Effect of Adrenergic Agonists on High-Fat Diet-Induced Hepatic Steatosis in Mice

 ,
,
Abstract
:1. Introduction
2. Results
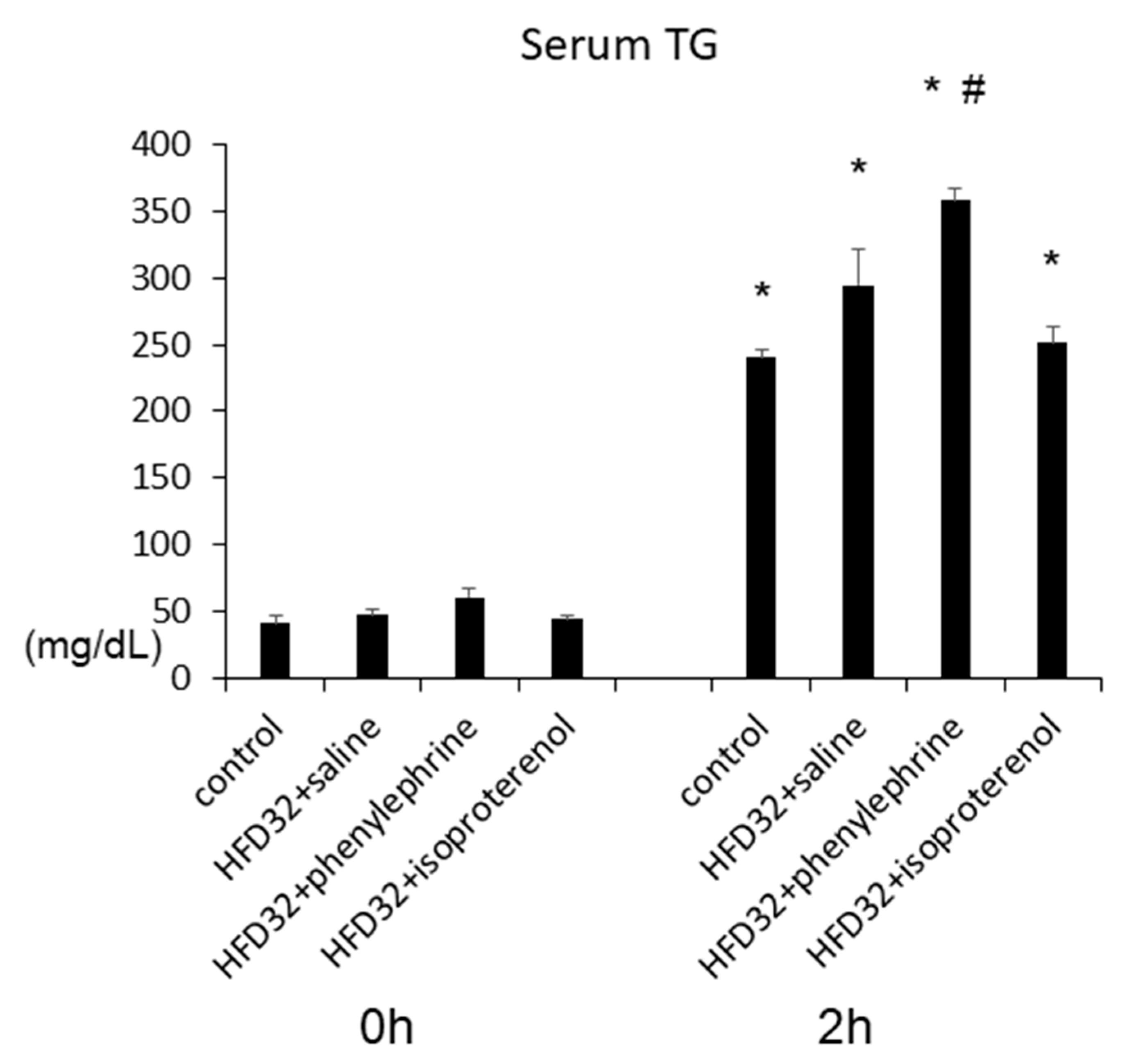
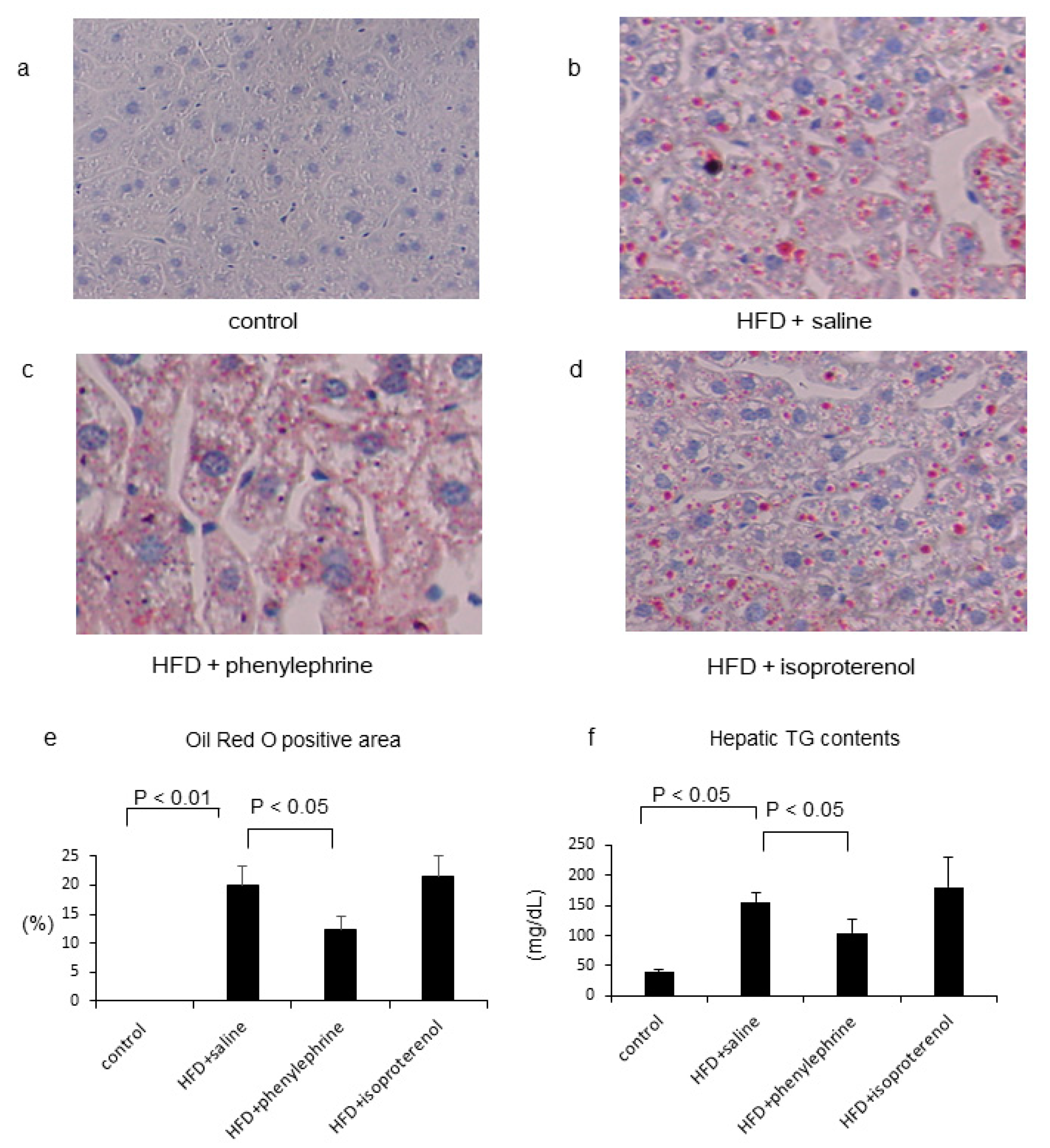
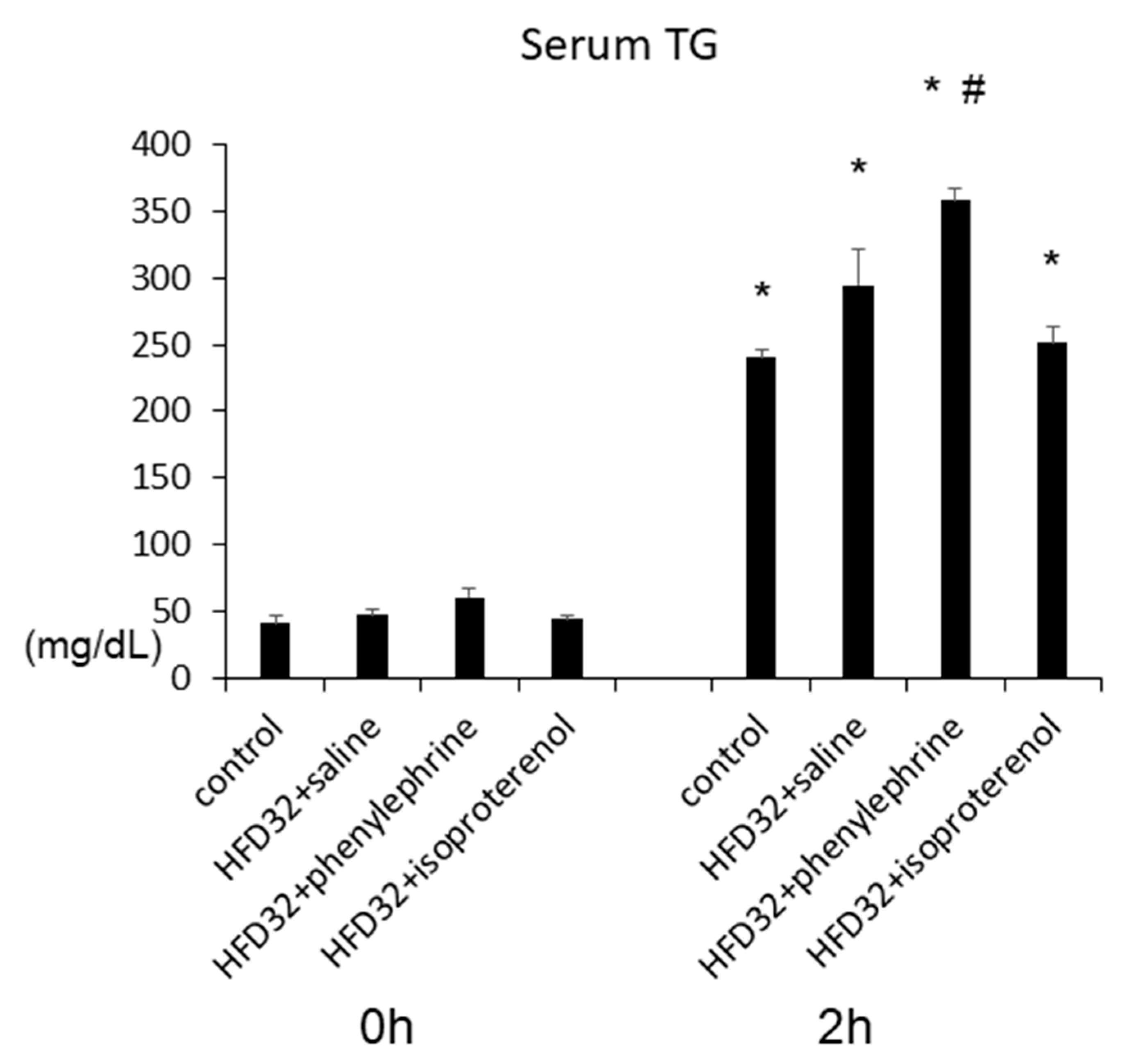
2.1. Effect of Phenylephrine and Isoproterenol on Hepatic Steatosis and VLDL–TG Secretion
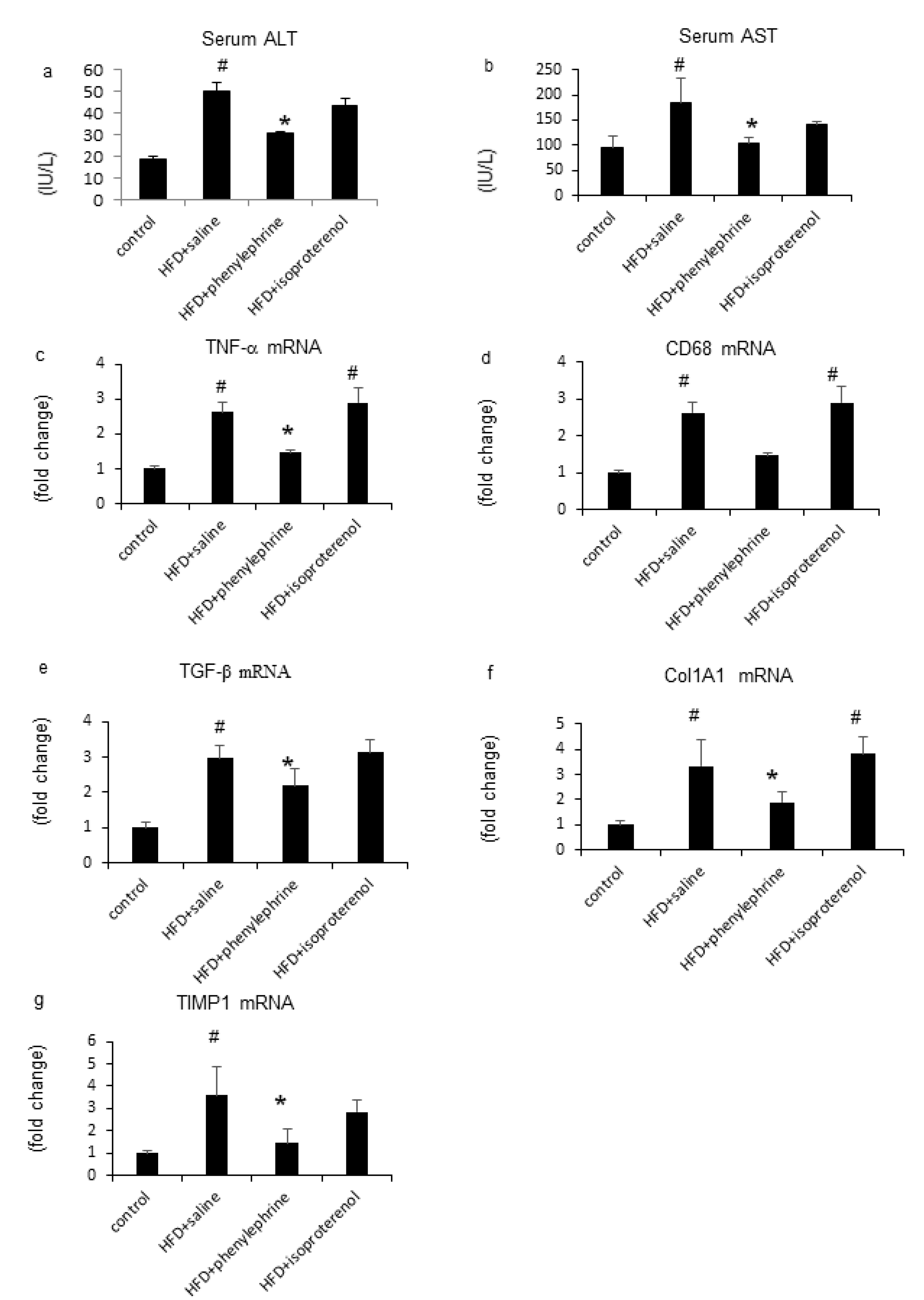
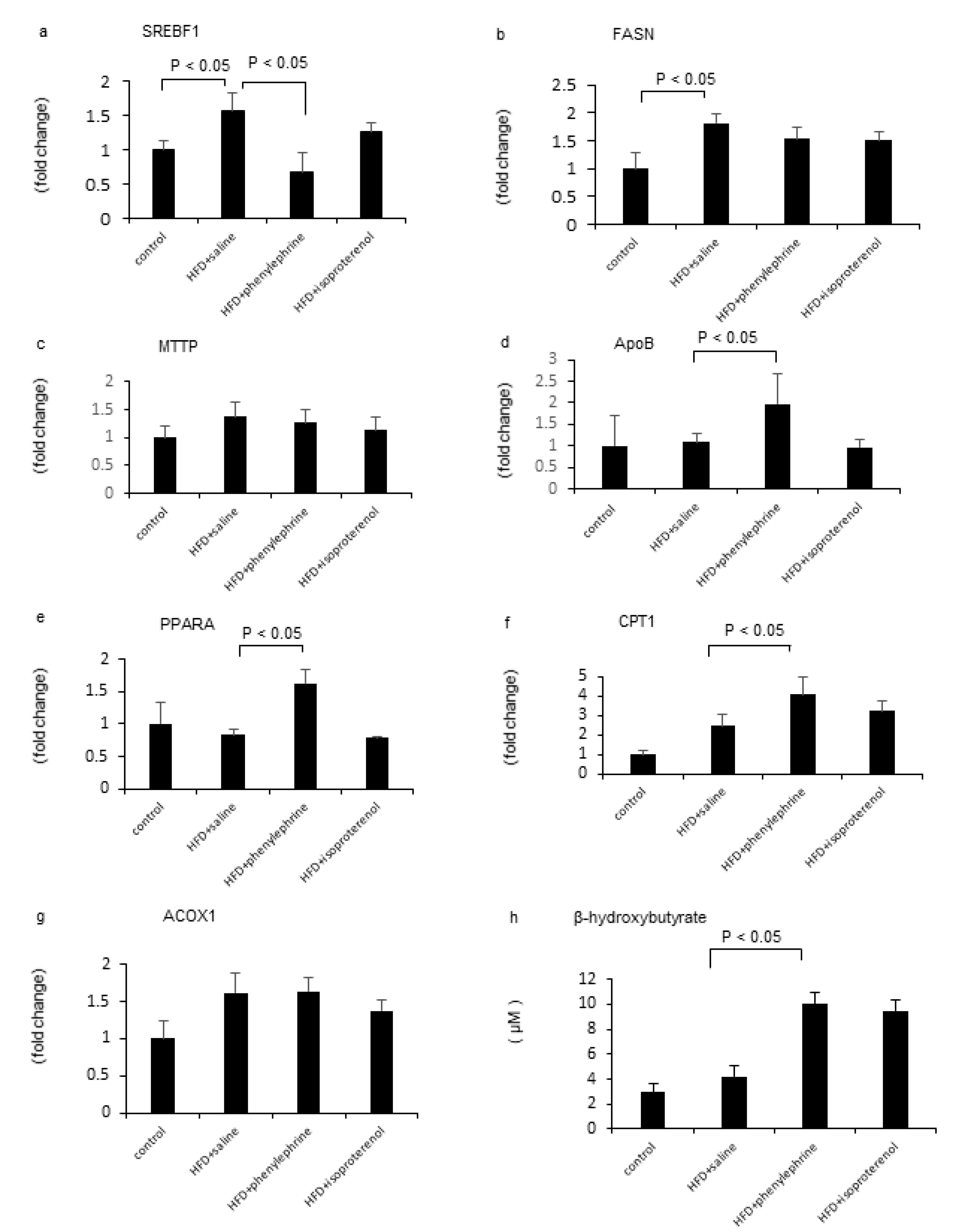
2.2. Effect of Phenylephrine or Isoproterenol on Hepatic Fat Metabolism-Related Genes
2.3. Effect of Phenylephrine and Isoproterenol on VLDL–TG Secretion
2.4. Hepatic Inflammatory Responses by Phenylephrine and Isoproterenol
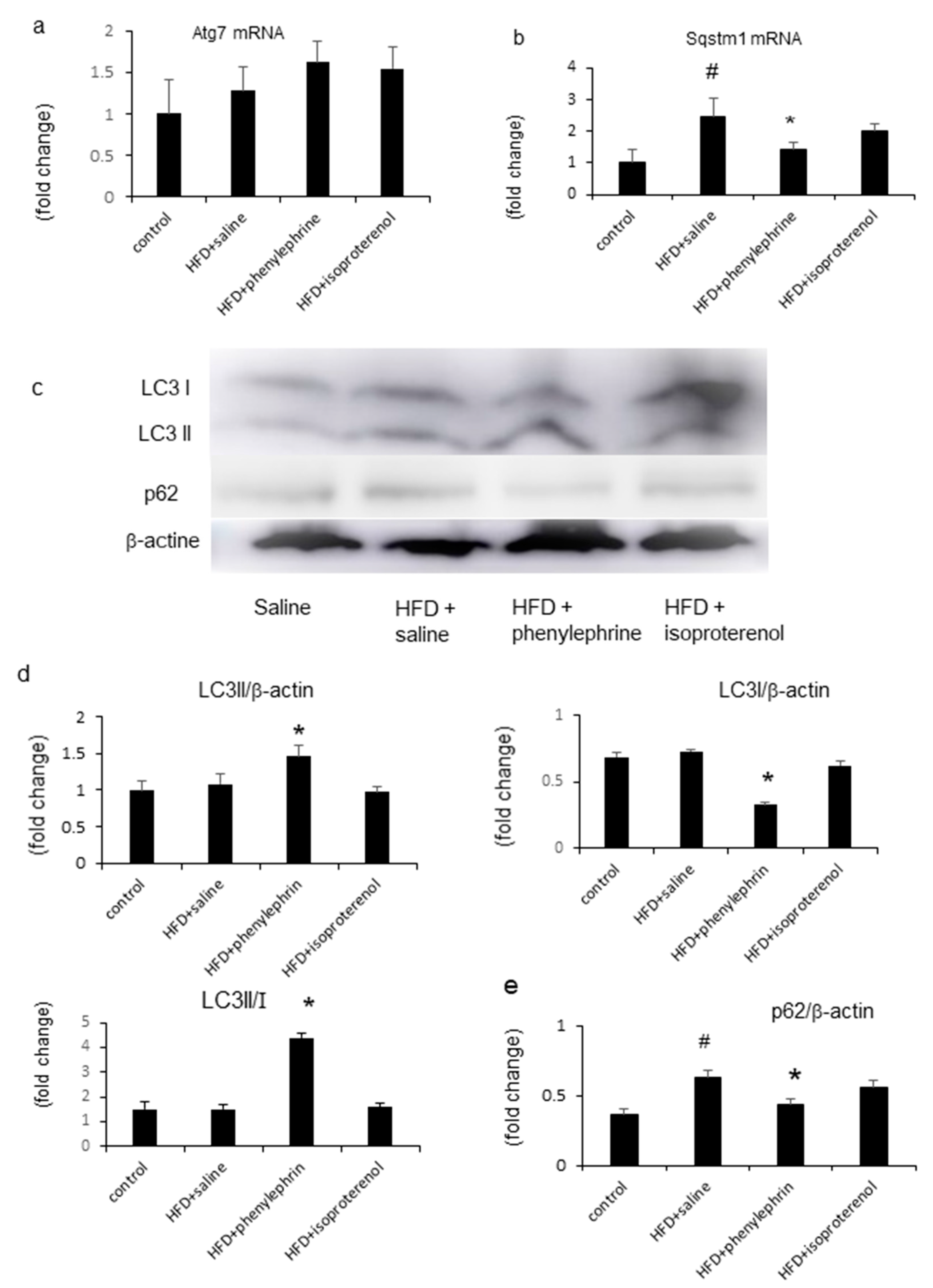
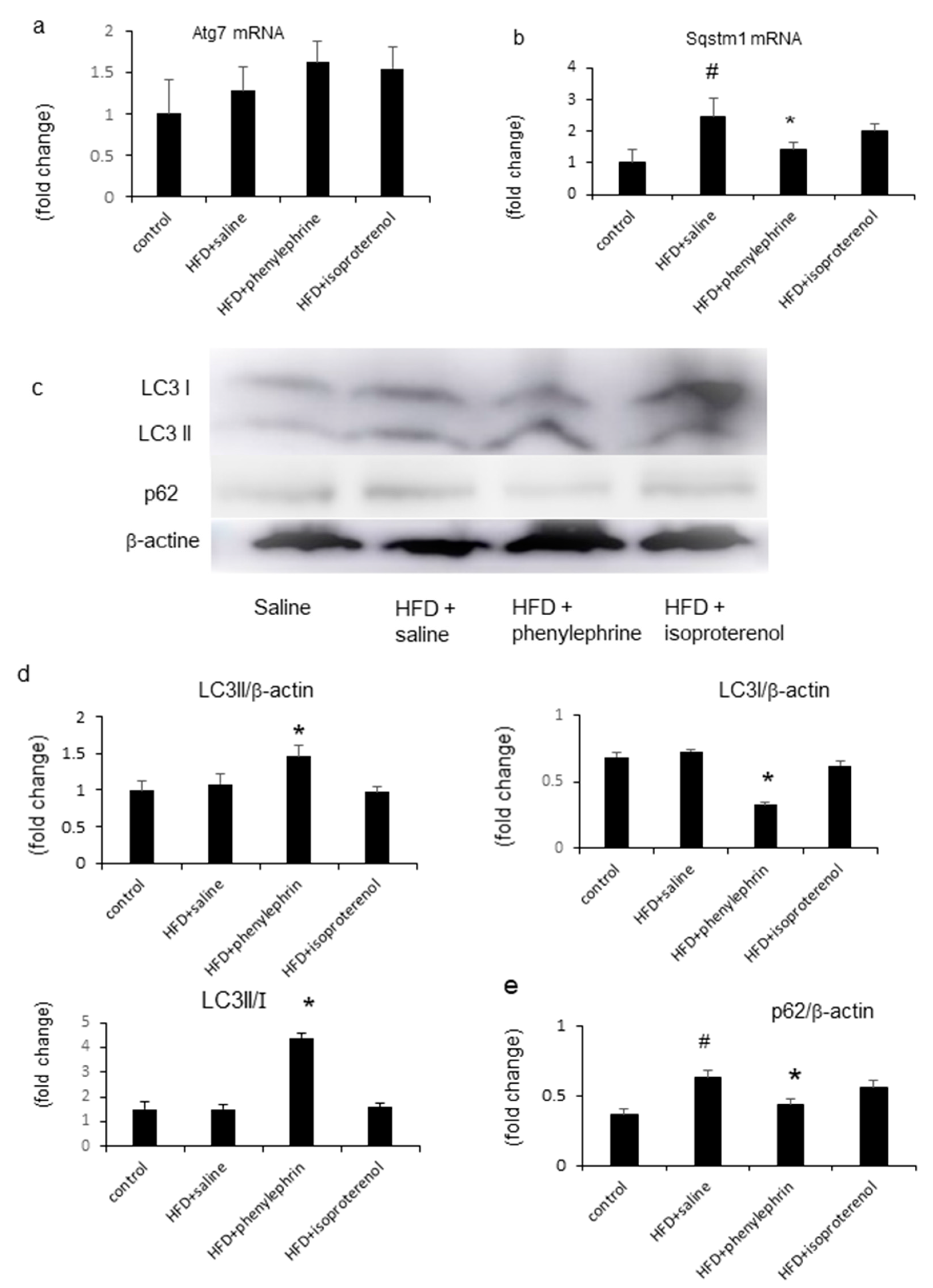
2.5. Enhancement of Autophagy by Phenylephrine and Isoproterenol Via Regulation of Autophagy-Related 7 (Atg7) and Sequestosome-1 (SQSTM1)
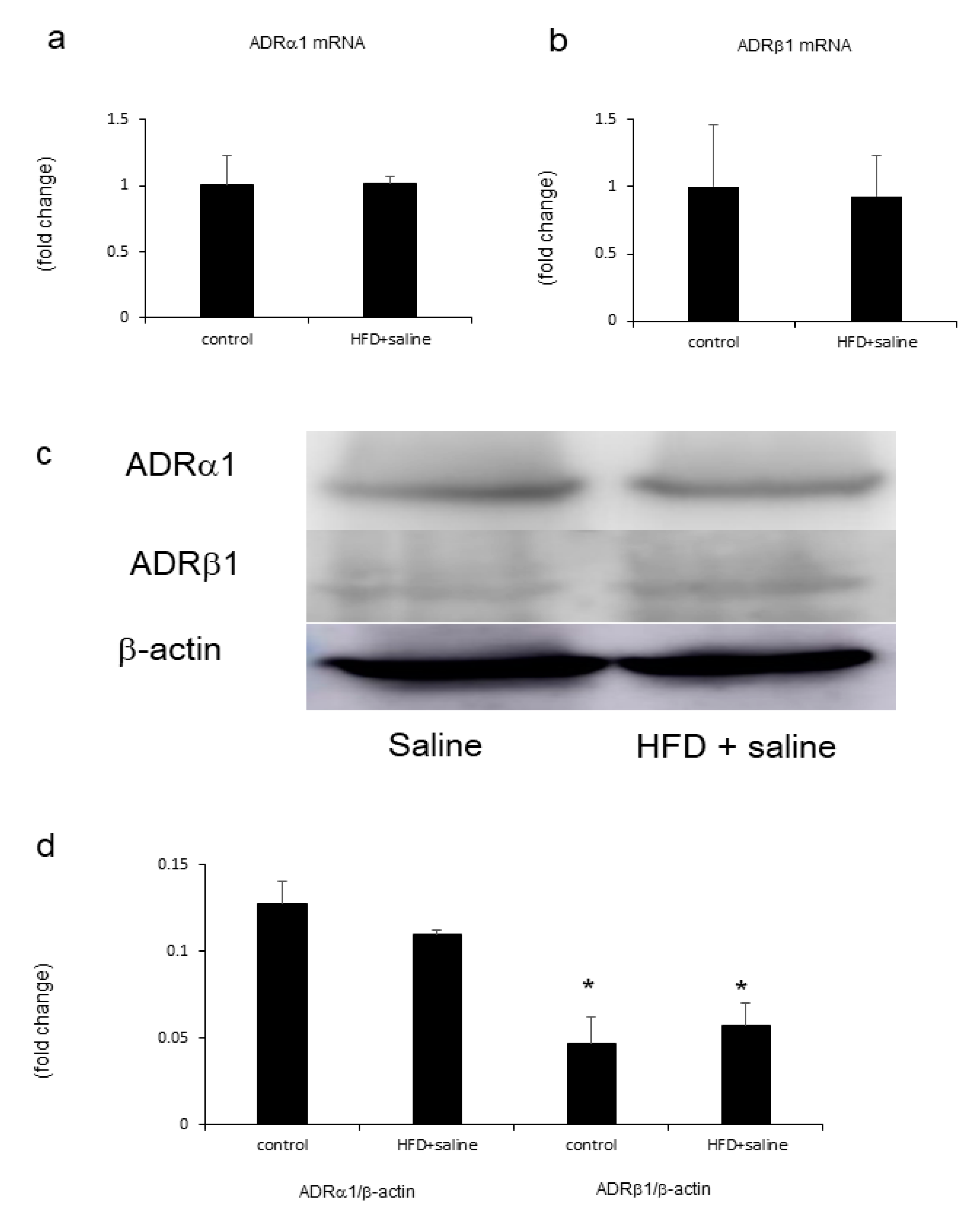
2.6. Hepatic Adrenergic Receptors mRNA and Proteins Mediated by Phenylephrine and Isoproterenol
3. Discussion
4. Materials and Methods
4.1. Substances and Treatments
4.2. Animal Model and Experimental Design
4.3. Biochemical Characterization of Serum and Tissue
4.4. Histopathological Examination
4.5. Real-Time Polymerase Chain Reaction (PCR)
4.6. Western Blotting
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The histological course of nonalcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Schlaich, M.P.; Straznicky, N.; Lambert, E.; Lambert, G. Metabolic syndrome: A sympathetic disease? Lancet Diabetes Endocrinol. 2015, 3, 148–157. [Google Scholar] [CrossRef]
- Geerling, J.J.; Boon, M.R.; Kooijman, S.; Parlevliet, E.T.; Havekes, L.M.; Romijn, J.A.; Meurs, I.M.; Rensen, P.C.N. Sympathetic nervous system control of triglyceride metabolism: Novel concepts derived from recent studies. J. Lipid Res. 2013, 55, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Bruinstroop, E.; Pei, L.; Ackermans, M.T.; Foppen, E.; Borgers, A.J.; Kwakkel, J.; Alkemade, A.; Fliers, E.; Kalsbeek, A. Hypothalamic Neuropeptide Y (NPY) Controls Hepatic VLDL-Triglyceride Secretion in Rats via the Sympathetic Nervous System. Diabetes 2012, 61, 1043–1050. [Google Scholar] [CrossRef] [Green Version]
- Sigala, B.; McKee, C.; Soeda, J.; Pazienza, V.; Morgan, M.; Lin, C.-I.; Selden, C.; Borght, S.V.; Mazzoccoli, G.; Roskams, T.; et al. Sympathetic Nervous System Catecholamines and Neuropeptide Y Neurotransmitters Are Upregulated in Human NAFLD and Modulate the Fibrogenic Function of Hepatic Stellate Cells. PLoS ONE 2013, 8, e72928. [Google Scholar] [CrossRef]
- Oben, J.A.; Roskams, T.; Yang, S.; Lin, H.; Sinelli, N.; Li, Z.; Torbenson, M.; Huang, J.; Guarino, P.; Kafrouni, M.; et al. Sympathetic nervous system inhibition increases hepatic progenitors and reduces liver injury. Hepatology 2003, 38, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Tietge, U.J.; Bakillah, A.; Maugeais, C.; Tsukamoto, K.; Hussain, M.; Rader, D.J. Hepatic overexpression of microsomal triglyceride transfer protein (MTP) results in increased in vivo secretion of VLDL triglycerides and apolipoprotein B. J. Lipid Res. 1999, 40, 2134–2139. [Google Scholar] [PubMed]
- McKee, C.; Soeda, J.; Asilmaz, E.; Sigalla, B.; Morgan, M.; Sinelli, N.; Roskams, T.; Oben, J.A. Propranolol, a β-adrenoceptor antagonist, worsens liver injury in a model of non-alcoholic steatohepatitis. Biochem. Biophys. Res. Commun. 2013, 437, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinella, M.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Fujimoto, Y.; Fujitake, M.; Endo, H.; Takahashi, H.; Inamori, M.; Kobayashi, N.; et al. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 2009, 50, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Charlton, M.; Sreekumar, R.; Rasmussen, D.; Lindor, K.; Nair, K.S. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology 2002, 35, 898–904. [Google Scholar] [CrossRef]
- Osumi, T.; Hashimoto, T.; Ui, N. Purification and properties of acyl-CoA oxidase from rat liver. J. Biochem. 1980, 87, 1735–1746. [Google Scholar] [CrossRef]
- Brandes, R.; Kaikaus, R.M.; Lysenko, N.; Ockner, R.K.; Bass, N.M. Induction of fatty acid binding protein by peroxisome proliferators in primary hepatocyte cultures and its relationship to the induction of peroxisomal beta-oxidation. Biochim. Biophys. Acta 1990, 1034, 53–61. [Google Scholar] [CrossRef]
- Rao, M.S.; Reddy, J.K. Peroxisomal beta-oxidation and steatohepatitis. Semin. Liver Dis. 2001, 21, 43–55. [Google Scholar] [CrossRef]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Barbosa-Da-Silva, S.; Souza-Mello, V.; Magliano, D.C.; Marinho, T.D.S.; Águila, M.B.; Mandarim-De-Lacerda, C.A. Singular effects of PPAR agonists on nonalcoholic fatty liver disease of diet-induced obese mice. Life Sci. 2015, 127, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.S.; Kim, J.H.; Jo, N.Y.; Choi, K.M.; Baik, S.H.; Park, J.-J.; Kim, J.S.; Byun, K.S.; Bak, Y.T.; Lee, C.H.; et al. PPAR agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. J. Gastroenterol. Hepatol. 2006, 23, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.M.; Shu, Z.J.; Zhu, B.; Lu, Z.; Ikeno, Y.; Barnes, J.L.; Yeh, C.K.; Zhang, B.X.; Katz, M.S.; Kamat, A. Role of beta-adrenergic receptors in regulation of hepatic fat accumulation during aging. J. Endocrinol. 2012, 213, 251–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gettys, T.W.; Rohlfs, E.M.; Prpic, V.; Daniel, K.W.; Taylor, I.L.; Collins, S. Age-dependent changes in beta-adrenergic receptor subtypes and adenylyl cyclase activation in adipocytes from Fischer 344 rats. Endocrinology 1995, 136, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Scheglmann, D.; Dettmer, D. Alpha-1 adrenergic receptor number and receptor density in isolated hepatocytes from foetal, juvenile and adult rats. Cell Signal. 1992, 4, 287–291. [Google Scholar] [CrossRef]
- Yang, L.; Roh, Y.S.; Song, J.; Zhang, B.; Liu, C.; Loomba, R.; Seki, E. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 2014, 59, 483–495. [Google Scholar] [CrossRef] [Green Version]
- McCuskey, R.S.; Ito, Y.; Robertson, G.R.; McCuskey, M.K.; Perry, M.; Farrell, G. Hepatic microvascular dysfunction during evolution of dietary steatohepatitis in mice. Hepatology 2004, 40, 386–393. [Google Scholar] [CrossRef]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Nakano, S.; Katsuno, K.; Isaji, M.; Nagasawa, T.; Buehrer, B.M.; Walker, S.; Wilkison, W.O.; Cheatham, B. Remogliflozin Etabonate Improves Fatty Liver Disease in Diet-Induced Obese Male Mice. J. Clin. Exp. Hepatol. 2015, 5, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Tejero-Taldo, M.I.; Gursoy, E.; Zhao, T.C.; Kukreja, R.C. Alpha-adrenergic receptor stimulation produces late preconditioning through inducible nitric oxide synthase in mouse heart. J. Mol. Cell Cardiol. 2002, 34, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Nakade, Y.; Sakamoto, K.; Yamauchi, T.; Inoue, T.; Kobayashi, Y.; Yamamoto, T.; Ishii, N.; Ohashi, T.; Sumida, Y.; Ito, K.; et al. Conophylline inhibits non-alcoholic steatohepatitis in mice. PLoS ONE 2017, 12, e0178436. [Google Scholar] [CrossRef] [PubMed]
- Vanpatten, S.; Karkanias, G.B.; Rossetti, L.; Cohen, D.E. Intracerebroventricular leptin regulates hepatic cholesterol metabolism. Biochem. J. 2004, 379, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Saito, R.; Hamano, K.; Nagasawa, M.; Aoki, F.; Takei, I.; Umezawa, K.; Kuwano, H.; Kojima, I. Conophylline suppresses hepatic stellate cells and attenuates thioacetamide-induced liver fibrosis in rats. Liver Int. 2014, 34, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Nakade, Y.; Yamauchi, T.; Makino, Y.; Yokohama, S.; Okada, M.; Aso, K.; Kanamori, H.; Ohashi, T.; Sato, K.; et al. Angiotensin II type 1 receptor antagonist prevents hepatic carcinoma in rats with nonalcoholic steatohepatitis. J. Gastroenterol. 2013, 48, 491–503. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | n | Body Weight Gain (g) | Subepididymal Fat Weight (g) | FBG (mg/dL) | IRI (µg/L) |
|---|---|---|---|---|---|
| Control | 5 | 2.23 ± 0.45 | 0.23 ± 0.03 | 178 ± 25 | 1.14 ± 0.34 |
| HFD | 6 | 7.69 ± 0.21 a | 0.31 ± 0.03 a | 215 ± 18 | 1.27 ± 0.36 |
| HFD + phenylephrine | 6 | 6.08 ± 0.93 | 0.37 ± 0.07 | 239 ± 6.5 | 1.19 ± 0.43 |
| HFD + isoproterenol | 7 | 8.4 ± 0.37 | 0.13 ± 0.03 b | 242 ± 11 | 0.45 ± 0.09 a |
| Group | n | Serum TG (mg/dL) | Serum FFA (mEq/L) | Hepatic FFA (mEq/L) | Serum Cholesterol (mg/dL) |
|---|---|---|---|---|---|
| Control | 5 | 30.1 ± 2.4 | 0.62 ± 0.05 | 0.23 ± 0.02 | 82.1 ± 4.8 |
| HFD | 6 | 40.2 ± 2.8 | 0.60 ± 0.03 | 1.13 ± 0.11 a | 156 ± 28 |
| HFD + phenylephrine | 6 | 53.4 ± 6.8 b | 0.98 ± 0.09 b | 0.60 ± 0.07 b | 140 ± 6.3 |
| HFD + isoproterenol | 7 | 37.8 ± 4.5 | 0.56 ± 0.03 | 0.90 ± 0.12 | 150 ± 3.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakade, Y.; Kitano, R.; Yamauchi, T.; Kimoto, S.; Sakamoto, K.; Inoue, T.; Kobayashi, Y.; Ohashi, T.; Sumida, Y.; Ito, K.; et al. Effect of Adrenergic Agonists on High-Fat Diet-Induced Hepatic Steatosis in Mice. Int. J. Mol. Sci. 2020, 21, 9392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249392
Nakade Y, Kitano R, Yamauchi T, Kimoto S, Sakamoto K, Inoue T, Kobayashi Y, Ohashi T, Sumida Y, Ito K, et al. Effect of Adrenergic Agonists on High-Fat Diet-Induced Hepatic Steatosis in Mice. International Journal of Molecular Sciences. 2020; 21(24):9392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249392
Chicago/Turabian StyleNakade, Yukiomi, Rena Kitano, Taeko Yamauchi, Satoshi Kimoto, Kazumasa Sakamoto, Tadahisa Inoue, Yuji Kobayashi, Tomohiko Ohashi, Yoshio Sumida, Kiyoaki Ito, and et al. 2020. "Effect of Adrenergic Agonists on High-Fat Diet-Induced Hepatic Steatosis in Mice" International Journal of Molecular Sciences 21, no. 24: 9392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249392