
- Gouw, A.A.; Alsema, A.M.; Tijms, B.M.; Scheltens, P.; Stam, C.J.; van der Flier, W.M. The relation between eeg spectral analysis and clinical progression in non-demented, amyloid-positive subjects. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2015, 11, P255–P256. [Google Scholar] [CrossRef]
- Ferreri, F.; Vecchio, F.; Vollero, L.; Guerra, A.; Petrichella, S.; Ponzo, D.; Määttä, S.; Mervaala, E.; Könönen, M.; Ursini, F.; et al. Sensorimotor cortex excitability and connectivity in Alzheimer’s disease: A TMS-EEG Co-registration study. Hum. Brain Mapp. 2016, 37, 2083–2096. [Google Scholar] [CrossRef]
- Lizio, R.; Vecchio, F.; Frisoni, G.B.; Ferri, R.; Rodriguez, G.; Babiloni, C. Electroencephalographic rhythms in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2011, 2011, 927573. [Google Scholar] [CrossRef] [Green Version]
- Filon, M.J.; Wallace, E.; Wright, S.; Douglas, D.J.; Steinberg, L.I.; Verkuilen, C.L.; Westmark, P.R.; Maganti, R.K.; Westmark, C.J. Sleep and Diurnal Rest-Activity Rhythm Disturbances in a Mouse Model of Alzheimer’s Disease. Sleep 2020. [Google Scholar] [CrossRef]
- Pase, M.P.; Himali, J.J.; Grima, N.A.; Beiser, A.S.; Satizabal, C.L.; Aparicio, H.J.; Thomas, R.J.; Gottlieb, D.J.; Auerbach, S.H.; Seshadri, S. Sleep architecture and the risk of incident dementia in the community. Neurology 2017, 89, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Tsolaki, A.; Kazis, D.; Kompatsiaris, I.; Kosmidou, V.; Tsolaki, M. Electroencephalogram and Alzheimer’s Disease: Clinical and Research Approaches. Int. J. Alzheimer’s Dis. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Bliwise, D.L. Sleep in normal aging and dementia. Sleep 1993, 16, 40–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, P.N.; Vitaliano, P.P.; Vitiello, M.V.; Bokan, J.; Raskind, M.; Peskind, E.; Gerber, C. Sleep, EEG and mental function changes in senile dementia of the Alzheimer’s type. Neurobiol. Aging 1983, 3, 361–370. [Google Scholar] [CrossRef]
- Liguori, C.; Romigi, A.; Nuccetelli, M.; Zannino, S.; Sancesario, G.; Martorana, A.; Albanese, M.; Mercuri, N.B.; Izzi, F.; Bernardini, S.; et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014, 71, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.-E.S.; Lucey, B.P.; Holtzman, D.M. Sleep and Alzheimer disease pathology—A bidirectional relationship. Nat. Rev. Neurol. 2014, 10, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.-E.S.; McLeland, J.S.; Toedebusch, C.D.; Xiong, C.; Fagan, A.M.; Duntley, S.P.; Morris, J.C.; Holtzman, D.M. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013, 70, 587–593. [Google Scholar] [CrossRef]
- Yan, M.; Xu, X.; Huang, Z.-L.; Yao, M.; Urade, Y.; Qu, W. Selection of optimal epoch duration in assessment of rodent sleep-wake profiles. Sleep Biol. Rhythm. 2011, 9, 46–55. [Google Scholar] [CrossRef]
- Fulda, S.; Romanowski, C.P.; Becker, A.; Wetter, T.C.; Kimura, M.; Fenzl, T. Rapid eye movements during sleep in mice: High trait-like stability qualifies rapid eye movement density for characterization of phenotypic variation in sleep patterns of rodents. BMC Neurosci. 2011, 12, 110. [Google Scholar] [CrossRef] [Green Version]
- Kreuzer, M.; Polta, S.; Gapp, J.; Schuler, C.; Kochs, E.; Fenzl, T. Sleep scoring made easy—Semi-automated sleep analysis software and manual rescoring tools for basic sleep research in mice. MethodsX 2015, 2, 232–240. [Google Scholar] [CrossRef]
- Zhang, F.; Zhong, R.; Li, S.; Fu, Z.; Wang, R.; Wang, T.; Huang, Z.; Le, W. Alteration in sleep architecture and electroencephalogram as an early sign of Alzheimer’s disease preceding the disease pathology and cognitive decline. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2019, 15, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Kent, B.A.; Michalik, M.; Marchant, E.G.; Yau, K.W.; Feldman, H.H.; Mistlberger, R.E.; Nygaard, H.B. Delayed daily activity and reduced NREM slow-wave power in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Neurobiol. Aging 2019, 78, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Van Erum, J.; Van Dam, D.; Sheorajpanday, R.; De Deyn, P.P. Sleep architecture changes in the APP23 mouse model manifest at onset of cognitive deficits. Behav. Brain Res. 2019, 373, 112089. [Google Scholar] [CrossRef] [PubMed]
- Coben, L.A.; Danziger, W.L.; Berg, L. Frequency analysis of the resting awake EEG in mild senile dementia of Alzheimer type. Electroencephalogr. Clin. Neurophysiol. 1983, 55, 372–380. [Google Scholar] [CrossRef]
- Liddell, B.J.; Paul, R.H.; Arns, M.; Gordon, N.; Kukla, M.; Rowe, D.; Cooper, N.; Moyle, J.; Williams, L.M. Rates of decline distinguish Alzheimer’s disease and mild cognitive impairment relative to normal aging: Integrating cognition and brain function. J. Integr. Neurosci. 2007, 6, 141–174. [Google Scholar] [CrossRef]
- Mander, B.A.; Rao, V.; Lu, B.; Saletin, J.M.; Lindquist, J.R.; Ancoli-Israel, S.; Jagust, W.; Walker, M.P. Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nat. Neurosci. 2013, 16, 357–364. [Google Scholar] [CrossRef]
- Schreiter-Gasser, U.; Gasser, T.; Ziegler, P. Quantitative EEG analysis in early onset Alzheimer’s disease: A controlled study. Electroencephalogr. Clin. Neurophysiol. 1993, 86, 15–22. [Google Scholar] [CrossRef]
- Perlis, M.L.; Smith, M.T.; Andrews, P.J.; Orff, H.; Giles, D.E. Beta/Gamma EEG activity in patients with primary and secondary insomnia and good sleeper controls. Sleep 2001, 24, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Krystal, A.D.; Edinger, J.D.; Wohlgemuth, W.K.; Marsh, G.R. NREM sleep EEG frequency spectral correlates of sleep complaints in primary insomnia subtypes. Sleep 2002, 25, 630–640. [Google Scholar]
- Berger, S.; Schneider, G.; Kochs, E.F.; Jordan, D. Permutation Entropy: Too Complex a Measure for EEG Time Series? Entropy 2017, 19, 692. [Google Scholar] [CrossRef] [Green Version]
- Gómez, C.; Hornero, R. Entropy and complexity analyses in alzheimer’s disease: An MEG study. Open Biomed. Eng. J. 2010, 4, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantzidis, C.A.; Ladas, A.; Diamantoudi, M.D.; Semertzidou, A.; Grigoriadou, E.; Tsolaki, A.; Liapi, D.; Papadopoulou, A.; Kounti, F.; Vivas, A.B.; et al. What Are the Symbols of Alzheimer? A Permutation Entropy Based Symbolic Analysis for the Detection of Early Changes of the Electroencephalographic Complexity due to Mild Alzheimer. In Proceedings of the 2012 IEEE 12th International Conference onBioinformatics & Bioengineering BIBE, Larnaca, Cyprus, 11–13 November 2012; pp. 282–287. [Google Scholar]
- Czigler, B.; Csikós, D.; Hidasi, Z.; Gaál, Z.A.; Csibri, É.; Kiss, É.; Salacz, P.; Molnár, M. Quantitative EEG in early Alzheimer’s disease patients—Power spectrum and complexity features. Int. J. Psychophysiol. 2008, 68, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Geula, C.; Mesulam, M.-M. Cortical cholinergic fibers in aging and Alzheimer’s disease: A morphometric study. Neuroscience 1989, 33, 469–481. [Google Scholar] [CrossRef]
- Berntson, G.; Shafi, R.; Sarter, M. Specific contributions of the basal forebrain corticopetal cholinergic system to electroencephalographic activity and sleep/waking behaviour. Eur. J. Neurosci. 2002, 16, 2453–2461. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Arai, H.; Kobayashi, K.; Ichimiya, Y.; Kosaka, K.; Iizuka, R. Free amino acids in post-mortem cerebral cortices from patients with Alzheimer-type dementia. Neurosci. Res. 1985, 2, 486–490. [Google Scholar] [CrossRef]
- Kashani, A.; Lepicard, E.; Poirel, O.; Videau, C.; David, J.P.; Fallet-Bianco, C.; Simon, A.; Delacourte, A.; Giros, B.; Epelbaum, J.; et al. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol. Aging 2008, 29, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Masliah, E.; Alford, M.; DeTeresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef]
- Harkany, T.; Abraham, I.; Timmerman, W.; Laskay, G.; Toth, B.; Sasvari, M.; Konya, C.; Sebens, J.B.; Korf, J.; Nyakas, C.; et al. β-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur. J. Neurosci. 2000, 12, 2735–2745. [Google Scholar] [CrossRef]
- Fuchsberger, T.; Martinez-Bellver, S.; Giraldo, E.; Teruel-Marti, V.; Lloret, A.; Vina, J. Aβ Induces Excitotoxicity Mediated by APC/C-Cdh1 Depletion That Can Be Prevented by Glutaminase Inhibition Promoting Neuronal Survival. Sci. Rep. 2016, 6, 31158. [Google Scholar] [CrossRef] [Green Version]
- Rissman, R.A.; De Blas, A.L.; Armstrong, D.M. GABA(A) receptors in aging and Alzheimer’s disease. J. Neurochem. 2007, 103, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Rissman, R.A.; Mobley, W.C. Implications for treatment: GABAA receptors in aging, Down syndrome and Alzheimer’s disease. J. Neurochem. 2011, 117, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Liu, D.; Yin, J.; Qian, T.; Shrestha, S.; Ni, H. Glutamate-glutamine and GABA in brain of normal aged and patients with cognitive impairment. Eur. Radiol. 2017, 27, 2698–2705. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, D.M.; Sheffield, R.; Mishizen-Eberz, A.J.; Carter, T.L.; Rissman, R.A.; Mizukami, K.; Ikonomovic, M.D. Plasticity of glutamate and GABAA receptors in the hippocampus of patients with Alzheimer’s disease. Cell. Mol. Neurobiol. 2003, 23, 491–505. [Google Scholar] [CrossRef]
- Stoiljkovic, M.; Kelley, C.; Horvath, T.L.; Hajós, M. Neurophysiological signals as predictive translational biomarkers for Alzheimer’s disease treatment: Effects of donepezil on neuronal network oscillations in TgF344-AD rats. Alzheimers Res. Ther. 2018, 10, 105. [Google Scholar] [CrossRef]
- Speigel, I.; Bichler, E.K.; Garcia, P.S. The Influence of Regional Distribution and Pharmacologic Specificity of GABAAR Subtype Expression on Anesthesia and Emergence. Front. Syst. Neurosci. 2017, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Hanes, J.; Zilka, N.; Bartkova, M.; Caletkova, M.; Dobrota, D.; Novak, M. Rat tau proteome consists of six tau isoforms: Implication for animal models of human tauopathies. J. Neurochem. 2009, 108, 1167–1176. [Google Scholar] [CrossRef]
- Platt, B.; Drever, B.; Koss, D.; Stoppelkamp, S.; Jyoti, A.; Plano, A.; Utan, A.; Merrick, G.; Ryan, D.; Melis, V.; et al. Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PLoS ONE 2011, 6, e27068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jyoti, A.; Plano, A.; Riedel, G.; Platt, B. EEG, activity, and sleep architecture in a transgenic AβPPswe/PSEN1A246E Alzheimer’s disease mouse. J. Alzheimer’s Dis. 2010, 22, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, G.L.; Kuhar, M.J.; Bliwise, D.L.; Rye, D.B. Wake promoting effects of cocaine and amphetamine-regulated transcript (CART). Neuropeptides 2010, 44, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Safavynia, S.A.; Keating, G.; Spiegel, I.; Fidler, J.A.; Kreuzer, M.; Rye, D.B.; Jenkins, A.; García, P.S. Effects of γ-Aminobutyric Acid Type A Receptor Modulation by Flumazenil on Emergence from General Anesthesia. Anesthesiology 2016, 125, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenzl, T.; Romanowski, C.P.; Flachskamm, C.; Honsberg, K.; Boll, E.; Hoehne, A.; Kimura, M. Fully automated sleep deprivation in mice as a tool in sleep research. J. Neurosci. Methods 2007, 166, 229–235. [Google Scholar] [CrossRef]
- Louis, R.P.; Lee, J.; Stephenson, R. Design and validation of a computer-based sleep-scoring algorithm. J. Neurosci. Methods 2004, 133, 71–80. [Google Scholar] [CrossRef]
- Petit, D.; Gagnon, J.F.; Fantini, M.L.; Ferini-Strambi, L.; Montplaisir, J. Sleep and quantitative EEG in neurodegenerative disorders. J. Psychosom. Res. 2004, 56, 487–496. [Google Scholar] [CrossRef]
- Nardone, P. Entropy of Difference. arXiv 2014, arXiv:1411.0506. Available online: https://arxiv.org/abs/1411.0506 (accessed on 3 December 2020).
- Bandt, C.; Pompe, B. Permutation Entropy: A Natural Complexity Measure for Time Series. Phys. Rev. Lett. 2002, 88, 174102. [Google Scholar] [CrossRef]
- Jordan, D.; Stockmanns, G.; Kochs, E.F.; Pilge, S.; Schneider, G. Electroencephalographic Order Pattern Analysis for the Separation of Consciousness and Unconsciousness: An Analysis of Approximate Entropy, Permutation Entropy, Recurrence Rate, and Phase Coupling of Order Recurrence Plots. Anesthesiology 2008, 109, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Olofsen, E.; Sleigh, J.W.; Dahan, A. Permutation entropy of the electroencephalogram: A measure of anaesthetic drug effect. Br. J. Anaesth. 2008, 101, 810–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, A.J.; Henson, K.; Dorsey, S.G.; Frank, M.G. The truncated TrkB receptor influences mammalian sleep. Am. J. Physiol. Integr. Comp. Physiol. 2014, 308, R199–R207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Härtner, L.; Keil, T.W.M.; Kreuzer, M.; Fritz, E.M.; Wenning, G.K.; Stefanova, N.; Fenzl, T. Distinct Parameters in the EEG of the PLP α-SYN Mouse Model for Multiple System Atrophy Reinforce Face Validity. Front. Behav. Neurosci. 2017, 10, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentschke, H.; Stüttgen, M.C. Computation of measures of effect size for neuroscience data sets. Eur. J. Neurosci. 2011, 34, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, J.N. Receiver Operating Characteristic Curve in Diagnostic Test Assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, C.J. An effect size primer: A guide for clinicians and researchers. Prof. Psychol. Res. Pract. 2009, 40, 532. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AC vs. AD | WAKE | NREMS | REMS | |
| Inactive | Caudal | 0.59 [0.27–0.88] | 0.12 [0–0.35] | 0.75 [0.44–1] |
| Active | Caudal | 0.96 [0.81–1] | 0.14 [0–0.43] | 0.86 [0.57–1] |
| WAKE vs. NREMS | AC | AD | ||
| Inactive | Caudal | 1 | 0.57 [0.24–0.88] | |
| Active | Caudal | 1 | 0.65 [0.33–0.96] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreuzer, M.; Keating, G.L.; Fenzl, T.; Härtner, L.; Sinon, C.G.; Hajjar, I.; Ciavatta, V.; Rye, D.B.; García, P.S. Sleep/Wake Behavior and EEG Signatures of the TgF344-AD Rat Model at the Prodromal Stage. Int. J. Mol. Sci. 2020, 21, 9290. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239290
Kreuzer M, Keating GL, Fenzl T, Härtner L, Sinon CG, Hajjar I, Ciavatta V, Rye DB, García PS. Sleep/Wake Behavior and EEG Signatures of the TgF344-AD Rat Model at the Prodromal Stage. International Journal of Molecular Sciences. 2020; 21(23):9290. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239290
Chicago/Turabian StyleKreuzer, Matthias, Glenda L. Keating, Thomas Fenzl, Lorenz Härtner, Christopher G. Sinon, Ihab Hajjar, Vincent Ciavatta, David B. Rye, and Paul S. García. 2020. "Sleep/Wake Behavior and EEG Signatures of the TgF344-AD Rat Model at the Prodromal Stage" International Journal of Molecular Sciences 21, no. 23: 9290. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239290