Molecular Features and Clinical Management of Hereditary Gynecological Cancers



1
Center for Medical Genetics, Keio Cancer Center, Keio University School of Medicine, 35 Shinanomachi, Shinjuku-ku, Tokyo 160-8582, Japan
2
Department of Clinical Genomic Medicine, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, 2-5-1 Shikata-cho, Kita-ku, Okayama 700-8558, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(24), 9504; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249504
Submission received: 19 November 2020
/
Revised: 8 December 2020
/
Accepted: 11 December 2020
/
Published: 14 December 2020
(This article belongs to the Special Issue Gynecologic Oncology: From Molecular Mechanisms to Targeted Therapies 2.0)
Abstract
:Hereditary gynecological cancers are caused by several inherited genes. Tumors that arise in the female reproductive system, such as ovaries and the uterus, overlap with hereditary cancers. Several hereditary cancer-related genes are important because they might lead to therapeutic targets. Treatment of hereditary cancers should be updated in line with the advent of various new methods of evaluation. Next-generation sequencing has led to rapid, economical genetic analyses that have prompted a concomitant and significant paradigm shift with respect to hereditary cancers. Molecular tumor profiling is an epochal method for determining therapeutic targets. Clinical treatment strategies are now being designed based on biomarkers based on tumor profiling. Furthermore, the National Comprehensive Cancer Network (NCCN) guidelines significantly changed the genetic testing process in 2020 to initially consider multi-gene panel (MGP) evaluation. Here, we reviewed the molecular features and clinical management of hereditary gynecological malignancies, such as hereditary breast and ovarian cancer (HBOC), and Lynch, Li–Fraumeni, Cowden, and Peutz–Jeghers syndromes. We also reviewed cancer-susceptible genes revealed by MGP tests.
1. Introduction
Cancers that accumulate in families have been regarded as familial cancers. However, recent advancements in medical research have led to the re-definition of some familial cancers that are closely associated with genetic factors as hereditary cancers. Many of these arise due to pathogenic germline variants of the causative genes. The two-hit theory was presented in 1971 as a carcinogenic mechanism of autosomal-dominant inherited retinoblastoma [1]. This theory states that a loss-of-function mutation in one copy of a tumor-suppressive, predisposing gene in the germline (first hit), is followed by a somatic mutation (second hit) in another copy of the gene.
The typical clinical features of hereditary cancers include intrafamily accumulation of specific cancers, juvenile onset, and simultaneous/metachronous multiple cancers such as those with bilateral onset. Most hereditary cancers have autosomal dominant inheritance, with a 50% probability that the pathogenic variant will be passed down to the next generation, regardless of gender. When an individual has a specific genotype, the probability that the trait will be expressed in the body is called penetrance, and this depends on the causative gene. The cumulative risk of hereditary cancers is rarely 100%, and cancers notably do not develop in all individuals harboring the pathogenic variant.
Medical geneticists and genetic counselors can provide support for patients and families as needed if a hereditary cancer is suspected [2,3,4,5]. After explaining the advantages and disadvantages of genetic diagnosis, and obtaining written, informed consent, patients can undergo genetic tests. When causative pathogenic are confirmed by the results of such tests, patients are considered as carriers of pathogenic variants. However, not all genetic tests result in a diagnosis, which might be due to methodological limitations of tests, the involvement of other causative genes, unknown genes, environmental factors, or the patient is negative for a pathological variant. However, even if genetic tests do not reveal pathogenic variants of a gene, the possibility of a hereditary cancer cannot be ruled out, and individuals should be evaluated considering their medical and family history [6].
Precision medicine has recently been advocated, and personalized treatment strategies based on tumor profiling have attracted attention. Vertical cancer treatment of a specific organ has become possible across organs in precision medicine. Molecular tumor profiling is an epochal means of identifying therapeutic targets, and the design of clinical treatment strategies according to biomarkers defined by tumor profiling is becoming a major trend. This review summarizes the characteristics of various genetic tests, current knowledge of gynecological hereditary cancers, and their characteristics and clinical management.
2. Hereditary Gynecological Cancers
Cancer can develop in multiple organs and the cause can be a wide variety of hereditary cancer-related genes. Cancers that develop in the ovaries and uterus often overlap with hereditary cancers. Gynecologists play a crucial role in the diagnosis, treatment, and subsequent management of hereditary cancers. This section outlines the typical gynecological hereditary cancers shown in Table 1.
2.1. Hereditary Breast and Ovarian Cancer Syndrome
Hereditary breast and ovarian cancer syndrome (HBOC) is diagnosed when a pathogenic variant is identified in the BRCA1 or BRCA2 (BRCA1/2) genes, which are involved in DNA damage repair. This syndrome tends to develop in younger persons, and cancers of the breast, ovaries, fallopian tubes, and peritoneum typically occur within families [7,8,9]. The syndrome includes high-grade serous ovarian, male breast, and bilateral breast cancers.
Kuchenbaecker et al. reported that the cumulative risk of developing breast and ovarian cancers by the age 80 years is 72% and 44% and 69% and 17% for carriers of the BRCA1 and BRCA2 pathogenic variants, respectively [7]. Notably, 10–15% of all ovarian cancers are associated with BRCA1/2 pathogenic variants. Hirasawa et al. reported that 8.3% and 3.5% of all patients with ovarian cancer in Japan had BRCA1 and BRCA2 pathogenic variants, respectively [8]. This indicated that patients with HBOC are being treated for sporadic ovarian cancer. Clinically assessing genetic risk of ovarian cancer is important to ensure the choice of appropriate treatment. Ovarian cancer in the context of HBOC is characterized by a high proportion of serous carcinoma and patients with advanced stage III or higher [9,10,11,12].
Carriers of the BRCA1/2 pathogenic variant should be managed by risk-reducing surgery, and by surveillance screening [6] for hereditary cancers at an early stage. Guidelines in various countries recommend salpingo-oophorectomy (RRSO) which reduce risk overall [6,13,14,15,16], and of developing ovarian and fallopian tube cancer, and improves prognosis. Rebbeck et al. reported that RRSO for BRCA1/2 pathogenic variant carriers reduces the risk of developing ovarian and fallopian tube cancer by 79% (HR, 0.21, 95% CI, 0.12–0.39) [17]. However, gynecological surveillance is required because the risk of developing peritoneal cancer persists even after RRSO. Harmsen et al. reported a 3.5% incidence of peritoneal cancer 10 years after RRSO for BRCA1/2 pathogenic variant carriers [18]. If RRSO is not selected, gynecological surveillance by transvaginal ultrasonography and serum tumor marker CA125 are options; but these have not yet been validated [19].
Poly ADP ribose polymerase (PARP) inhibitors comprise a promising therapeutic strategy for HBOC-related cancers [20]. Both BRCA1/2 and PARP1 are involved in DNA damage repair. If the BRCA1/2 genes are dysfunctional, DNA repair then depends on PARP1. A PARP inhibitor inhibits the action of PARP1 in HBOC-related cancers in which the BRCA1/2 gene is dysfunctional, and specifically leads cancer cells to apoptosis. This mechanism (synthetic lethality) is gaining attention. With the introduction of PARP inhibitors for treating breast and ovarian cancers, the presence or absence of the germline BRCA1/2 pathogenic variant is being determined for appropriate drug selection [21,22,23,24,25,26,27,28,29,30,31,32,33,34]. The BRCA1 and BRCA2 proteins repair DNA double-strand breaks via the homologous recombination repair (HRR) pathway. A homologous recombination deficiency (HRD) is a target for PARP inhibitors, and HRD status now serves as a biomarker for indicating the appropriate time to apply these agents [25,35,36,37]. Various guidelines recommend BRCA1/2 genetic tests of all ovarian cancers [6,16,38]. Appropriate genetic care should be available to unaffected relatives of a family member who tested positive for BRCA1/2.
2.2. Lynch Syndrome
Lynch syndrome is a hereditary cancer syndrome caused by germline pathogenic variants in DNA mismatch repair genes (MMR) such as MLH1, MSH2, MSH6, PMS2, and EPCAM. Families with Lynch syndrome have a high lifetime risk of developing colorectal, endometrial, ovarian, small intestine, ureteral, and renal pelvis cancer, and tend to develop cancer at a young age. The risk of developing cancer in Lynch syndrome differs depending on the causative gene [39].
Lynch syndrome accounts for ~3% of all colorectal cancers and is one of the most common hereditary cancers [40]. The lifetime risk of developing endometrial cancer is comparable to that of colorectal cancer in women with Lynch syndrome. The average age of onset of endometrial cancer in women with Lynch syndrome is 47–55 years, which is younger than in the general population [41]. Therefore, endometrial cancer in a woman with Lynch syndrome becomes a “sentinel cancer,” which is the first diagnosed cancer in that individual [42]. After treatment for endometrial cancer, measures against other cancers such as colorectal cancer might be needed and members of a family in which one person has Lynch syndrome, should also be appropriately surveilled. Although surveillance for endometrial cancer in Lynch syndrome is not supported by evidence, the diagnostic utility of endometrial histology is high, and implementation every 1–2 years is considered [38,43]. In addition, endometrial cancer develops at a younger age in patients with Lynch syndrome, and the prognosis is good. The cumulative lifetime incidence of ovarian cancer in women with Lynch syndrome is 8–20% [44,45], but few reports have described ovarian cancer related to Lynch syndrome. Watson et al. characterized ovarian cancer in Lynch syndrome as follows: prevalent in various histological types, early stage (61% in stage I), average age at diagnosis is 43 years, and comorbid with endometrial cancer in 22% of patients [46,47].
Cancer cells with impaired function caused by two hits on MMR genes characteristically have abnormal replication of repetitive sequences, namely microsatellite instability (MSI) [48]. Tumors with MSI in two or more microsatellite regions are MSI-high (MSI-H), with only one MSI-low (MSI-L) region, and tumors without MSI are classified as microsatellite stable (MSS). Lynch syndrome has been identified in 16.3% of patients with MSI-H tumors [48]. That study also found that most patients with Lynch syndrome had MSI-H/I, and that 36% had MSS tumors. Indeed, among these patients with Lynch syndrome, 71.2% and 78.4% of germline pathogenic variants were detected in MLH1, MSH2, or EPCAM genes in MSI-H/I tumors, but in the lower-penetrance PMS2 or MSH6 genes in MSS tumors. Not only the risk of developing cancer, but also the frequency of MSI-H notably differs among MMR genes in Lynch syndrome. If Lynch syndrome is suspected, primary screening should determine whether it meets the Amsterdam II [49], or revised Bethesda criteria [50]. If these criteria are met, secondary screening for MSI or immunohistochemical tests should proceed to confirm MSI-H or the loss of protein expression by MMR genes [38]. Lynch syndrome is diagnosed when subsequent genetic testing reveals a pathogenic germline variant in MMR genes. Many genes involved in carcinogenesis contain microsatellite regions, and the accumulation of abnormalities in these regions results in MSI-H. Immune checkpoint inhibitors (ICI) are particularly effective against tumors with MSI-H and should be effective in Lynch syndrome [51,52].
2.3. Li–Fraumeni Syndrome
Li–Fraumeni syndrome is an autosomal-dominant hereditary cancer of juvenile onset caused by the TP53 gene [53]. Soft tissue sarcoma, osteosarcoma, adrenocortical carcinoma, brain tumors, as well as premenopausal breast, colorectal, and gastric cancer can develop due to Li–Fraumeni syndrome, and the lifetime risk for all cancers almost 100% [54,55,56,57]. Among them, soft tissue sarcoma, brain tumors, and adrenocortical carcinoma develop in childhood; therefore, intervention for family members is important. According to a French study, 14% of patients harbor de novo TP53 mutations [58]. Even if they do not have a familial history of cancer, Li–Fraumeni syndrome should be considered in specific pediatric patients with adrenocortical carcinomas or choroid plexus tumors, women with breast cancers before reaching the age of 31 years, or multiple primary cancers within the Li–Fraumeni spectrum.
Breast cancer in Li–Fraumeni syndrome affects 54% of the women by the age of 70 years [54,57]. Among women with breast cancer aged < 30 years without a family history of breast cancer, 3–8% have a TP53 pathogenic germline variant [58,59,60,61], suggesting that Li–Fraumeni syndrome is not uncommon. The average age of onset of breast cancer in Li–Fraumeni syndrome is the early 30s, and surveillance using breast MRI with contrast is recommended from the age of 20 years [6]. In addition, exposure to radiation including therapy might cause secondary cancer in Li–Fraumeni syndrome [6,53]. Total mastectomy is recommended to avoid radiation therapy for breast cancer patients with Li–Fraumeni syndrome [62], and should be considered when deciding surgical procedures. The NCCN guidelines for Li–Fraumeni syndrome describe that the TP53 pathogenic variant does not increase the risk of ovarian cancer [6], but ovarian cancer has developed in patients with Li–Fraumeni syndrome [57,63].
The TP53 gene is referred to as, “the guardian of the genome,” and its function is lost in various cancers. Although TP53 is one of the most frequently encountered genes in tumor profiles, it rarely has pathogenic variants in the germline [64]. Therefore, it is difficult to diagnose only by tumor profiling, and individual measures should be taken according to the medical and family history of each patient. Even if tumor profiling reveals TP53, no treatment is yet available. However, clinical trials of TP53-targeting compounds are underway [65].
2.4. Cowden Syndrome
Cowden syndrome is a multiple hamartoma syndrome caused by a germline pathogenic variant of PTEN, with a 77–85% lifetime risk of breast cancer in women [66]. The major criteria for clinical diagnosis include breast cancer, epithelial (especially follicular) thyroid cancer, gastrointestinal hamartoma, macrocephaly, and endometrial cancer [6,67]. Risk of endometrial cancer in Cowden syndrome is increased within the ages of 30 and 49 years, and the lifetime risk is 28% [68]. In addition, benign uterine fibroids often occur, but they are not included in the clinical diagnostic criteria due to insufficient evidence [6]. The utility of endometrial cancer screening as gynecological surveillance for Cowden syndrome has not been established. However, although the value of surveillance is limited, endometrial cytology/histology and transvaginal ultrasonography every 1–2 years should be considered for women with Cowden syndrome after the age of 30–35 years [6].
2.5. Peutz–Jeghers Syndrome (PJS)
The autosomal dominant genetic disorder, PJS, is characterized by multiple hamartoma polyps in the gastrointestinal tract and pigmentation of the skin mucosa [74], and STK11 is the causative gene [75,76]. Hamartomatous polyps of PJS are found in the stomach, small intestine, large intestine, and elsewhere, and melena and intestinal obstruction can occur. Epithelial, malignant colorectal, gastric, pancreatic, breast and other types of cancer are related to PJS [77]. Pigmentation is often found on the fingers, lips, and oral cavity of children; hence, a clinical diagnosis is relatively straightforward. However, pigmentation disappears with age [77].
Gynecological ovarian tumors are PJS-related. Ovarian tumors with PJS are mainly of the sex-cord with annular tubules (SCAT), but they can also be associated with other ovarian malignancies. The risk of SCAT in PJS is 21%, and the average age at diagnosis is 28 years [78], which cannot be overlooked. Minimal deviation adenocarcinoma (MDA) and lobular endocervical glandular hyperplasia (LEGH) in PJS are referred to as PJS-related cervical tumors [79]. Both MDA and LEGH are malignant tumors of the cervix formerly known as adenoma malignum, and they have a poor prognosis. The 5-year survival rate of MDA is 42% [80]. The risk of MDA morbidity in the general population is < 1%, but the lifetime risk in patients with PJS is 10%, and the average age of onset is 34–40 years [77,78,81]. The fact that MDA can be difficult to diagnose should be considered in the surveillance of female patients with PJS. Since PJS is often diagnosed in childhood, surveillance should be continued in collaboration with various clinical departments.
2.6. Other Cancer-Susceptible Genes
Genes with high and moderate susceptibility for hereditary cancers have recently been identified and classified. Genes with low susceptibility are also classified as causes of multifactorial carcinogenesis. The risk of developing cancer is high in patients with highly and moderately susceptible genes [11,84,85], and many genes in the NCCN guidelines can be referred to for management [6,43]. Although the penetration rate is low compared with HBOC and Lynch syndrome, hereditary cancers caused by other genes associated with cancer susceptibility cannot be overlooked. For example, the BRIP1, PALB2, RAD51C, RAD51D, and BARD1, as well as the BRCA1/2, MLH1, MSH2, MSH6, and PMS2 genes are associated with hereditary ovarian cancer [11]. These genes are also thought to be involved in HRD, and PARP inhibitors should be effective in patients with these cancer-susceptible genes [28,35,37,86,87].
DICER1 causes a cancer predisposition syndrome and DICER1 syndrome is an autosomal dominant genetic disorder characterized by pleuropulmonary blastoma (PPB), multinodular goiter, cystic nephroma, Sertoli–Leydig cell tumors of the ovary (SLCT), and other rare tumors. Pleuropulmonary blastoma is the most prevalent primary lung malignancy in children, and those at highest risk for clinically significant PPB are aged < 7 years [88]. Ovarian tumors associated with DICER1 diagnosed at a median age of 16.9 years, sometimes develop synchronous or metachronous contralateral tumors [88]. Stewart et al. reported that 24 (21.2%) of 113 female carriers of DICER1 developed SLCT [89]. The DICER1 gene encodes DICER protein, which is an RNase III enzyme that functions in micro-RNA (miRNA) processing [90]. Targeted therapy against DICER1 is not yet available. However, a few reports have described these rare cancer-predisposing genes, and the latest information on clinical management should be reviewed, because caution is required when dealing with moderate- and low-grade susceptibility genes. The numbers of rare hereditary cancer-related genes judged as variants of uncertain significance (VUS) is likely to increase [84,91,92], and additional confirmation might be required for interpretation.
3. Genetic Testing for Hereditary Cancers
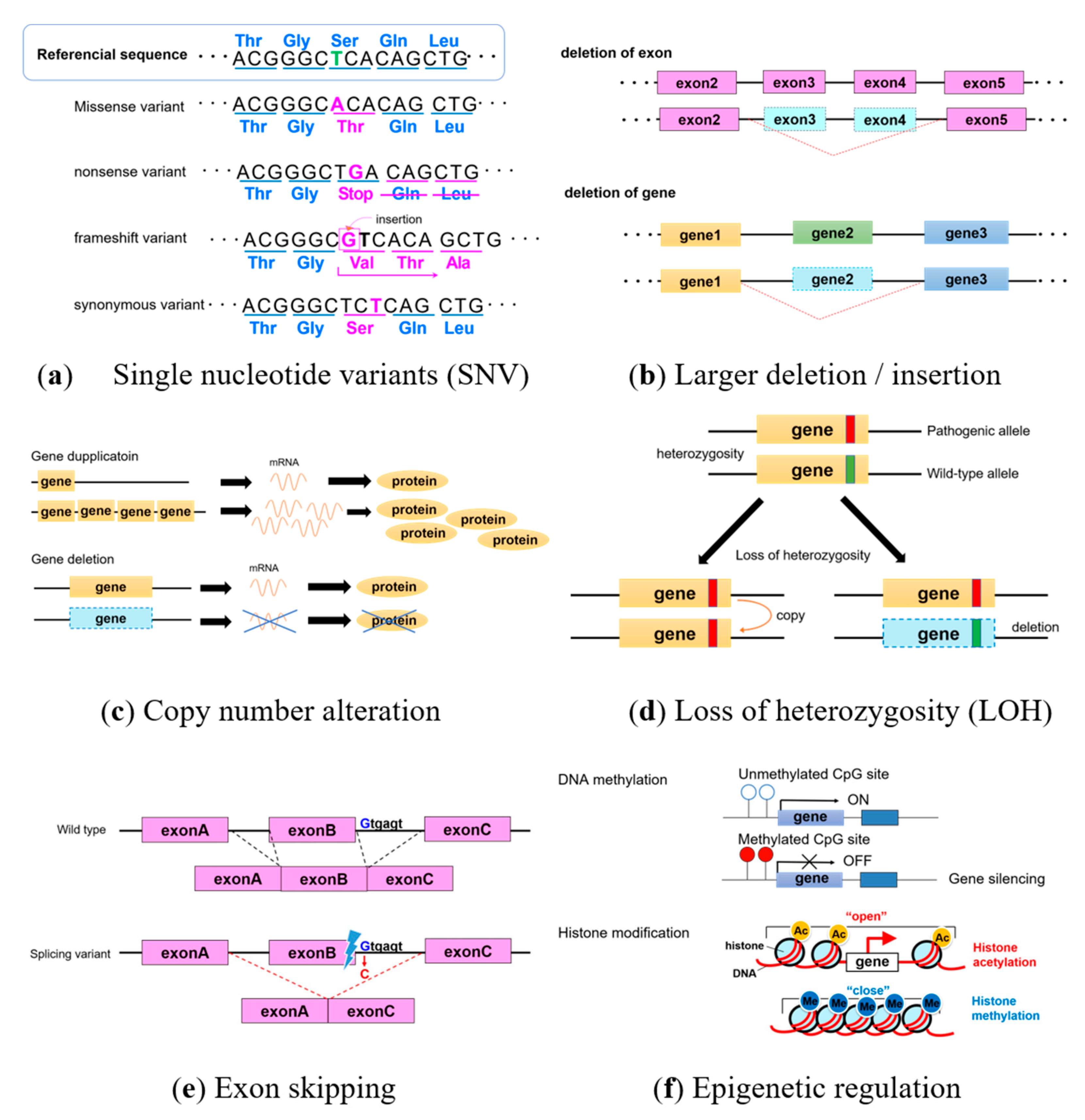
Genetic test results targeting hereditary tumor-related genes are usually analyzed using next-generation sequencing. However, further investigation is occasionally needed to conclude a correct diagnosis. Figure 1 shows that genetic alterations include single nucleotide variants (SNV), insertion and deletion of bases (indels), splicing variants in non-coding intron regions, copy number variants (CNV), and epigenetic changes, such as DNA hypermethylation and histone modification. Here, we outline genetic alterations and analyses.
Single nucleotide variants have a change in one base (Figure 1a). Some synonymous SNV encode the same amino acid and have no significant effects, whereas some missense variants have significant effects due to amino acid substitutions. A variant is pathogenic if changes affect the three-dimensional structure of amino acids. When encoded stop codon and produces a truncated protein with altered amino acids, they are called nonsense variants and are often pathogenic. Some frameshift variants move the reading frames of encoded amino acids by inserting or deleting several bases.
In addition, if a slightly larger deletion/insertion in the exon or gene (Figure 1b) causes difficulties with detecting changes by next-generation sequencing, Multiplex Ligation-dependent Probe Amplification (MLPA) can detect changes in copy numbers. Gene amplification (Figure 1c) and the loss of heterozygosity (LOH; Figure 1d) also occur in cancers. Analyzing copy numbers is useful for detecting LOH, and gene deletions and duplications can be analyzed using fluorescence in situ hybridization (FISH). Even if a change occurs in a non-coding intron, when variants affect the splice site, exon skipping leads to pathogenic protein formation (Figure 1e). Targeted RNA analysis might be required in the event of exon-skipping.
The hypermethylation of DNA and histone modification are epigenetic alterations (Figure 1f) that control gene expression without changing the DNA sequence. Many genes have a region of CpG islands near the promotor region upstream of a target gene. Hypermethylated DNA is often identified in cancers, and gene expression can be controlled by methylating CPG islands. Because DNA hypermethylation cannot be detected by next-generation sequencing, the methylation status of the promoter region should be analyzed. Histone modification is a complex mechanism in which histone acetylation and methylation respectively opens and closes the chromatin structure to activate and switch off transcription.
Consequently, genetic testing using various means can not only diagnose hereditary tumors, but also facilitate pharmacogenomics and the design of personalized therapy for cancer patients. The number of hereditary cancer-related genes diagnosed by tumor profiling as germline findings is increasing. Here, the characteristics of each genetic test are described.
3.1. Genetic Tests for Diagnosing Hereditary Cancers
Single genes have historically been tested based on the most likely hereditary cancer of a patient. With the widespread advent of next-generation sequencing, MGP has become the mainstream genetic test as is more rapid and cost-effective. Judgments were issued in the USA to invalidate the patent for BRCA1/2 genetic tests monopolized by Myriad Genetics during 2013 [93,94]. Thereafter, several companies have entered the market and now provide various MGP tests.
A set of genes that are considered to be related to hereditary cancers can be simultaneously analyzed using MGP. The introduction of MGP tests should increase the numbers of individuals diagnosed with pathogenic variants in genes associated with hereditary cancers that hitherto could not be identified by conventional single-gene tests. In fact, MGP tests replaced BRCA1/2-only tests in 2014 [91]. The spread of MGP tests will reduce the number of misdiagnosed hereditary cancers.
On the other hand, the number of patients with rare hereditary cancers will increase, even though they were not suspected before genetic tests. Therefore, MGP tests should be applied with reference to the most recent NCCN guidelines for the management of rare hereditary cancer-related genes. The revised NCCN guidelines (2020) [43,95], caused a major paradigm shift as the description changed to consider MGP tests first among genetic tests.
According to the guidelines of the American College of Medical Genetics and Genomics (ACMG) [96], the results of genetic tests are classified as: pathogenic, likely pathogenic, benign, likely benign, and variant of uncertain significance (VUS). The VUS classification means that pathogenicity cannot be determined, despite some variants in the gene. Genetic management based on VUS results is not recommended.
Multi-gene panels target multiple hereditary cancer-related genes. As the number of MGP tests increase, the number of rare hereditary cancer-related genes judged as VUS will also probably increase, and additional confirmation might be required for appropriate interpretation [84,85,97,98]. Furthermore, whether targeted genes can be analyzed by MGP should be confirmed because individual manufacturers will have different offerings, and not all targeted genes are clinically compatible. Thus, MGP tests must be accompanied by pre- and post-test genetic counseling based on genetic expertise [6,43].
3.2. Genetic Tests for Pharmacogenetics and Personalized Therapy
Several genetic tests can be conducted to decide appropriate treatment for patients. In December 2014, the PARP inhibitor, Olaparib, was approved by the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA), after another drug for BRCA1/2 mutation-positive ovarian cancer [23]. Accordingly, individuals with germline pathogenic variants in the BRCA1/2 gene are indicated for PARP inhibitors, and HBOC can be simultaneously diagnosed, so it is essential to provide appropriate information to patients and family members.
Indications for ICI, regardless of the type of cancer, have been determined by tests of MSI, and ICI are indicated treating for MSI-H solid tumors [52,99]. However, Lynch syndrome is also a possibility in patients with MSI-H [48]. Thus, careful assessment based on family and medical history is required.
The features of a BRCA1/2 functional deletion have been called BRCAness, the definition of which is ambiguous. Thus many tests have been proposed, such as the HRD score [100], the COSMIC mutational signature #3 [101], and the LOH status of the BRCA1 or BRCA2 locus [102]. The PARP inhibitor, Niraparib, is useful for late-line treatment of ovarian cancer, when HRD status serves as a biomarker [103]. Germline pathogenic variants in BRCA1/2 result in HRD, which can be exploited by PARP inhibitor as a matter of course. Although the HRR pathway involves numerous genes, HRR pathway genes other than BRCA1/2 such as PALB2, RAD51, and ATM, are candidates that might be effective for PARP inhibitor [36,104,105,106,107]. Furthermore, direct sequencing of HRR pathway genes can predict responsiveness to platinum and PARP [108]. Confirmation of HRD might reveal hereditary tumors.
Thus, the important issue is the appropriate approach to the possibility of hereditary cancers revealed by treatment indications. Collaboration with genetic experts is important, as is linking appropriate genetic counseling and hereditary cancer management when conducting genetic tests [43].
3.3. Tumor Profiling for Precision Medicine
The purpose of tumor and gene profiling in individual cancer tissues is to control cancers with personalized therapeutic strategies targeting driver genes [109,110]. On the other hand, the possibility of hereditary cancer-related genes can be clarified by comparison with normal sites as controls during tumor profiling. The amount of incidental germline findings discovered through tumor profiling is increasing [111,112]; a 5–15% chance of germline findings is associated with hereditary cancers by tumor profiling [113,114].
The ACMG (2020) issued a statement on presumed germline pathogenic variants (PGPV) that can be revealed from tumor tests [111]. With respect to the importance of germline findings in tissues, they state that, “Identifying germline pathogenic variants can inform future cancer risks, cancer surveillance, and prevention options for the patient and family members. In addition, germline genetic information, independent of somatic variation, can influence the choice of targeted therapy for a tumor.” Germline pathogenic variants in BRCA1/2 are informative as they confirm eligibility for treatment with PARP inhibitors.
Germline findings identified by tumor tests indicate that cancers are caused by genetic germline variants that might be shared by the families of patients. The European Society of Medical Oncology (ESMO) Precision Medicine Working Group (PMWG) recommended germline-focused analysis of tumor-only sequences in 2019 [64]. Thresholds of 20% and 30% VAF for small insertions/deletions and SNV, respectively, and limiting target genes for germline-focused tumor analysis to 27 (BRCA1, BRCA2, BRIP1, MLH1, MSH2, MSH6, PALB2, PMS2, VHL, RAD51C, RAD51D, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TSC2, MUTYH, RB1, APC, FLCN, FH, BAP1, POLE, TP53, and NF1), were proposed to narrow variants requiring follow-up germline tests.
By disclosing these germline findings, useful information can be provided to patients and their families, appropriate surveillance methods for early detection and early treatment for cancers can be suggested, indications for risk-reducing surgery can be discussed, and appropriate treatment for individual patients can be selected [114]. When germline alterations are suggested, genetic counselors can organize information considering treatment strategies and their impact on family members. Co-operation with genetic experts is necessary for additional confirmation.
4. Conclusions
The introduction of multigene panels has enabled the simultaneous analysis of multiple genes. In addition, a wider range of analyses using tumor profiling to target germline and somatic variants, has facilitated fewer omissions. Whole exome and genome sequencing will become routine in the near future, as analytical technology rapidly advances. However, consensus about target genes for MGP tests and tumor profiling has not been reached, so the situation established by each test institution or facility should be determined. Recognizing common target genes will become essential, and if somatic variants are revealed by tumor profiling, appropriate treatment methods should be designed.
A definitive diagnosis of the causative gene of a hereditary cancer is characterized as being lifelong, affecting the family, and predictive of cancer onset. In addition, patients with hereditary cancer must be carefully managed. However, genetic information could be useful for subsequent treatment selection and actionable information about diseases. When a germline pathogenic variant associated with a hereditary cancer-related gene is detected by tumor profiling, treatment should be individualized for each patient. Clinicians might be reluctant to definitively diagnose hereditary cancers, given the effects on family members. However, medical intervention will be appropriate if clinicians and patients both recognize that hereditary cancer-related genes are important to know as they will lead to primary cancer prevention for family members. A thorough understanding of hereditary cancers should allow clinicians to use tumor profiling information as a useful tool and provide their patients with optimal medical care.
Author Contributions
Both authors contributed to the research, drafting, and editing of the manuscript. Both authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Number 17K16881.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| DICER1 | Ribonuclease III; double-stranded RNA-specific endoribonuclease |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| HBOC | hereditary breast and ovarian cancer syndrome |
| HRD | homologous recombination deficiency |
| HRR | homologous recombination repair |
| ICI | immune checkpoint inhibitor |
| LEGH | lobular endocervical glandular hyperplasia |
| MDA | minimal deviation adenocarcinoma |
| MGP | multi-gene panel |
| MMR | mismatch repair |
| MRI | magnetic resonance imaging |
| MSI | microsatellite instability |
| MSI-H | microsatellite instability-high |
| MSI-L | microsatellite instability-low |
| MSS | microsatellite stable |
| NCCN | National Comprehensive Cancer Network |
| PARP | poly ADP ribose polymerase |
| PJS | Peutz–Jeghers syndrome |
| RRSO | risk-reducing salpingo-oophorectomy |
| SCAT | sex-cord tumor with annular tubules |
| VUS | variant of uncertain significance |
References
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Fournier, D.M.; Bazzell, A.F.; Dains, J.E. Comparing Outcomes of Genetic Counseling Options in Breast and Ovarian Cancer: An Integrative Review. Oncol. Nurs. Forum 2018, 45, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H. Genetic counseling and cascade genetic testing in Lynch syndrome. Fam. Cancer 2016, 15, 423–427. [Google Scholar] [CrossRef]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef] [Green Version]
- Hooker, G.W.; Clemens, K.R.; Quillin, J.; Vogel Postula, K.J.; Summerour, P.; Nagy, R.; Buchanan, A.H. Cancer Genetic Counseling and Testing in an Era of Rapid Change. J. Genet. Couns. 2017, 26, 1244–1253. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guidelines in Oncology. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version1, 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 19 November 2020).
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [Green Version]
- Hirasawa, A.; Imoto, I.; Naruto, T.; Akahane, T.; Yamagami, W.; Nomura, H.; Masuda, K.; Susumu, N.; Tsuda, H.; Aoki, D. Prevalence of pathogenic germline variants detected by multigene sequencing in unselected Japanese patients with ovarian cancer. Oncotarget 2017, 8, 112258–112267. [Google Scholar] [CrossRef] [Green Version]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012, 307, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, T.; Aoki, D.; Hattori, K.; Jinushi, M.; Kigawa, J.; Takeshima, N.; Tsuda, H.; Watanabe, Y.; Yoshihara, K.; Sugiyama, T. The first Japanese nationwide multicenter study of BRCA mutation testing in ovarian cancer: CHARacterizing the cross-sectionaL approach to Ovarian cancer geneTic TEsting of BRCA (CHARLOTTE). Int. J. Gynecol. Cancer 2019, 29, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Norquist, B.; Lacchetti, C.; Armstrong, D.; Grisham, R.N.; Goodfellow, P.J.; Kohn, E.C.; Levine, D.A.; Liu, J.F.; Lu, K.H.; et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 1222–1245. [Google Scholar] [CrossRef]
- NICE Guideline: Familial Breast Cancer: Classification, Care and Managing Breast Cancer and Related Risks in People with a Family History of Breast Cancer. Available online: www.nice.org.uk/guidance/cg164 (accessed on 19 November 2020).
- Owens, D.K.; Davidson, K.W.; Krist, A.H.; Barry, M.J.; Cabana, M.; Caughey, A.B.; Doubeni, C.A.; Epling, J.W., Jr.; Kubik, M.; Landefeld, C.S.; et al. Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2019, 322, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Paluch-Shimon, S.; Cardoso, F.; Sessa, C.; Balmana, J.; Cardoso, M.J.; Gilbert, F.; Senkus, E.; Committee, E.G. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol. 2016, 27, v103–v110. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Kauff, N.D.; Domchek, S.M. Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J. Natl. Cancer Inst. 2009, 101, 80–87. [Google Scholar] [CrossRef]
- Harmsen, M.G.; Piek, J.M.J.; Bulten, J.; Casey, M.J.; Rebbeck, T.R.; Mourits, M.J.; Greene, M.H.; Slangen, B.F.M.; van Beurden, M.; Massuger, L.; et al. Peritoneal carcinomatosis after risk-reducing surgery in BRCA1/2 mutation carriers. Cancer 2018, 124, 952–959. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K.; Amso, N.N.; Apostolidou, S.; Benjamin, E.; Cruickshank, D.; et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A randomised controlled trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Iglehart, J.D.; Silver, D.P. Synthetic lethality—A new direction in cancer-drug development. N. Engl. J. Med. 2009, 361, 189–191. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- George, A.; Kaye, S.; Banerjee, S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nat. Rev. Clin. Oncol. 2017, 14, 284–296. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.J.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Han, H.S.; Diéras, V.; Robson, M.; Palácová, M.; Marcom, P.K.; Jager, A.; Bondarenko, I.; Citrin, D.; Campone, M.; Telli, M.L.; et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: Randomized phase II study. Ann. Oncol. 2018, 29, 154–161. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Rugo, H.S.; Olopade, O.I.; DeMichele, A.; Yau, C.; van’t Veer, L.J.; Buxton, M.B.; Hogarth, M.; Hylton, N.M.; Paoloni, M.; Perlmutter, J.; et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N. Engl. J. Med. 2016, 375, 23–34. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous recombination deficiency and ovarian cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann. Oncol. 2016, 27, 1449–1455. [Google Scholar] [CrossRef]
- Practice Bulletin No 182: Hereditary Breast and Ovarian Cancer Syndrome. Obstet. Gynecol. 2017, 130, e110–e126. [CrossRef]
- Win, A.K.; Lindor, N.M.; Young, J.P.; Macrae, F.A.; Young, G.P.; Williamson, E.; Parry, S.; Goldblatt, J.; Lipton, L.; Winship, I.; et al. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J. Natl. Cancer Inst. 2012, 104, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Sinicrope, F.A. Lynch Syndrome-Associated Colorectal Cancer. N. Engl. J. Med. 2018, 379, 764–773. [Google Scholar] [CrossRef]
- Lu, K.H.; Daniels, M. Endometrial and ovarian cancer in women with Lynch syndrome: Update in screening and prevention. Fam. Cancer 2013, 12, 273–277. [Google Scholar] [CrossRef]
- Lu, K.H.; Dinh, M.; Kohlmann, W.; Watson, P.; Green, J.; Syngal, S.; Bandipalliam, P.; Chen, L.M.; Allen, B.; Conrad, P.; et al. Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet. Gynecol. 2005, 105, 569–574. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guideline in Oncology, Genetic / Familial High-Risk Assessment: Colorectal, Version1. 2020. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf (accessed on 19 November 2020).
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.A.J.; Evans, D.G.; Green, K.; Crosbie, E.J. Pathological features and clinical behavior of Lynch syndrome-associated ovarian cancer. Gynecol. Oncol. 2017, 144, 491–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.; Bützow, R.; Lynch, H.T.; Mecklin, J.P.; Järvinen, H.J.; Vasen, H.F.; Madlensky, L.; Fidalgo, P.; Bernstein, I. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol. Oncol. 2001, 82, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Vasen, H.F.A.; Mecklin, J.P.; Bernstein, I.; Aarnio, M.; Järvinen, H.J.; Myrhøj, T.; Sunde, L.; Wijnen, J.T.; Lynch, H.T. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int. J. Cancer 2008, 123, 444–449. [Google Scholar] [CrossRef] [Green Version]
- Latham, A.; Srinivasan, P.; Kemel, Y.; Shia, J.; Bandlamudi, C.; Mandelker, D.; Middha, S.; Hechtman, J.; Zehir, A.; Dubard-Gault, M.; et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol. 2019, 37, 286–295. [Google Scholar] [CrossRef]
- Vasen, H.F. Clinical diagnosis and management of hereditary colorectal cancer syndromes. J. Clin. Oncol. 2000, 18, 81s–92s. [Google Scholar]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Dudley, J.C.; Lin, M.T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Schneider KA, G.J. Li-Fraumeni Syndrome. In GeneReviews. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1311/ (accessed on 19 November 2020).
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef]
- Kratz, C.P.; Achatz, M.I.; Brugières, L.; Frebourg, T.; Garber, J.E.; Greer, M.C.; Hansford, J.R.; Janeway, K.A.; Kohlmann, W.K.; McGee, R.; et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin. Cancer Res. 2017, 23, e38–e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, K.A.; Ballinger, M.L.; Killick, E.; Kirk, J.; Tattersall, M.H.; Eeles, R.A.; Thomas, D.M.; Mitchell, G. Li-Fraumeni syndrome: Cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014, 11, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122, 3673–3681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical characteristics of families with p53 germline mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Lalloo, F.; Varley, J.; Moran, A.; Ellis, D.; O’Dair, L.; Pharoah, P.; Antoniou, A.; Hartley, R.; Shenton, A.; Seal, S.; et al. BRCA1, BRCA2 and TP53 mutations in very early-onset breast cancer with associated risks to relatives. Eur. J. Cancer 2006, 42, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- McCuaig, J.M.; Armel, S.R.; Novokmet, A.; Ginsburg, O.M.; Demsky, R.; Narod, S.A.; Malkin, D. Routine TP53 testing for breast cancer under age 30: Ready for prime time? Fam. Cancer 2012, 11, 607–613. [Google Scholar] [CrossRef]
- Mouchawar, J.; Korch, C.; Byers, T.; Pitts, T.M.; Li, E.; McCredie, M.R.; Giles, G.G.; Hopper, J.L.; Southey, M.C. Population-based estimate of the contribution of TP53 mutations to subgroups of early-onset breast cancer: Australian Breast Cancer Family Study. Cancer Res. 2010, 70, 4795–4800. [Google Scholar] [CrossRef] [Green Version]
- Kamihara, J.; Rana, H.Q.; Garber, J.E. Germline TP53 mutations and the changing landscape of Li-Fraumeni syndrome. Hum. Mutat. 2014, 35, 654–662. [Google Scholar] [CrossRef]
- Valdez, J.M.; Nichols, K.E.; Kesserwan, C. Li-Fraumeni syndrome: A paradigm for the understanding of hereditary cancer predisposition. Br. J. Haematol. 2017, 176, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Mandelker, D.; Donoghue, M.; Talukdar, S.; Bandlamudi, C.; Srinivasan, P.; Vivek, M.; Jezdic, S.; Hanson, H.; Snape, K.; Kulkarni, A.; et al. Germline-focussed analysis of tumour-only sequencing: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. 2019, 30, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Bubien, V.; Bonnet, F.; Brouste, V.; Hoppe, S.; Barouk-Simonet, E.; David, A.; Edery, P.; Bottani, A.; Layet, V.; Caron, O.; et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J. Med. Genet. 2013, 50, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilarski, R.; Burt, R.; Kohlman, W.; Pho, L.; Shannon, K.M.; Swisher, E. Cowden syndrome and the PTEN hamartoma tumor syndrome: Systematic review and revised diagnostic criteria. J. Natl. Cancer Inst. 2013, 105, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y.; et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA 2017, 318, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef]
- Komiya, T.; Blumenthal, G.M.; DeChowdhury, R.; Fioravanti, S.; Ballas, M.S.; Morris, J.; Hornyak, T.J.; Wank, S.; Hewitt, S.M.; Morrow, B.; et al. A Pilot Study of Sirolimus in Subjects with Cowden Syndrome or Other Syndromes Characterized by Germline Mutations in PTEN. Oncologist 2019, 24, 1510-e1265. [Google Scholar] [CrossRef] [Green Version]
- Marsh, D.J.; Trahair, T.N.; Martin, J.L.; Chee, W.Y.; Walker, J.; Kirk, E.P.; Baxter, R.C.; Marshall, G.M. Rapamycin treatment for a child with germline PTEN mutation. Nat. Clin. Pract. Oncol. 2008, 5, 357–361. [Google Scholar] [CrossRef]
- Schmid, G.L.; Kässner, F.; Uhlig, H.H.; Körner, A.; Kratzsch, J.; Händel, N.; Zepp, F.P.; Kowalzik, F.; Laner, A.; Starke, S.; et al. Sirolimus treatment of severe PTEN hamartoma tumor syndrome: Case report and in vitro studies. Pediatr. Res. 2014, 75, 527–534. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, I.P.; Houlston, R.S. Peutz-Jeghers syndrome. J. Med. Genet. 1997, 34, 1007–1011. [Google Scholar] [CrossRef] [Green Version]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Höglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Müller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef] [PubMed]
- McGarrity, T.J.; Amos, C.I.; Frazier, M.L.; Wei, C. Peutz-Jeghers Syndrome. In GeneReviews. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1266/ (accessed on 10 November 2020).
- Giardiello, F.M.; Trimbath, J.D. Peutz-Jeghers syndrome and management recommendations. Clin. Gastroenterol. Hepatol. 2006, 4, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, A.; Akahane, T.; Tsuruta, T.; Kobayashi, Y.; Masuda, K.; Banno, K.; Fujii, T.; Susumu, N.; Itsubo, T.; Kameyama, K.; et al. Lobular endocervical glandular hyperplasia and peritoneal pigmentation associated with Peutz-Jeghers syndrome due to a germline mutation of STK11. Ann. Oncol. 2012, 23, 2990–2992. [Google Scholar] [CrossRef]
- Karamurzin, Y.S.; Kiyokawa, T.; Parkash, V.; Jotwani, A.R.; Patel, P.; Pike, M.C.; Soslow, R.A.; Park, K.J. Gastric-type Endocervical Adenocarcinoma: An Aggressive Tumor With Unusual Metastatic Patterns and Poor Prognosis. Am. J. Surg. Pathol. 2015, 39, 1449–1457. [Google Scholar] [CrossRef] [Green Version]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Xu, L.; Seki, M.; Yokoyama, T.T.; Kasahara, M.; Kashima, Y.; Ohashi, A.; Shimada, Y.; Motoi, N.; Tsuchihara, K.; et al. Long-read sequencing for non-small-cell lung cancer genomes. Genome Res. 2020, 30, 1243–1257. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Tsaousis, G.N.; Papadopoulou, E.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Kampouri, S.; Diamantopoulos, N.; Floros, T.; Iosifidou, R.; Katopodi, O.; et al. Analysis of hereditary cancer syndromes by using a panel of genes: Novel and multiple pathogenic mutations. BMC Cancer 2019, 19, 535. [Google Scholar] [CrossRef] [Green Version]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.R.; Syngal, S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613.e620. [Google Scholar] [CrossRef] [Green Version]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.R.; Best, A.F.; Williams, G.M.; Harney, L.A.; Carr, A.G.; Harris, A.K.; Kratz, C.P.; Dehner, L.P.; Messinger, Y.H.; Rosenberg, P.S.; et al. Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1. J. Clin. Oncol. 2019, 37, 668–676. [Google Scholar] [CrossRef]
- Bailey, K.M.; Jacobs, M.F.; Anderson, B.; Rabah, R.; Wu, Y.M.; Else, T.; Mody, R.J. DICER1 Mutations in the Era of Expanding Integrative Clinical Sequencing in Pediatric Oncology. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef] [PubMed]
- Kurian, A.W.; Ward, K.C.; Hamilton, A.S.; Deapen, D.M.; Abrahamse, P.; Bondarenko, I.; Li, Y.; Hawley, S.T.; Morrow, M.; Jagsi, R.; et al. Uptake, Results, and Outcomes of Germline Multiple-Gene Sequencing After Diagnosis of Breast Cancer. JAMA Oncol. 2018, 4, 1066–1072. [Google Scholar] [CrossRef]
- LaDuca, H.; Polley, E.C.; Yussuf, A.; Hoang, L.; Gutierrez, S.; Hart, S.N.; Yadav, S.; Hu, C.; Na, J.; Goldgar, D.E.; et al. A clinical guide to hereditary cancer panel testing: Evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet. Med. 2020, 22, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Conley, J.M.; Cook-Deegan, R.; Lázaro-Muñoz, G. MYRIAD AFTER MYRIAD: THE PROPRIETARY DATA DILEMMA. North Carol. J. Law Technol. 2014, 15, 597–637. [Google Scholar]
- Cook-Deegan, R.; Niehaus, A. After Myriad: Genetic Testing in the Wake of Recent Supreme Court Decisions about Gene Patents. Curr. Genet. Med. Rep. 2014, 2, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Seng, S.; Yoo, J.; Equivel, P.; Lum, S.S. Clinical Management of Patients at Risk for Hereditary Breast Cancer with Variants of Uncertain Significance in the Era of Multigene Panel Testing. Ann. Surg. Oncol. 2019, 26, 3389–3396. [Google Scholar] [CrossRef] [PubMed]
- Ndugga-Kabuye, M.K.; Issaka, R.B. Inequities in multi-gene hereditary cancer testing: Lower diagnostic yield and higher VUS rate in individuals who identify as Hispanic, African or Asian and Pacific Islander as compared to European. Fam. Cancer 2019, 18, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast. Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef] [Green Version]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.H.; Chen, M.; Tsai, L.W.; Lo, C.; Yen, T.C.; Huang, T.Y.; Chen, C.K.; Fan, S.C.; Kuo, S.H.; Huang, C.S. Using next-generation sequencing to redefine BRCAness in triple-negative breast cancer. Cancer Sci. 2020, 111, 1375–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavin, T.P.; Coffee, B.; Bernhisel, R.; Logan, J.; Cox, H.C.; Marcucci, G.; Weitzel, J.; Neuhausen, S.L.; Mancini-DiNardo, D. Prevalence and characteristics of likely-somatic variants in cancer susceptibility genes among individuals who had hereditary pan-cancer panel testing. Cancer Genet. 2019, 235–236, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Li, M.M.; Chao, E.; Esplin, E.D.; Miller, D.T.; Nathanson, K.L.; Plon, S.E.; Scheuner, M.T.; Stewart, D.R. Points to consider for reporting of germline variation in patients undergoing tumor testing: A statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 1142–1148. [Google Scholar] [CrossRef] [Green Version]
- Pujol, P.; Vande Perre, P.; Faivre, L.; Sanlaville, D.; Corsini, C.; Baertschi, B.; Anahory, M.; Vaur, D.; Olschwang, S.; Soufir, N.; et al. Guidelines for reporting secondary findings of genome sequencing in cancer genes: The SFMPP recommendations. Eur. J. Hum. Genet. 2018, 26, 1732–1742. [Google Scholar] [CrossRef]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef]
- Seifert, B.A.; O’Daniel, J.M.; Amin, K.; Marchuk, D.S.; Patel, N.M.; Parker, J.S.; Hoyle, A.P.; Mose, L.E.; Marron, A.; Hayward, M.C.; et al. Germline Analysis from Tumor-Germline Sequencing Dyads to Identify Clinically Actionable Secondary Findings. Clin. Cancer Res. 2016, 22, 4087–4094. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Genetic alterations. (a) Single nucleotide variants, (b) large deletion/insertion in exon or gene, (c) copy number alteration, (d) loss of heterozygosity, (e) exon skipping, and (f) epigenetic regulation.
Figure 1.
Genetic alterations. (a) Single nucleotide variants, (b) large deletion/insertion in exon or gene, (c) copy number alteration, (d) loss of heterozygosity, (e) exon skipping, and (f) epigenetic regulation.


{kind=link}
Table 1.
Molecular feature and clinical management of hereditary gynecological cancers.
| Hereditary Gynecological Cancer | Causative Genes | Related Gynecological Cancers | Related Non-Gynecological Cancers | Drug Sensitivity |
|---|---|---|---|---|
| HBOC | BRCA1, BRCA2 | Ovarian cancer | Breast cancer Prostate cancer Pancreatic cancer | PARP inhibitors |
| Lynch syndrome | MLH1, MSH2, MSH6, PMS2, EPCAM | Endometrial cancer Ovarian cancer | Colorectal cancer Small intestine cancer Ureteral cancer Renal pelvis cancer | ICI |
| Li–Fraumeni syndrome | TP53 | Insufficient evidence of ovarian cancer | Soft tissue sarcoma Osteosarcoma Premenopausal breast cancer Colorectal cancer Gastric cancer Adrenocortical carcinoma Brain tumor | - |
| Cowden syndrome | PTEN | Endometrial cancer | Breast cancer Epithelial thyroid cancer 1 Gastrointestinal hamartoma | - |
| Peutz–Jeghers syndrome | STK11 | Ovarian tumor (SCAT) Cervical tumor (MDA, LEGH) | Colorectal cancer Gastric cancer Pancreatic cancer Breast cancer | - |
| Other cancer-susceptible genes | RAD51C, RAD51D Etc. | Ovarian cancer | Various cancers | HRD status→PARP inhibitors |
| DICER1 | Sertoli–Leydig cell tumor (SLCT) of the ovary | Pleuropulmonary blastoma | - |
1 Particularly follicular thyroid cancer. Abbreviations: HBOC, hereditary breast and ovarian cancer syndrome; HRD, homologous recombination deficiency; ICI, immune checkpoint inhibitor; LEGH, lobular endocervical glandular hyperplasia; MDA, minimal deviation adenocarcinoma; PARP, poly ADP ribose polymerase; SCAT, sex-cord tumor with annular tubules; SLCT, Sertoli–Leydig cell tumor.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ueki, A.; Hirasawa, A. Molecular Features and Clinical Management of Hereditary Gynecological Cancers. Int. J. Mol. Sci. 2020, 21, 9504. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249504
AMA Style
Ueki A, Hirasawa A. Molecular Features and Clinical Management of Hereditary Gynecological Cancers. International Journal of Molecular Sciences. 2020; 21(24):9504. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249504
Chicago/Turabian StyleUeki, Arisa, and Akira Hirasawa. 2020. "Molecular Features and Clinical Management of Hereditary Gynecological Cancers" International Journal of Molecular Sciences 21, no. 24: 9504. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249504
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.