The Regulation of Intestinal Inflammation and Cancer Development by Type 2 Immune Responses

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
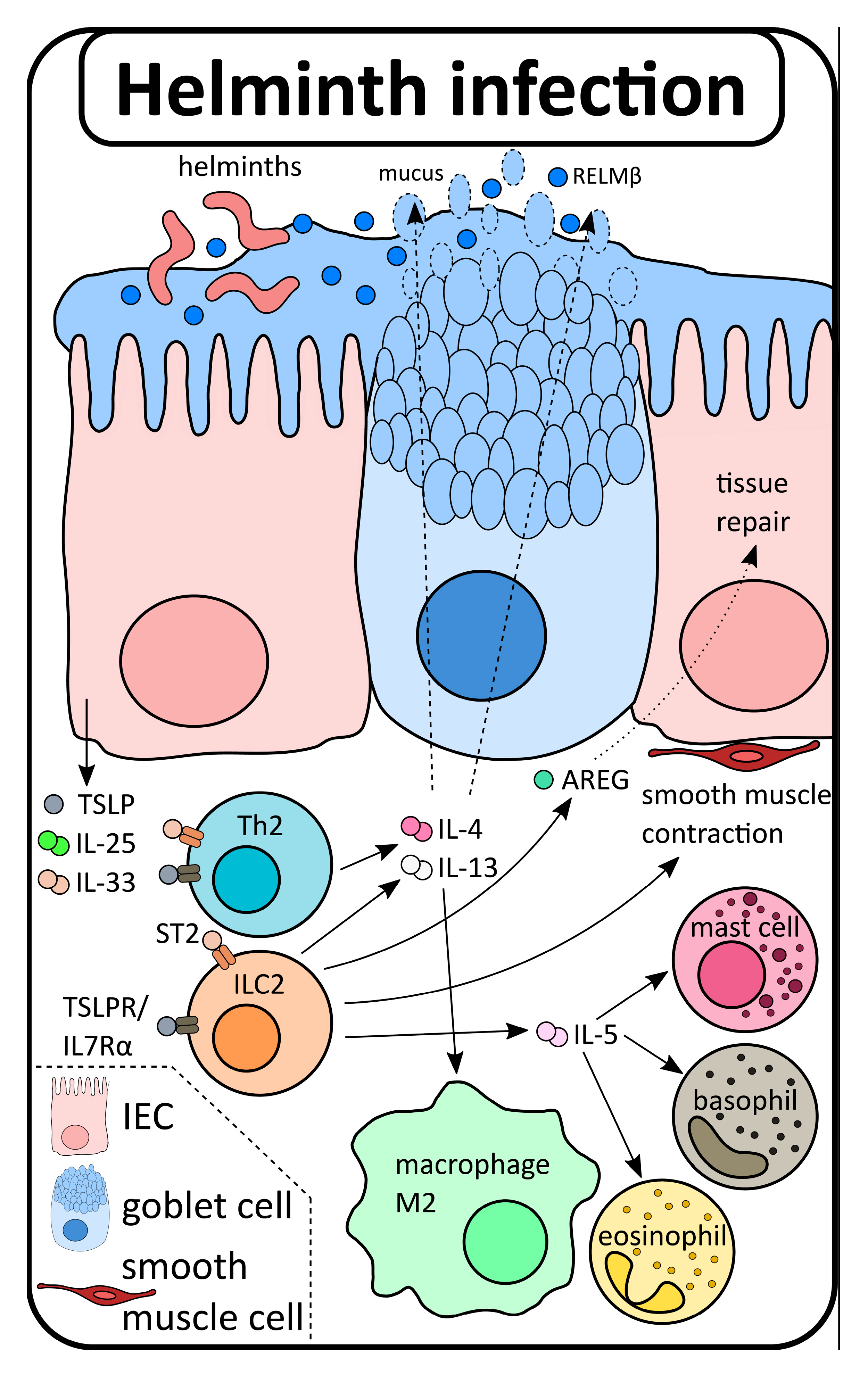
2. The Role of Type 2 Immunity in the Gut: From Homeostasis to Disease
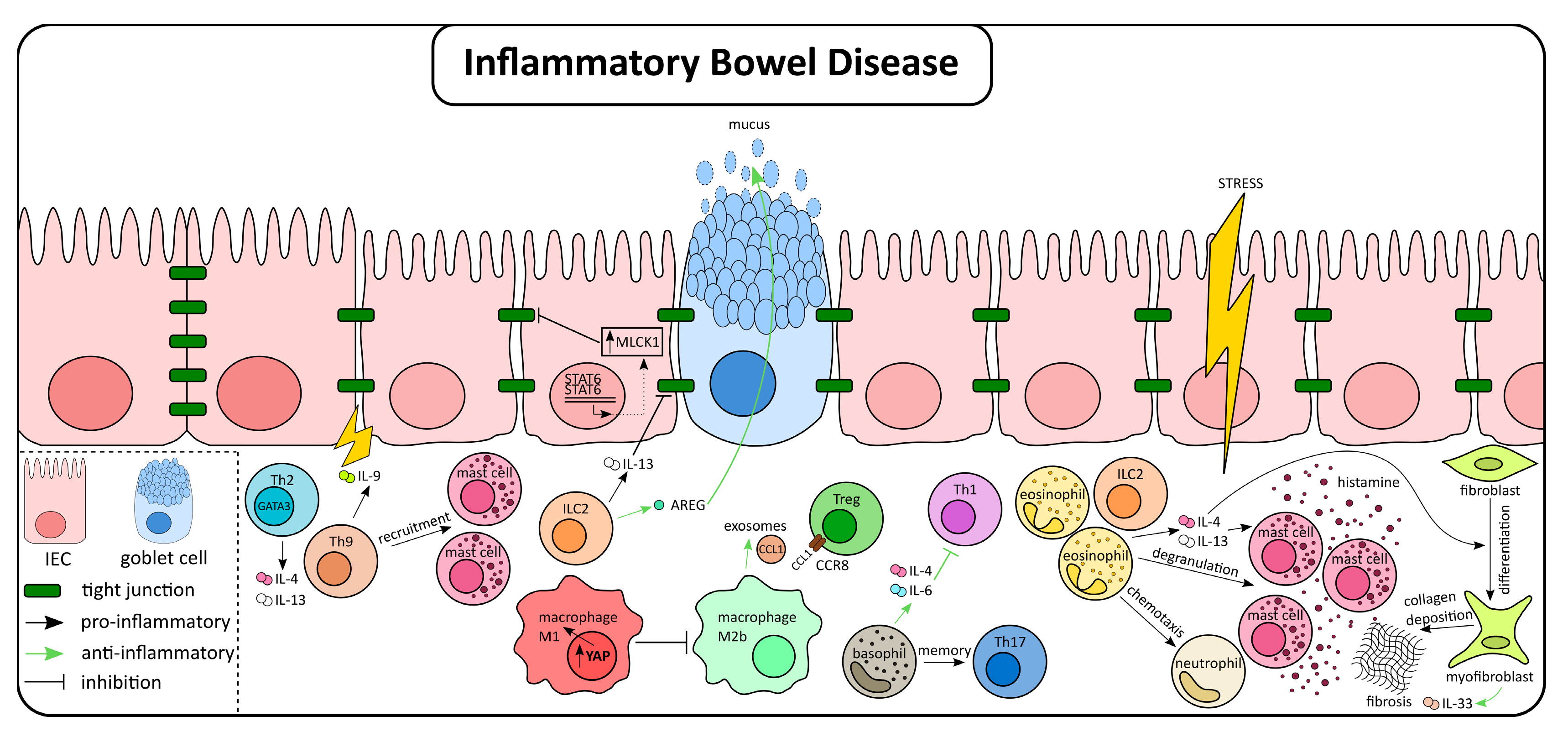
3. The Role of Type 2 Immunity in IBD
4. Type 2 Associated Immune Cells in IBD
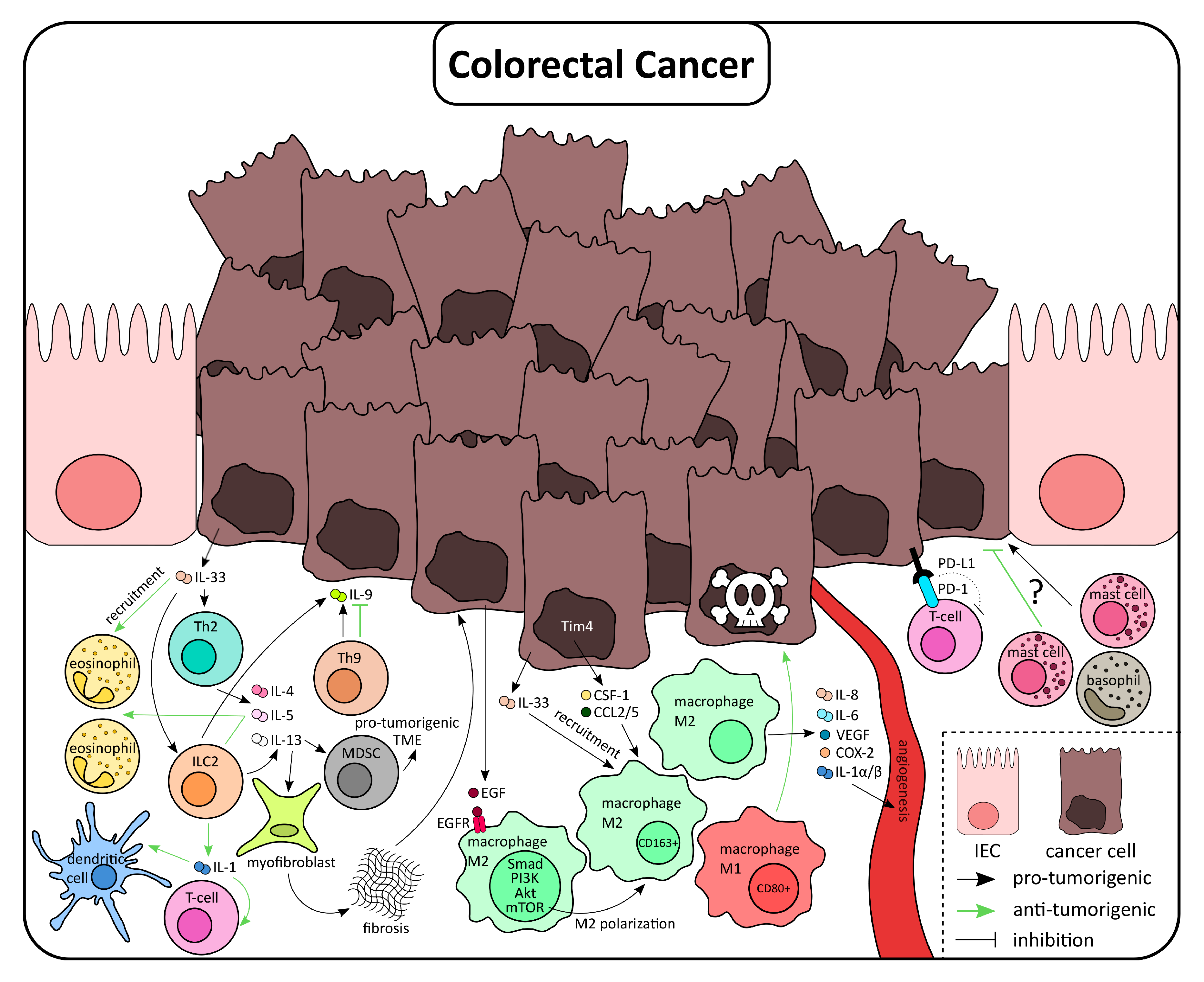
5. Type 2 Immunity Responses in the Context of Colorectal Cancer
6. Type 2 Immune Cells in CRC
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieseck, R.L., 3rd; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef]
- Lloyd, C.M.; Snelgrove, R.J. Type 2 immunity: Expanding our view. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Caminati, M.; Pham, D.L.; Bagnasco, D.; Canonica, G.W. Type 2 immunity in asthma. World Allergy Organ. J. 2018, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Micosse, C.; von Meyenn, L.; Steck, O.; Kipfer, E.; Adam, C.; Simillion, C.; Seyed Jafari, S.M.; Olah, P.; Yawlkar, N.; Simon, D.; et al. Human “TH9” cells are a subpopulation of PPAR-gamma(+) TH2 cells. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Henry, E.K.; Inclan-Rico, J.M.; Siracusa, M.C. Type 2 cytokine responses: Regulating immunity to helminth parasites and allergic inflammation. Curr. Pharmacol. Rep. 2017, 3, 346–359. [Google Scholar] [CrossRef]
- Zhu, P.; Zhu, X.; Wu, J.; He, L.; Lu, T.; Wang, Y.; Liu, B.; Ye, B.; Sun, L.; Fan, D.; et al. IL-13 secreted by ILC2s promotes the self-renewal of intestinal stem cells through circular RNA circPan3. Nat. Immunol. 2019, 20, 183–194. [Google Scholar] [CrossRef]
- Jung, Y.; Wen, T.; Mingler, M.K.; Caldwell, J.M.; Wang, Y.H.; Chaplin, D.D.; Lee, E.H.; Jang, M.H.; Woo, S.Y.; Seoh, J.Y.; et al. IL-1beta in eosinophil-mediated small intestinal homeostasis and IgA production. Mucosal Immunol. 2015, 8, 930–942. [Google Scholar] [CrossRef] [Green Version]
- Von Moltke, J.; Ji, M.; Liang, H.E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipriani, G.; Gibbons, S.J.; Kashyap, P.C.; Farrugia, G. Intrinsic Gastrointestinal Macrophages: Their Phenotype and Role in Gastrointestinal Motility. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhner, S.; Schemann, M. Mast cell-nerve axis with a focus on the human gut. Biochim. Biophys. Acta 2012, 1822, 85–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamias, G.; Cominelli, F. Role of type 2 immunity in intestinal inflammation. Curr. Opin. Gastroenterol. 2015, 31, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Gerbe, F.; Sidot, E.; Smyth, D.J.; Ohmoto, M.; Matsumoto, I.; Dardalhon, V.; Cesses, P.; Garnier, L.; Pouzolles, M.; Brulin, B.; et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016, 529, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Loser, S.; Smith, K.A.; Maizels, R.M. Innate Lymphoid Cells in Helminth Infections-Obligatory or Accessory? Front. Immunol. 2019, 10, 620. [Google Scholar] [CrossRef]
- Gause, W.C.; Rothlin, C.; Loke, P. Heterogeneity in the initiation, development and function of type 2 immunity. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Pelly, V.S.; Kannan, Y.; Coomes, S.M.; Entwistle, L.J.; Ruckerl, D.; Seddon, B.; MacDonald, A.S.; McKenzie, A.; Wilson, M.S. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol. 2016, 9, 1407–1417. [Google Scholar] [CrossRef] [Green Version]
- Halim, T.Y.F.; Rana, B.M.J.; Walker, J.A.; Kerscher, B.; Knolle, M.D.; Jolin, H.E.; Serrao, E.M.; Haim-Vilmovsky, L.; Teichmann, S.A.; Rodewald, H.R.; et al. Tissue-Restricted Adaptive Type 2 Immunity Is Orchestrated by Expression of the Costimulatory Molecule OX40L on Group 2 Innate Lymphoid Cells. Immunity 2018, 48, 1195–1207. [Google Scholar] [CrossRef] [Green Version]
- Oliphant, C.J.; Hwang, Y.Y.; Walker, J.A.; Salimi, M.; Wong, S.H.; Brewer, J.M.; Englezakis, A.; Barlow, J.L.; Hams, E.; Scanlon, S.T.; et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 2014, 41, 283–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gause, W.C.; Wynn, T.A.; Allen, J.E. Type 2 immunity and wound healing: Evolutionary refinement of adaptive immunity by helminths. Nat. Rev. Immunol 2013, 13, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, D.R.; Douglas, B.; Zullo, K. Group 2 Innate Lymphoid Cells (ILC2): Type 2 Immunity and Helminth Immunity. Int. J. Mol. Sci. 2019, 20, 2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motran, C.C.; Silvane, L.; Chiapello, L.S.; Theumer, M.G.; Ambrosio, L.F.; Volpini, X.; Celias, D.P.; Cervi, L. Helminth Infections: Recognition and Modulation of the Immune Response by Innate Immune Cells. Front. Immunol. 2018, 9, 664. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Ding, J.; Zhang, L.; Zhang, Y.; Yang, Y.; Bai, X.; Liu, X.; Jin, X.; Guo, H.; Yang, Y.; et al. Effect of recombinant serine protease from adult stage of Trichinella spiralis on TNBS-induced experimental colitis in mice. Int. Immunopharmacol. 2020, 86, 106699. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Jin, X.; Wang, Y.; Yang, Y.; Yang, L.; Bai, X.; Yang, Y.; Xu, N.; Wang, X.; Liu, M. Effect of recombinant serine protease from newborn larval stage of Trichinella spiralis on 2,4,6-trinitrobenzene sulfonic acid-induced experimental colitis in mice. Acta Trop 2020, 211, 105553. [Google Scholar] [CrossRef]
- Xu, J.; Liu, M.; Yu, P.; Wu, L.; Lu, Y. Effect of recombinant Trichinella spiralis cysteine proteinase inhibitor on TNBS-induced experimental inflammatory bowel disease in mice. Int. Immunopharmacol. 2019, 66, 28–40. [Google Scholar] [CrossRef]
- Xu, J.; Wu, L.; Yu, P.; Liu, M.; Lu, Y. Effect of two recombinant Trichinella spiralis serine protease inhibitors on TNBS-induced experimental colitis of mice. Clin. Exp. Immunol. 2018, 194, 400–413. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yu, P.; Wu, L.; Liu, M.; Lu, Y. Effect of Trichinella spiralis intervention on TNBS-induced experimental colitis in mice. Immunobiology 2019, 224, 147–153. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, L.; Liu, X.; Zhang, Y.; Shi, H.; Jia, W.; Zhu, H.; Jia, H.; Liu, M.; Bai, X. Extracellular Vesicles Derived From Trichinella spiralis Muscle Larvae Ameliorate TNBS-Induced Colitis in Mice. Front. Immunol. 2020, 11, 1174. [Google Scholar] [CrossRef]
- Sarazin, A.; Dendooven, A.; Delbeke, M.; Gatault, S.; Pagny, A.; Standaert, A.; Rousseaux, C.; Desreumaux, P.; Dubuquoy, L.; Capron, M. Treatment with P28GST, a schistosome-derived enzyme, after acute colitis induction in mice: Decrease of intestinal inflammation associated with a down regulation of Th1/Th17 responses. PLoS ONE 2018, 13, e0209681. [Google Scholar] [CrossRef] [PubMed]
- Wangchuk, P.; Shepherd, C.; Constantinoiu, C.; Ryan, R.Y.M.; Kouremenos, K.A.; Becker, L.; Jones, L.; Buitrago, G.; Giacomin, P.; Wilson, D.; et al. Hookworm-Derived Metabolites Suppress Pathology in a Mouse Model of Colitis and Inhibit Secretion of Key Inflammatory Cytokines in Primary Human Leukocytes. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.R.; Jeong, Y.I.; Kim, H.J.; Shin, H.E.; Lee, S.E.; Ju, J.W.; Sohn, W.M.; Cho, S.H. Metagonimus miyatai ameliorates dextran sodium sulfate-induced colitis in mice. Parasitol. Int. 2020, 74, 101924. [Google Scholar] [CrossRef] [PubMed]
- Taghipour, N.; Mosaffa, N.; Aghdaei, H.A.; Rostami-Nejad, M.; Weinstock, J.V.; Shahnavaz, S.; Zali, M.R. Immunomodulatory effect of Syphacia obvelata in treatment of experimental DSS-induced colitis in mouse model. Sci. Rep. 2019, 9, 19127. [Google Scholar] [CrossRef] [Green Version]
- Sipahi, A.M.; Baptista, D.M. Helminths as an alternative therapy for intestinal diseases. World J. Gastroenterol. 2017, 23, 6009–6015. [Google Scholar] [CrossRef]
- Koloski, N.A.; Bret, L.; Radford-Smith, G. Hygiene hypothesis in inflammatory bowel disease: A critical review of the literature. World J. Gastroenterol. 2008, 14, 165–173. [Google Scholar] [CrossRef]
- Maruszewska-Cheruiyot, M.; Donskow-Lysoniewska, K.; Doligalska, M. Helminth Therapy: Advances in the use of Parasitic Worms Against Inflammatory Bowel Diseases and its Challenges. Helminthologia 2018, 55, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Muzes, G.; Molnar, B.; Tulassay, Z.; Sipos, F. Changes of the cytokine profile in inflammatory bowel diseases. World J. Gastroenterol. 2012, 18, 5848–5861. [Google Scholar] [CrossRef] [Green Version]
- Boden, E.K.; Lord, J.D. CD4 T Cells in IBD: Crossing the Line? Dig. Dis. Sci. 2017, 62, 2208–2210. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, Z.H.; Bogdanovski, D.A.; Barratt-Stopper, P.; Paglinco, S.R.; Antonioli, L.; Rolandelli, R.H. Crohn’s Disease and Ulcerative Colitis Show Unique Cytokine Profiles. Cureus 2017, 9, e1177. [Google Scholar] [CrossRef] [Green Version]
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; de la Motte, C.; Strong, S.A.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. [Google Scholar] [PubMed]
- Giuffrida, P.; Caprioli, F.; Facciotti, F.; Di Sabatino, A. The role of interleukin-13 in chronic inflammatory intestinal disorders. Autoimmun. Rev. 2019, 18, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Biancheri, P.; Di Sabatino, A.; Ammoscato, F.; Facciotti, F.; Caprioli, F.; Curciarello, R.; Hoque, S.S.; Ghanbari, A.; Joe-Njoku, I.; Giuffrida, P.; et al. Absence of a role for interleukin-13 in inflammatory bowel disease. Eur. J. Immunol. 2014, 44, 370–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoving, J.C. Targeting IL-13 as a Host-Directed Therapy Against Ulcerative Colitis. Front. Cell Infect. Microbiol. 2018, 8, 395. [Google Scholar] [CrossRef]
- Kasaian, M.T.; Page, K.M.; Fish, S.; Brennan, A.; Cook, T.A.; Moreira, K.; Zhang, M.; Jesson, M.; Marquette, K.; Agostinelli, R.; et al. Therapeutic activity of an interleukin-4/interleukin-13 dual antagonist on oxazolone-induced colitis in mice. Immunology 2014, 143, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Lv, X.; Zhu, S.; Li, T.; Cheng, H.; Chen, J. Critical Roles of Balanced Innate Lymphoid Cell Subsets in Intestinal Homeostasis, Chronic Inflammation, and Cancer. J. Immunol. Res. 2019, 2019, 1325181. [Google Scholar] [CrossRef]
- Karmele, E.P.; Pasricha, T.S.; Ramalingam, T.R.; Thompson, R.W.; Gieseck, R.L., 3rd; Knilans, K.J.; Hegen, M.; Farmer, M.; Jin, F.; Kleinman, A.; et al. Anti-IL-13Ralpha2 therapy promotes recovery in a murine model of inflammatory bowel disease. Mucosal Immunol. 2019, 12, 1174–1186. [Google Scholar] [CrossRef] [Green Version]
- Okamura, M.; Yoh, K.; Ojima, M.; Morito, N.; Takahashi, S. Overexpression of GATA-3 in T cells accelerates dextran sulfate sodium-induced colitis. Exp. Anim. 2014, 63, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Kaser, A. Failure of interleukin 13 blockade in ulcerative colitis. Gut 2015, 64, 857–858. [Google Scholar] [CrossRef]
- Imam, T.; Park, S.; Kaplan, M.H.; Olson, M.R. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front. Immunol. 2018, 9, 1212. [Google Scholar] [CrossRef]
- Ohtani, K.; Ohtsuka, Y.; Ikuse, T.; Baba, Y.; Yamakawa, Y.; Aoyagi, Y.; Fujii, T.; Kudo, T.; Nagata, S.; Shimizu, T. Increased mucosal expression of GATA-3 and STAT-4 in pediatric ulcerative colitis. Pediatr. Int. 2010, 52, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Lin, Y.H.; Bi, L.H.; Wang, J.D.; Bai, Y.; Liu, S.D. Effects of interleukin-4 or interleukin-10 gene therapy on trinitrobenzenesulfonic acid-induced murine colitis. BMC Gastroenterol. 2013, 13, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satitsuksanoa, P.; Jansen, K.; Globinska, A.; van de Veen, W.; Akdis, M. Regulatory Immune Mechanisms in Tolerance to Food Allergy. Front. Immunol. 2018, 9, 2939. [Google Scholar] [CrossRef] [PubMed]
- Li, L.J.; Zeng, L.; Li, X.X.; Mo, L.H.; Geng, X.R.; Zheng, P.Y.; Liu, Z.G.; Feng, B.S.; Yang, P.C. Induction of colitis in mice with food allergen-specific immune response. Sci. Rep. 2016, 6, 32765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlyar, D.S.; Shum, M.; Hsieh, J.; Blonski, W.; Greenwald, D.A. Non-pulmonary allergic diseases and inflammatory bowel disease: A qualitative review. World J. Gastroenterol. 2014, 20, 11023–11032. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Liu, F.; Zhou, G.; Sun, M.; Ai, F.; Liu, Z. Food-specific IgGs Are Highly Increased in the Sera of Patients with Inflammatory Bowel Disease and Are Clinically Relevant to the Pathogenesis. Int. Med. 2018, 57, 2787–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, K.B.; Whitman, L.; Sullivan, C.; Gause, W.C.; Urban, J.F., Jr.; Katona, I.M.; Finkelman, F.D.; Shea-Donohue, T. Role of STAT6 and mast cells in IL-4- and IL-13-induced alterations in murine intestinal epithelial cell function. J. Immunol. 2002, 169, 4417–4422. [Google Scholar] [CrossRef] [Green Version]
- Dames, P.; Bergann, T.; Fromm, A.; Bucker, R.; Barmeyer, C.; Krug, S.M.; Fromm, M.; Schulzke, J.D. Interleukin-13 affects the epithelial sodium channel in the intestine by coordinated modulation of STAT6 and p38 MAPK activity. J. Physiol. 2015, 593, 5269–5282. [Google Scholar] [CrossRef] [Green Version]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Burgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Fukui, H. Increased Intestinal Permeability and Decreased Barrier Function: Does It Really Influence the Risk of Inflammation? Inflamm. Intest. Dis. 2016, 1, 135–145. [Google Scholar] [CrossRef]
- Michielan, A.; D’Inca, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediators Inflamm 2015, 2015, 628157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popp, V.; Gerlach, K.; Mott, S.; Turowska, A.; Garn, H.; Atreya, R.; Lehr, H.A.; Ho, I.C.; Renz, H.; Weigmann, B.; et al. Rectal Delivery of a DNAzyme That Specifically Blocks the Transcription Factor GATA3 and Reduces Colitis in Mice. Gastroenterology 2017, 152, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.P.; Goswami, R. A Decade of Th9 Cells: Role of Th9 Cells in Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shohan, M.; Sabzevary-Ghahfarokhi, M.; Bagheri, N.; Shirzad, H.; Rahimian, G.; Soltani, A.; Ghatreh-Samani, M.; Deris, F.; Tahmasbi, K.; Shahverdi, E.; et al. Intensified Th9 Response is Associated with the Immunopathogenesis of Active Ulcerative Colitis. Immunol. Invest. 2018, 47, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat. Immunol. 2014, 15, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Forkel, M.; van Tol, S.; Hoog, C.; Michaelsson, J.; Almer, S.; Mjosberg, J. Distinct Alterations in the Composition of Mucosal Innate Lymphoid Cells in Newly Diagnosed and Established Crohn’s Disease and Ulcerative Colitis. J. Crohns. Colitis. 2019, 13, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Arancibia-Carcamo, C.V. Innate Lymphoid Cells in Intestinal Inflammation. Front. Immunol. 2017, 8, 1296. [Google Scholar] [CrossRef]
- Lin, Y.; Li, B.; Yang, X.; Liu, T.; Shi, T.; Deng, B.; Zhang, Y.; Jia, L.; Jiang, Z.; He, R. Non-hematopoietic STAT6 induces epithelial tight junction dysfunction and promotes intestinal inflammation and tumorigenesis. Mucosal Immunol. 2019, 12, 1304–1315. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Hunter, M.M.; Wang, A.; Parhar, K.S.; Johnston, M.J.; Van Rooijen, N.; Beck, P.L.; McKay, D.M. In vitro-derived alternatively activated macrophages reduce colonic inflammation in mice. Gastroenterology 2010, 138, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Leung, G.; Wang, A.; Fernando, M.; Phan, V.C.; McKay, D.M. Bone marrow-derived alternatively activated macrophages reduce colitis without promoting fibrosis: Participation of IL-10. Am. J. Physiol. Gastrointest Liver Physiol. 2013, 304, G781–G792. [Google Scholar] [CrossRef] [PubMed]
- Enderlin Vaz da Silva, Z.; Lehr, H.A.; Velin, D. In vitro and in vivo repair activities of undifferentiated and classically and alternatively activated macrophages. Pathobiology 2014, 81, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARgamma and the Innate Immune System Mediate the Resolution of Inflammation. PPAR Res. 2015, 2015, 549691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Li, W.; Wang, S.; Zhang, P.; Wang, Q.; Xiao, J.; Zhang, C.; Zheng, X.; Xu, X.; Xue, S.; et al. YAP Aggravates Inflammatory Bowel Disease by Regulating M1/M2 Macrophage Polarization and Gut Microbial Homeostasis. Cell Rep. 2019, 27, 1176–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhou, J.; Feng, Y.; Chen, L.; Zhang, L.; Yang, F.; Zha, H.; Wang, X.; Han, X.; Shu, C.; et al. Control of Intestinal Inflammation, Colitis-Associated Tumorigenesis, and Macrophage Polarization by Fibrinogen-Like Protein 2. Front. Immunol. 2018, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Liao, Y.; Wang, L.; He, P.; Hu, Y.; Yuan, D.; Wu, Z.; Sun, X. Exosomes Derived From M2b Macrophages Attenuate DSS-Induced Colitis. Front. Immunol. 2019, 10, 2346. [Google Scholar] [CrossRef] [Green Version]
- Isidro, R.A.; Appleyard, C.B. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am. J. Physiol. Gastrointest Liver Physiol. 2016, 311, G59–G73. [Google Scholar] [CrossRef]
- Wakahara, K.; Baba, N.; Van, V.Q.; Begin, P.; Rubio, M.; Ferraro, P.; Panzini, B.; Wassef, R.; Lahaie, R.; Caussignac, Y.; et al. Human basophils interact with memory T cells to augment Th17 responses. Blood 2012, 120, 4761–4771. [Google Scholar] [CrossRef] [Green Version]
- Sarfati, M.; Wakahara, K.; Chapuy, L.; Delespesse, G. Mutual Interaction of Basophils and T Cells in Chronic Inflammatory Diseases. Front. Immunol. 2015, 6, 399. [Google Scholar] [CrossRef] [Green Version]
- Gomez, M.R.; Talke, Y.; Hofmann, C.; Ketelsen, I.; Hermann, F.; Reich, B.; Goebel, N.; Schmidbauer, K.; Dunger, N.; Bruhl, H.; et al. Basophils control T-cell responses and limit disease activity in experimental murine colitis. Mucosal Immunol. 2014, 7, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egawa, M.; Mukai, K.; Yoshikawa, S.; Iki, M.; Mukaida, N.; Kawano, Y.; Minegishi, Y.; Karasuyama, H. Inflammatory monocytes recruited to allergic skin acquire an anti-inflammatory M2 phenotype via basophil-derived interleukin-4. Immunity 2013, 38, 570–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loktionov, A. Eosinophils in the gastrointestinal tract and their role in the pathogenesis of major colorectal disorders. World J. Gastroenterol. 2019, 25, 3503–3526. [Google Scholar] [CrossRef] [PubMed]
- Abedin, N.; Seemann, T.; Kleinfeld, S.; Ruehrup, J.; Roseler, S.; Trautwein, C.; Streetz, K.; Sellge, G. Fecal Eosinophil Cationic Protein Is a Diagnostic and Predictive Biomarker in Young Adults with Inflammatory Bowel Disease. J. Clin. Med. 2019, 8, 2025. [Google Scholar] [CrossRef] [Green Version]
- Amcoff, K.; Cao, Y.; Zhulina, Y.; Lampinen, M.; Halfvarson, J.; Carlson, M. Prognostic significance of faecal eosinophil granule proteins in inflammatory bowel disease. Scand. J. Gastroenterol. 2019, 54, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Katinios, G.; Casado-Bedmar, M.; Walter, S.A.; Vicario, M.; Gonzalez-Castro, A.M.; Bednarska, O.; Soderholm, J.D.; Hjortswang, H.; Keita, A.V. Increased Colonic Epithelial Permeability and Mucosal Eosinophilia in Ulcerative Colitis in Remission Compared With Irritable Bowel Syndrome and Health. Inflamm. Bowel Dis. 2020. [Google Scholar] [CrossRef]
- Alhmoud, T.; Gremida, A.; Colom Steele, D.; Fallahi, I.; Tuqan, W.; Nandy, N.; Ismail, M.; Aburajab Altamimi, B.; Xiong, M.J.; Kerwin, A.; et al. Outcomes of inflammatory bowel disease in patients with eosinophil-predominant colonic inflammation. BMJ Open Gastroenterol. 2020, 7, e000373. [Google Scholar] [CrossRef]
- Zammit, S.C.; Cachia, M.; Sapiano, K.; Gauci, J.; Montefort, S.; Ellul, P. Eosinophilic gastrointestinal disorder: Is it what it seems to be? Ann. Gastroenterol 2018, 31, 475–479. [Google Scholar] [CrossRef]
- Kurashima, Y.; Amiya, T.; Nochi, T.; Fujisawa, K.; Haraguchi, T.; Iba, H.; Tsutsui, H.; Sato, S.; Nakajima, S.; Iijima, H.; et al. Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat. Commun. 2012, 3, 1034. [Google Scholar] [CrossRef]
- De Zuani, M.; Dal Secco, C.; Frossi, B. Mast cells at the crossroads of microbiota and IBD. Eur. J. Immunol 2018, 48, 1929–1937. [Google Scholar] [CrossRef] [Green Version]
- Boeckxstaens, G. Mast cells and inflammatory bowel disease. Curr Opin. Pharmacol. 2015, 25, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.N.; Lee, O.Y. the role of mast cells in irritable bowel syndrome. Gastroenterol. Res. Pract. 2016, 2016, 2031480. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C. mast cells in irritable bowel syndrome and ulcerative colitis: Function not numbers is what makes all the difference. Dig. Dis. Sci. 2014, 59, 897–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florea, A.; Sangare, L.; Lowe, K. A multinational assessment of gastric, esophageal, and colorectal cancer burden: A report of disease incidence, prevalence, and fatality. J. Gastrointest Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.M.; Wei, C.; Ensor, J.E.; Smolenski, D.J.; Amos, C.I.; Levin, B.; Berry, D.A. Meta-analyses of colorectal cancer risk factors. Cancer Causes Control. 2013, 24, 1207–1222. [Google Scholar] [CrossRef]
- Biancone, L.; Armuzzi, A.; Scribano, M.L.; Castiglione, F.; D’Inca, R.; Orlando, A.; Papi, C.; Daperno, M.; Vecchi, M.; Riegler, G.; et al. Cancer Risk in Inflammatory Bowel Disease: A 6-Year Prospective Multicenter Nested Case-Control IG-IBD Study. Inflamm. Bowel Dis. 2020, 26, 450–459. [Google Scholar] [CrossRef]
- Rhodes, J.M.; Campbell, B.J. Inflammation and colorectal cancer: IBD-associated and sporadic cancer compared. Trends Mol. Med. 2002, 8, 10–16. [Google Scholar] [CrossRef]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, Y.; Watanabe, M. The Th1, Th2, and Th17 Paradigm in Inflammatory Bowel Disease. In Crohn’s Disease and Ulcerative Colitis; Baumgart, D., Ed.; Springer: Boston, MA, USA, 2012. [Google Scholar]
- Markman, J.L.; Shiao, S.L. Impact of the immune system and immunotherapy in colorectal cancer. J. Gastrointest Oncol. 2015, 6, 208–223. [Google Scholar] [CrossRef]
- Shamoun, L.; Skarstedt, M.; Andersson, R.E.; Wagsater, D.; Dimberg, J. Association study on IL-4, IL-4Ralpha and IL-13 genetic polymorphisms in Swedish patients with colorectal cancer. Clin. Chim. Acta 2018, 487, 101–106. [Google Scholar] [CrossRef]
- Jayakumar, A.; Bothwell, A.L.M. Stat6 Promotes Intestinal Tumorigenesis in a Mouse Model of Adenomatous Polyposis by Expansion of MDSCs and Inhibition of Cytotoxic CD8 Response. Neoplasia 2017, 19, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Barderas, R.; Bartolome, R.A.; Fernandez-Acenero, M.J.; Torres, S.; Casal, J.I. High expression of IL-13 receptor alpha2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 2012, 72, 2780–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formentini, A.; Braun, P.; Fricke, H.; Link, K.H.; Henne-Bruns, D.; Kornmann, M. Expression of interleukin-4 and interleukin-13 and their receptors in colorectal cancer. Int. J. Colorectal. Dis. 2012, 27, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Wasmer, M.H.; Rau, T.T.; Krebs, P. Cytokine-Induced Modulation of Colorectal Cancer. Front. Oncol. 2016, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Matsui, S.; Okabayashi, K.; Tsuruta, M.; Shigeta, K.; Seishima, R.; Ishida, T.; Kondo, T.; Suzuki, Y.; Hasegawa, H.; Shimoda, M.; et al. Interleukin-13 and its signaling pathway is associated with obesity-related colorectal tumorigenesis. Cancer Sci. 2019, 110, 2156–2165. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, J.; Liu, H.; Wan, L.; Zhang, H.; Huang, Q.; Xu, E.; Lai, M. IL-13/STAT6 signaling plays a critical role in the epithelial-mesenchymal transition of colorectal cancer cells. Oncotarget 2016, 7, 61183–61198. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Tao, W.; Su, Z. Propofol prevents IL-13-induced epithelial-mesenchymal transition in human colorectal cancer cells. Cell Biol. Int. 2018, 42, 985–993. [Google Scholar] [CrossRef]
- Bartolome, R.A.; Jaen, M.; Casal, J.I. An IL13Ralpha2 peptide exhibits therapeutic activity against metastatic colorectal cancer. Br. J. Cancer 2018, 119, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Bartolome, R.A.; Garcia-Palmero, I.; Torres, S.; Lopez-Lucendo, M.; Balyasnikova, I.V.; Casal, J.I. IL13 Receptor alpha2 Signaling Requires a Scaffold Protein, FAM120A, to Activate the FAK and PI3K Pathways in Colon Cancer Metastasis. Cancer Res. 2015, 75, 2434–2444. [Google Scholar] [CrossRef] [Green Version]
- Gharib, A.F.; Shalaby, S.M.; Raafat, N.; Fawzy, W.M.S.; Abdel Hakim, N.H. Assessment of neutralizing interleukin-4 effect on CD133 gene expression in colon cancer cell line. Cytokine 2017, 97, 66–72. [Google Scholar] [CrossRef]
- Liu, H.; Antony, S.; Roy, K.; Juhasz, A.; Wu, Y.; Lu, J.; Meitzler, J.L.; Jiang, G.; Polley, E.; Doroshow, J.H. Interleukin-4 and interleukin-13 increase NADPH oxidase 1-related proliferation of human colon cancer cells. Oncotarget 2017, 8, 38113–38135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Konate, M.M.; Lu, J.; Makhlouf, H.; Chuaqui, R.; Antony, S.; Meitzler, J.L.; Difilippantonio, M.J.; Liu, H.; Juhasz, A.; et al. IL-4 and IL-17A Cooperatively Promote Hydrogen Peroxide Production, Oxidative DNA Damage, and Upregulation of Dual Oxidase 2 in Human Colon and Pancreatic Cancer Cells. J. Immunol. 2019, 203, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Atreya, I.; Kindermann, M.; Wirtz, S. Innate lymphoid cells in intestinal cancer development. Semin Immunol 2019, 41, 101267. [Google Scholar] [CrossRef] [PubMed]
- Pastille, E.; Wasmer, M.H.; Adamczyk, A.; Vu, V.P.; Mager, L.F.; Phuong, N.N.T.; Palmieri, V.; Simillion, C.; Hansen, W.; Kasper, S.; et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. 2019, 12, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Yuan, A.; Pang, Z.; Zheng, W.; Li, Z.; Goll, R. Contribution of IL-33 to the Pathogenesis of Colorectal Cancer. Front. Oncol. 2018, 8, 561. [Google Scholar] [CrossRef]
- Zhou, Y.; Ji, Y.; Wang, H.; Zhang, H.; Zhou, H. IL-33 Promotes the Development of Colorectal Cancer Through Inducing Tumor-Infiltrating ST2L(+) Regulatory T Cells in Mice. Technol. Cancer Res. Treat. 2018, 17, 1533033818780091. [Google Scholar] [CrossRef] [Green Version]
- Long, A.; Dominguez, D.; Qin, L.; Chen, S.; Fan, J.; Zhang, M.; Fang, D.; Zhang, Y.; Kuzel, T.M.; Zhang, B. Type 2 Innate Lymphoid Cells Impede IL-33-Mediated Tumor Suppression. J. Immunol. 2018, 201, 3456–3464. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Shi, J.; Qi, S.; Zhang, J.; Peng, D.; Chen, Z.; Wang, G.; Wang, Z.; Wang, L. IL-33 facilitates proliferation of colorectal cancer dependent on COX2/PGE2. J. Exp. Clin. Cancer Res. 2018, 37, 196. [Google Scholar] [CrossRef]
- Chen, X.; Lu, K.; Timko, N.J.; Weir, D.M.; Zhu, Z.; Qin, C.; Mann, J.D.; Bai, Q.; Xiao, H.; Nicholl, M.B.; et al. IL-33 notably inhibits the growth of colon cancer cells. Oncol. Lett. 2018, 16, 769–774. [Google Scholar] [CrossRef] [Green Version]
- Niccolai, E.; Ricci, F.; Russo, E.; Nannini, G.; Emmi, G.; Taddei, A.; Ringressi, M.N.; Melli, F.; Miloeva, M.; Cianchi, F.; et al. The Different Functional Distribution of "Not Effector" T Cells (Treg/Tnull) in Colorectal Cancer. Front. Immunol. 2017, 8, 1900. [Google Scholar] [CrossRef] [Green Version]
- Hoang, T.H.; Zhao, Y.; Lam, Y.; Piekos, S.; Han, Y.C.; Reilly, C.; Joshi, P.; Hong, S.H.; Sung, C.O.; Giardina, C.; et al. BioTarget: A Computational Framework Identifying Cancer Type Specific Transcriptional Targets of Immune Response Pathways. Sci. Rep. 2019, 9, 9029. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Pallone, F.; Monteleone, G.; Fantini, M.C. Intestinal inflammation and colorectal cancer: A double-edged sword? World J. Gastroenterol. 2011, 17, 3092–3100. [Google Scholar] [CrossRef]
- Cui, G. TH9, TH17, and TH22 Cell Subsets and Their Main Cytokine Products in the Pathogenesis of Colorectal Cancer. Front. Oncol. 2019, 9, 1002. [Google Scholar] [CrossRef] [PubMed]
- Loyon, R.; Jary, M.; Salome, B.; Gomez-Cadena, A.; Galaine, J.; Kroemer, M.; Romero, P.; Trabanelli, S.; Adotevi, O.; Borg, C.; et al. Peripheral Innate Lymphoid Cells Are Increased in First Line Metastatic Colorectal Carcinoma Patients: A Negative Correlation With Th1 Immune Responses. Front. Immunol. 2019, 10, 2121. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Ghiringhelli, F. Deciphering the Roles of Innate Lymphoid Cells in Cancer. Front. Immunol. 2019, 10, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabo, I.; Olsson, H.; Elkarim, R.; Sun, X.F.; Svanvik, J. Macrophage Infiltration in Tumor Stroma is Related to Tumor Cell Expression of CD163 in Colorectal Cancer. Cancer Microenviron. 2014, 7, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Cortese, N.; Soldani, C.; Franceschini, B.; Barbagallo, M.; Marchesi, F.; Torzilli, G.; Donadon, M. Macrophages in Colorectal Cancer Liver Metastases. Cancers 2019, 11, 633. [Google Scholar] [CrossRef] [Green Version]
- Yahaya, M.A.F.; Lila, M.A.M.; Ismail, S.; Zainol, M.; Afizan, N. Tumour-Associated Macrophages (TAMs) in Colon Cancer and How to Reeducate Them. J. Immunol. Res. 2019, 2019, 2368249. [Google Scholar] [CrossRef]
- He, Z.; Chen, L.; Souto, F.O.; Canasto-Chibuque, C.; Bongers, G.; Deshpande, M.; Harpaz, N.; Ko, H.M.; Kelley, K.; Furtado, G.C.; et al. Epithelial-derived IL-33 promotes intestinal tumorigenesis in Apc (Min/+) mice. Sci. Rep. 2017, 7, 5520. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Ge, X.; Zhao, W.; Xue, L.; Dai, C.; Lin, F.; Peng, W. PIPKIgamma Regulates CCL2 Expression in Colorectal Cancer by Activating AKT-STAT3 Signaling. J. Immunol. Res. 2019, 2019, 3690561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Huang, W.; Ge, X.; Xue, L.; Zhao, W.; Xue, J. Type Igamma phosphatidylinositol phosphate kinase promotes tumor growth by facilitating Warburg effect in colorectal cancer. EBioMedicine 2019, 44, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Zhang, Z.; Yao, H.; Shen, L. Tim-4 promotes the growth of colorectal cancer by activating angiogenesis and recruiting tumor-associated macrophages via the PI3K/AKT/mTOR signaling pathway. Cancer Lett. 2018, 436, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Huang, T.W.; Hsieh, Y.T.; Wang, Y.F.; Yen, C.C.; Lee, G.L.; Yeh, C.C.; Peng, Y.J.; Kuo, Y.Y.; Wen, H.T.; et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Chen, S.; Ouyang, M.; Li, F.; Chen, L.; Yang, J. Colon Cancer Cell Secretes EGF to Promote M2 Polarization of TAM Through EGFR/PI3K/AKT/mTOR Pathway. Technol. Cancer Res. Treat. 2019, 18, 1533033819849068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019, 10, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.; Chen, B.; Yang, Z. The Role of Tumor-Associated Macrophages in Colorectal Carcinoma Progression. Cell Physiol. Biochem. 2018, 45, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.L.; Rios, E.; Duraes, C.; Ribeiro, R.; Machado, J.C.; Mantovani, A.; Barbosa, M.A.; Carneiro, F.; Oliveira, M.J. The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front. Immunol. 2019, 10, 1875. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Ge, X.; Xu, X.; Yu, S.; Wang, J.; Sun, L. Prognostic value and clinicopathological roles of phenotypes of tumour-associated macrophages in colorectal cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 3005–3019. [Google Scholar] [CrossRef]
- Dong, R.; Gong, Y.; Meng, W.; Yuan, M.; Zhu, H.; Ying, M.; He, Q.; Cao, J.; Yang, B. The involvement of M2 macrophage polarization inhibition in fenretinide-mediated chemopreventive effects on colon cancer. Cancer Lett. 2017, 388, 43–53. [Google Scholar] [CrossRef]
- Suarez-Lopez, L.; Sriram, G.; Kong, Y.W.; Morandell, S.; Merrick, K.A.; Hernandez, Y.; Haigis, K.M.; Yaffe, M.B. MK2 contributes to tumor progression by promoting M2 macrophage polarization and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E4236–E4244. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-beta Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Yao, Z.; Wang, J.; Zhang, W.; Yang, Y.; Zhang, Y.; Qu, X.; Zhu, Y.; Zou, J.; Peng, S.; et al. Macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Li, Q.; Egilmez, N.K. IFNbeta-producing CX3CR1(+) macrophages promote T-regulatory cell expansion and tumor growth in the APC(min/+) / Bacteroides fragilis colon cancer model. Oncoimmunology 2019, 8, e1665975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.B.; Zhao, Z.B.; Liu, Q.Z.; Hu, T.D.; Long, J.; Yan, K.; Lian, Z.X. FoxO1 is a regulator of MHC-II expression and anti-tumor effect of tumor-associated macrophages. Oncogene 2018, 37, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wang, J.; Ding, N.; Zhang, Y.; Zhu, Y.; Dong, S.; Wang, X.; Peng, C.; Zhou, C.; Zhou, L.; et al. Inflammation induced by incomplete radiofrequency ablation accelerates tumor progression and hinders PD-1 immunotherapy. Nat. Commun. 2019, 10, 5421. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wei, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Dou, R.; Xiong, B. Elevated CD163(+)/CD68(+) Ratio at Tumor Invasive Front is Closely Associated with Aggressive Phenotype and Poor Prognosis in Colorectal Cancer. Int. J. Biol. Sci. 2019, 15, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 64. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Prizment, A.E.; Vierkant, R.A.; Smyrk, T.C.; Tillmans, L.S.; Lee, J.J.; Sriramarao, P.; Nelson, H.H.; Lynch, C.F.; Thibodeau, S.N.; Church, T.R.; et al. Tumor eosinophil infiltration and improved survival of colorectal cancer patients: Iowa Women’s Health Study. Mod. Pathol. 2016, 29, 516–527. [Google Scholar] [CrossRef]
- Reichman, H.; Itan, M.; Rozenberg, P.; Yarmolovski, T.; Brazowski, E.; Varol, C.; Gluck, N.; Shapira, S.; Arber, N.; Qimron, U.; et al. Activated Eosinophils Exert Antitumorigenic Activities in Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 388–400. [Google Scholar] [CrossRef] [Green Version]
- Hayes, M.D.; Ward, S.; Crawford, G.; Seoane, R.C.; Jackson, W.D.; Kipling, D.; Voehringer, D.; Dunn-Walters, D.; Strid, J. Inflammation-induced IgE promotes epithelial hyperplasia and tumour growth. Elife 2020, 9. [Google Scholar] [CrossRef]
- Ma, W.; Yang, J.; Li, P.; Lu, X.; Cai, J. Association between allergic conditions and colorectal cancer risk/mortality: A meta-analysis of prospective studies. Sci Rep. 2017, 7, 5589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, E.J.; Gapstur, S.M.; Newton, C.C.; Turner, M.C.; Campbell, P.T. Hay Fever and asthma as markers of atopic immune response and risk of colorectal cancer in three large cohort studies. Cancer Epidemiol Biomarkers Prev. 2013, 22, 661–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Talaiti, A.; Ma, Y.; Zhang, Q.; Ma, L.; Zheng, H. Allergies and risk of colorectal cancer: A systematic review and meta-analysis of observational studies. Oncotarget 2017, 8, 14646–14654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prizment, A.E.; Anderson, K.E.; Visvanathan, K.; Folsom, A.R. Inverse association of eosinophil count with colorectal cancer incidence: Atherosclerosis risk in communities study. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 1861–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubowska, K.; Koda, M.; Kisielewski, W.; Kanczuga-Koda, L.; Famulski, W. Prognostic significance of inflammatory cell response in patients with colorectal cancer. Oncol. Lett. 2019, 18, 783–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreone, S.; Spadaro, F.; Buccione, C.; Mancini, J.; Tinari, A.; Sestili, P.; Gambardella, A.R.; Lucarini, V.; Ziccheddu, G.; Parolini, I.; et al. IL-33 Promotes CD11b/CD18-Mediated Adhesion of Eosinophils to Cancer Cells and Synapse-Polarized Degranulation Leading to Tumor Cell Killing. Cancers 2019, 11, 1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Zhang, X.; Wang, G.; Zhou, Y.; Luo, M.; Wang, S.; Hong, C. The impacts of pretreatment circulating eosinophils and basophils on prognosis of stage - colorectal cancer. Asia Pac. J. Clin. Oncol. 2018, 14, e243–e251. [Google Scholar] [CrossRef]
- Wu, J.; Ge, X.X.; Zhu, W.; Zhi, Q.; Xu, M.D.; Duan, W.; Chen, K.; Gong, F.R.; Tao, M.; Shou, L.M.; et al. Values of applying white blood cell counts in the prognostic evaluation of resectable colorectal cancer. Mol. Med. Rep. 2019, 19, 2330–2340. [Google Scholar] [CrossRef] [Green Version]
- Komi, D.E.A.; Redegeld, F.A. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin. Rev. Allergy Immunol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehdawi, L.; Osman, J.; Topi, G.; Sjolander, A. High tumor mast cell density is associated with longer survival of colon cancer patients. Acta Oncol. 2016, 55, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Giusca, S.E.; Caruntu, I.D.; Cimpean, A.M.; Avadanei, R.E.; Balica, A.R.; Jitariu, A.A.; Raica, M. Tryptase-positive and CD117 Positive Mast Cells Correlate with Survival in Patients with Liver Metastasis. Anticancer Res. 2015, 35, 5325–5331. [Google Scholar] [PubMed]
- Xia, Q.; Ding, Y.; Wu, X.J.; Peng, R.Q.; Zhou, Q.; Zeng, J.; Hou, J.H.; Zhang, X.; Zeng, Y.X.; Zhang, X.S.; et al. Mast Cells in Adjacent Normal Colon Mucosa rather than Those in Invasive Margin are Related to Progression of Colon Cancer. Chin. J. Cancer Res. 2011, 23, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, L.; Shi, R.; Liu, X.; Zhang, J.; Zou, Z.; Hao, Z.; Tao, A. Mast Cell Targeted Chimeric Toxin Can Be Developed as an Adjunctive Therapy in Colon Cancer Treatment. Toxins 2016, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Yin, G.; Ma, Y.; Xu, K.; Liu, J.; Li, J. The critical role of mast cell-derived hypoxia-inducible factor-1alpha in regulating mast cell function. J. Pharm. Pharmacol. 2016, 68, 1409–1416. [Google Scholar] [CrossRef]
- Hu, G.; Wang, S.; Cheng, P. Tumor-infiltrating tryptase(+) mast cells predict unfavorable clinical outcome in solid tumors. Int. J. Cancer 2018, 142, 813–821. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Feng, Q.; Zheng, P.; Yang, L.; Zhu, D.; Chang, W.; Ji, M.; He, G.; Xu, J. Low tumor infiltrating mast cell density confers prognostic benefit and reflects immunoactivation in colorectal cancer. Int. J. Cancer 2018, 143, 2271–2280. [Google Scholar] [CrossRef] [Green Version]
- Ko, E.A.; Sanders, K.M.; Zhou, T. A transcriptomic insight into the impacts of mast cells in lung, breast, and colon cancers. Oncoimmunology 2017, 6, e1360457. [Google Scholar] [CrossRef]
- Iwanaga, K.; Nakamura, T.; Maeda, S.; Aritake, K.; Hori, M.; Urade, Y.; Ozaki, H.; Murata, T. Mast cell-derived prostaglandin D2 inhibits colitis and colitis-associated colon cancer in mice. Cancer Res. 2014, 74, 3011–3019. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Churchill, M.J.; Nagar, K.K.; Tailor, Y.H.; Chu, T.; Rush, B.S.; Jiang, Z.; Wang, E.B.; Renz, B.W.; Wang, H.; et al. IL-17 producing mast cells promote the expansion of myeloid-derived suppressor cells in a mouse allergy model of colorectal cancer. Oncotarget 2015, 6, 32966–32979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustino-Rocha, A.I.; Ferreira, R.; Gama, A.; Oliveira, P.A.; Ginja, M. Antihistamines as promising drugs in cancer therapy. Life Sci. 2017, 172, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yi, H.G.; Wu, Z.; Han, W.; Chen, K.; Zang, M.; Wang, D.; Zhao, X.; Wang, H.; Qu, C. Activation of mucosal mast cells promotes inflammation-related colon cancer development through recruiting and modulating inflammatory CD11b(+)Gr1(+) cells. Cancer Lett. 2015, 364, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danelli, L.; Frossi, B.; Gri, G.; Mion, F.; Guarnotta, C.; Bongiovanni, L.; Tripodo, C.; Mariuzzi, L.; Marzinotto, S.; Rigoni, A.; et al. Mast cells boost myeloid-derived suppressor cell activity and contribute to the development of tumor-favoring microenvironment. Cancer Immunol. Res. 2015, 3, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Blokhuis, B.; Derks, Y.; Kumari, S.; Garssen, J.; Redegeld, F. Human mast cells promote colon cancer growth via bidirectional crosstalk: Studies in 2D and 3D coculture models. Oncoimmunology 2018, 7, e1504729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadalla, A.; Lima, M.M.; Tsai, F.; Osman, A.; Singh, M.P.; Linden, D.R.; Dennis, K.L.; Haeryfar, S.M.M.; Gurish, M.F.; Gounari, F.; et al. Cell Intrinsic Deregulated ss-Catenin Signaling Promotes Expansion of Bone Marrow Derived Connective Tissue Type Mast Cells, Systemic Inflammation, and Colon Cancer. Front. Immunol. 2019, 10, 2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigoni, A.; Bongiovanni, L.; Burocchi, A.; Sangaletti, S.; Danelli, L.; Guarnotta, C.; Lewis, A.; Rizzo, A.; Silver, A.R.; Tripodo, C.; et al. Mast Cells Infiltrating Inflamed or Transformed Gut Alternatively Sustain Mucosal Healing or Tumor Growth. Cancer Res. 2015, 75, 3760–3770. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamez-Belmonte, R.; Erkert, L.; Wirtz, S.; Becker, C. The Regulation of Intestinal Inflammation and Cancer Development by Type 2 Immune Responses. Int. J. Mol. Sci. 2020, 21, 9772. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249772
Gamez-Belmonte R, Erkert L, Wirtz S, Becker C. The Regulation of Intestinal Inflammation and Cancer Development by Type 2 Immune Responses. International Journal of Molecular Sciences. 2020; 21(24):9772. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249772
Chicago/Turabian StyleGamez-Belmonte, Reyes, Lena Erkert, Stefan Wirtz, and Christoph Becker. 2020. "The Regulation of Intestinal Inflammation and Cancer Development by Type 2 Immune Responses" International Journal of Molecular Sciences 21, no. 24: 9772. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249772