1. Introduction
Soybean (
Glycine max L.) is one of the most economically important crops, being a rich source of both edible oil and protein, and can fix atmospheric nitrogen through a symbiotic association with microorganisms in the soil, and are used as a model plant for legume research [
1]. However, over the past five decades, a continuous decline in soybean production in China has been recorded [
2]. Besides, annually, China imports more than 80% of soybeans and its products to meet its domestic demands; hence, there is an immediate need to increase the domestic production of soybean to make the country self-sufficient [
2]. Yield-related traits are the key target of plant breeders to improve soybean yield/production. In this regard, traits related to seed size and shape are the crucial parameters determining seed-weight and yield in soybean [
3,
4]. In soybean, seed size traits such as length (SL); width (SW) and thickness (ST); and seed shape traits, viz., length-to-width (SLW), length-to-thickness (SLT), and width-to-thickness (SWT) ratios determine seed appearance, quality, and yield in soybeans [
5]. Seed size is also a vital fitness trait in flowering plants and plays a crucial role in adaptation to a particular environment [
6]. However, seed size and shape are complex quantitative traits governed by polygenes and highly influenced by the environment (E) and genotype × environment (G × E) interactions [
7,
8]. Specific soy-based food products made from soybean are also determined mainly by seed size and shape [
9,
10]. For example, for the production of fermented soybeans (natto) and sprouts, small-seeded cultivars are suitable, while for soymilk, green soybeans (edamame), boiled soybeans (nimame), and soybean curd (tofu), large-seeded varieties are used [
11,
12,
13]. Additionally, these traits influence the germination ability and seedling vigor, and that, in turn, plays an essential role in determining the competitive strength of the seedlings for light, nutrient resources, and stress tolerance [
14,
15,
16].
Quantitative trait loci (QTL) analysis has proved as a powerful technique to elucidate complex trait architecture. Over the past two decades, recent advances in marker technology and statistical methods have allowed the identification of many QTLs related to seed size and shape traits. The USDA Soybean Genome Database (SoyBase,
http://www.soybase.org) presently document more than 400 QTLs for seed size and shape, and the majority of them are not confirmed (
http://www.soybase.org). The previous studies used mostly low-resolution and low-density molecular markers such as simple sequence repeats (SSRs) that often result in larger confidence intervals and make the use of these QTLs less effective in crop improvement [
3,
5,
17,
18]. For example, Mian et al. [
19] reported 16 QTLs for seed size and shape on 12 different chromosomes of soybean. Hoeck et al. [
20] identified 27 QTLs associated with seed size distributed on 16 soybean chromosomes, and Li et al. [
21] detected three QTLs for SL on Chr07, Chr13, and Chr16. Lü et al. [
18] identified 19 main-effect QTLs (M-QTLs) and three epistatic-effect QTLs (E-QTLs) for SL on eight chromosomes. Xie et al. [
22] finely mapped QTLs for soybean seed size traits on Chr06 in the recombinant inbred line (RIL) population derived from a cross between Lishuizhongzihuang and Nannong493-1. Likewise, Che et al. [
17] identified 16 QTLs for seed shape, distributed on seven linkage groups in soybeans by using the RIL population. Hu et al. [
7] mapped 10 QTLs for seed shape on six chromosomes in soybeans. However, only a few yield-related stable QTLs have been identified in different genetic backgrounds and environments [
23]. Hence, it is vital to identify and validate QTLs in multiple backgrounds and environments for their potential use in marker-assisted breeding (MAB). Lastly, the earlier studies mostly focused on the identification of main-effect QTLs for seed size/shape in soybean; however, minimal efforts have been made to understand complex genetic interaction effects, such as epistasis and environment effects [
24,
25,
26].
The inheritance of quantitative traits varies from simple to complex; however, the phenotypic variation of most quantitative traits is complex, governed by many factors [
27]. In addition to main-effect QTLs, phenotypic variation (PV) of complex traits is also governed by QTL by QTL (epistatic) and QTL by environment (QTL × E) interactions, which contribute significantly to complex trait variations [
28]. By considering these QTL interactions in the QTL mapping model of complex traits will lead to increased precision of QTL mapping [
29]. Therefore, these factors cannot be considered only as the main obstacles to dissect the genetic architecture of complex traits, but they also affect the accuracy of breeding value estimation, and thus, hinder the efficiency of breeding programs. Hence, it is imperative to consider these factors while dissecting the genetic basis of complex traits and their uses in improving plant performance. In recent years, epistatic and QTL × E interaction effects are under consideration in several crop species, including soybeans, for QTL mapping [
30]. Therefore, extensive efforts are required to study such QTL interaction effects for their effective exploitation in soybean breeding.
Development of high-density genetic maps, and their use in the detection of QTLs/genes, have allowed a detailed and broader understanding of the genetic basis underlying complex quantitative traits. Furthermore, the analysis of genes has partitioned the related traits into individual Mendelian factors [
31]. Nevertheless, limited reports are targeting the mapping of QTLs related to seed size and shape based on the high-density map in different genetic backgrounds. Besides, to mine candidate genes for seed size and shape in soybeans, negligible efforts were made. By keeping the above in view, the present study has used a high-density linkage map of two RIL populations, viz., ZY and K3N, evaluated in multiple environments to identify main and epistatic-effect QTLs, as well as their interactions with the environment, to mine candidate genes for seed size and shape in soybeans. These results will be helpful in MAB for developing soybean varieties with improved yield and quality, as well as to clone underlying genes for seed size and shape in soybean.
3. Discussion
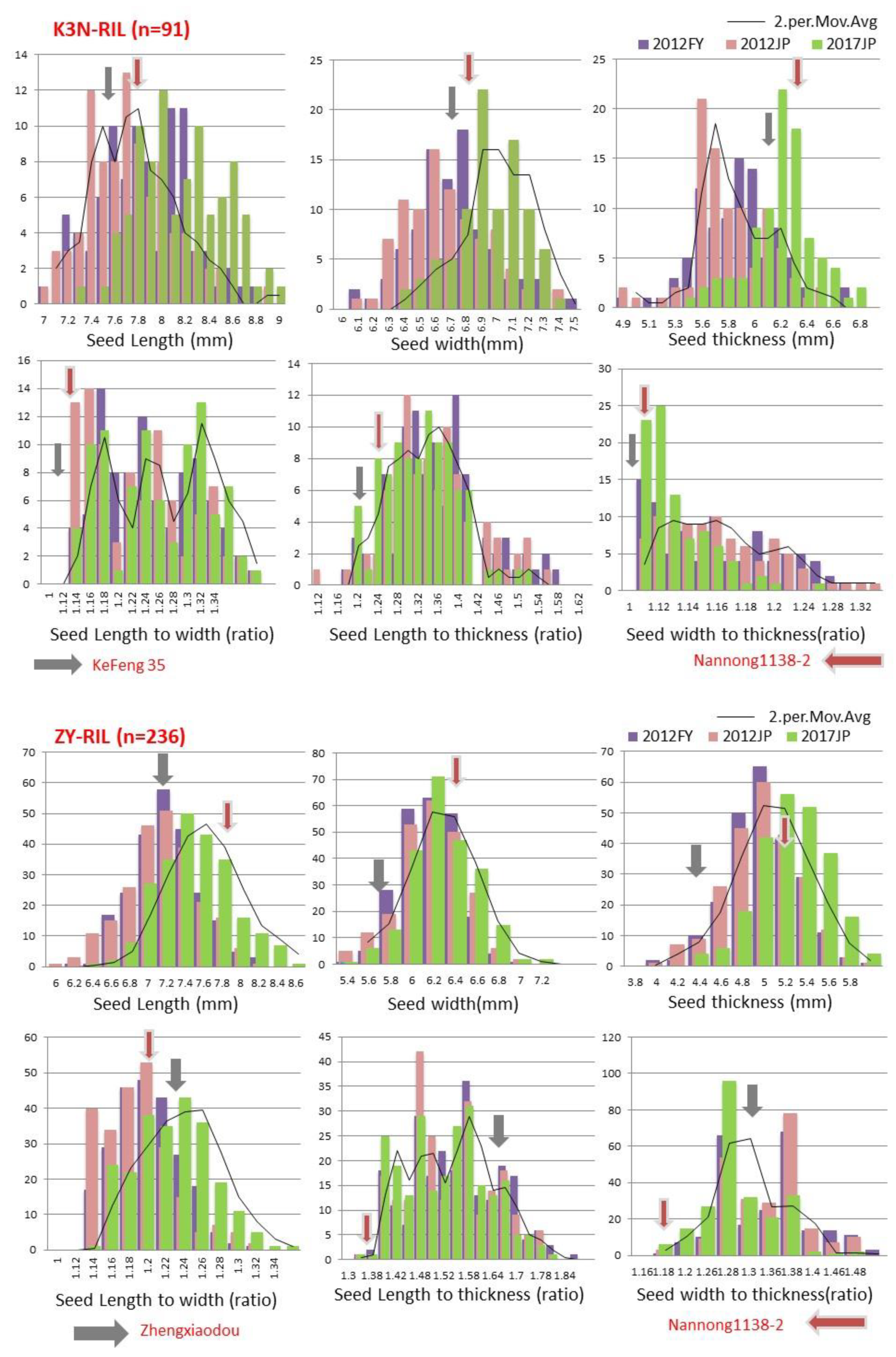
Seed shape and size is an economically important trait determining the yield and quality in soybeans. Hence, developing soybean cultivars with improved seed shapes and sizes is considered as a critical objective of soybean breeding programs. However, to develop improved cultivars, it is a prerequisite to have a detailed understanding of genetic architecture, as well as a mechanism underlying the trait of interest. Both seed shape and size are complex quantitative traits, governed by multiple genes and are highly environmentally sensitive. Although, over the past decades, many QTLs related to soybean seed shape/size have been reported but not stable and confirmed, due to small-sized mapping populations and low-density genetic maps, and, hence, not implied for breeding improved seed shapes and sizes in soybean. Thus, the present study aimed to utilize high-density intraspecific linkage maps of ZY and K3N populations, evaluated in three different environments, to identify the stable significant main-effect QTLs, “QTL Hotspots”, and epistatic-effect QTLs, as well as their interactions with the environment; additionally, find possible candidate genes for soybean seed sizes and shape traits. In this study, ANOVA results revealed a significant difference among the RILs of both ZY and K3N populations for all six traits (
P < 0.01,
Table S2). Similar to previous studies, our study also reported that all six traits related to seed size/shape were significantly affected by G, E, and G × E [
7,
36]. Frequency distribution of all six traits (SL, SW, ST, SLW, SLT, and SWT) showed the characteristics of continuous variations, and all these traits have transgressive segregation in both directions, which indicates that both parents contributed favorable alleles for these traits (
Figure 1). These findings are in agreement with the prior findings, which also stated continuous distribution and transgressive segregation for seed size/shape traits among RILs of soybeans in multiple environments [
3,
17,
22]. In our study, the estimated heritability of all six traits was high (>60%) in both RIL populations across all three environments (
Table S1), which was consistent with previous studies [
7]. The high heritability suggests that if the trial repeated in the same growing/environment conditions, there would be a high possibility of achieving the same kind of phenotypic results. A highly significant correlation (either positive or negative) between any two seed shapes or seed size traits and between seed size and shape traits is in accordance, as previously reported by Xu et al. [
5].
QTL mapping is a practical approach and has been frequently employed for the detection of QTLs/genes underlying the quantitative traits in crop plants. However, the efficiency and accuracy of QTL mappings are influenced, mainly by parental diversity and marker density [
26]. The quality of genetic maps has a significant impact on the accuracy of QTL detection, and, consequently, increasing marker density can intensify the resolution of QTL mapping [
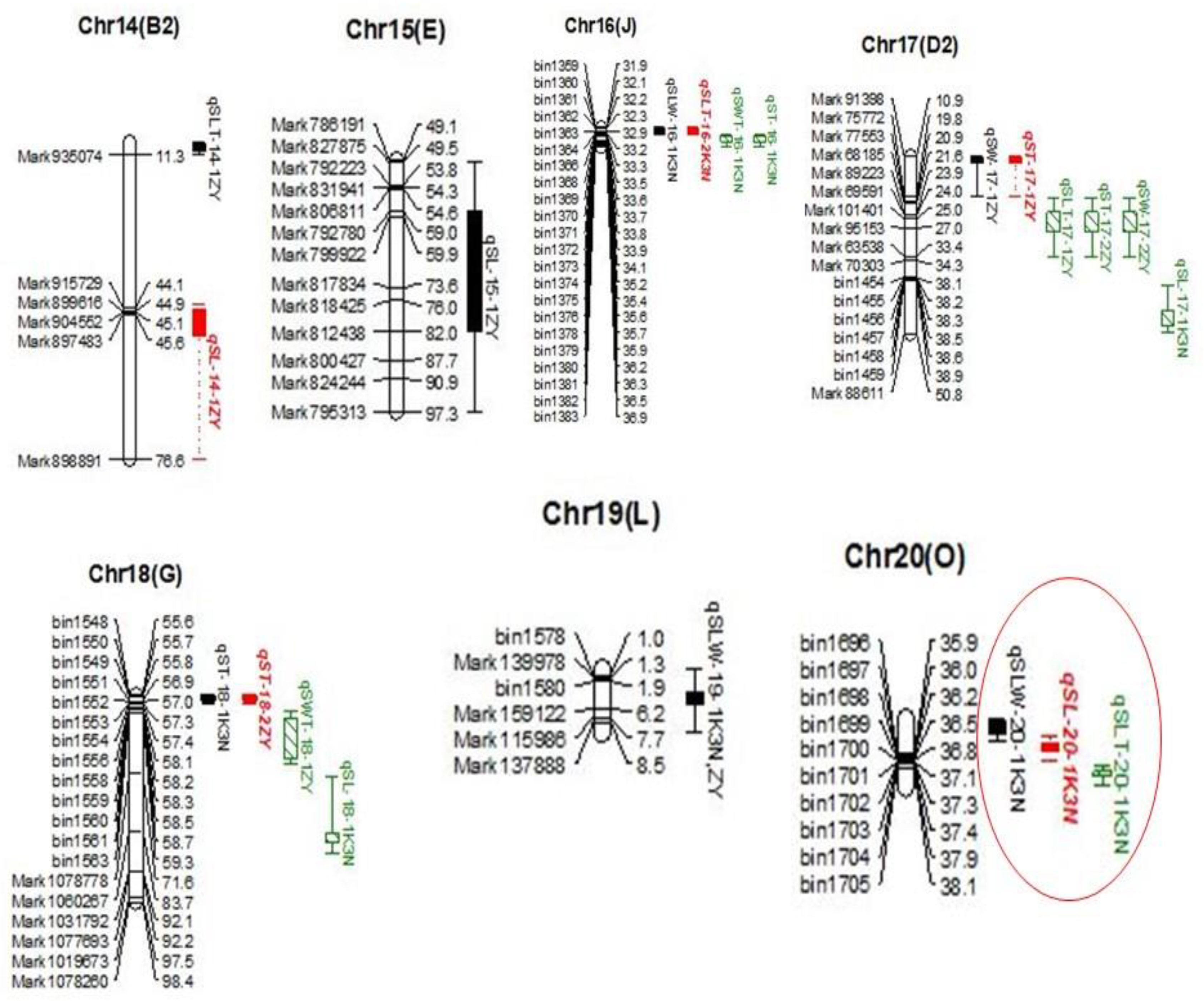
37]. Hence, it is a prerequisite to utilize high-density linkage maps for improving the efficiency and accuracy of linkage mapping and MAS. In this study, high-density genetic maps of ZY and K3N populations were used, consisting of 3255 SLAF and 1733 bin markers, respectively. The markers in both linkage maps, viz., ZY and K3N, were integrated to all 20 LGs and covered the total length of 2144.85 and 2362.44 cM, respectively, with an average distance between adjacent markers of 0.66 cM and 1.36 cM, respectively.
The use of high-density bin-maps assisting in QTL identification with tightly linked markers provided a good foundation for analyzing quantitative traits. However, to reduce environmental errors, RILs were planted in three environments (consisting of different locations and years), and each of the environments was statistically different. Jansen et al. [
38] described that the QTL position and effects could be accurately evaluated if the phenotypic data collected in various environments were different from a statistical perspective. Although, markers associated with the QTLs regulating the seed sizes and shapes in soybeans have been mapped on all linkage groups (Soybase,
www.soybase.org). However, for cross-validation and improving the accuracy of QTL mapping results, we used two different methods for QTL mapping, viz., CIM and MCIM. A total of 88 and 48 QTLs were detected by CIM and MCIM methods, respectively, associated with all six traits related to seed size and shape (
Table 1,
Table 2 and
Table 3). Among these QTLs, 15 common QTLs were verified through both CIM and MCIM, indicating that these QTLs were stable and utilized effectively as potential candidate regions for enhancing seed sizes and shapes in soybeans. The QTL results of our study revealed better matches with the SoyBase database (
www.soybase.org;
Table 1 and
Table 2); however, 51 (CIM) and 27 (MCIM) QTLs were identified for the very first time (
Table 1,
Table 2 and
Table 3). These novel QTLs collectively expressed more than 90% of PV for seed size and shape, suggesting their potential value for the development of improved soybean cultivars. Among these novel QTLs,
qSL-9-1ZY,
K3N,
qST-6-2ZY,
qSLW-6-1ZY,
qSLW-20-1K3N,
qSLT-10-1ZY, and
qSLT-20-1K3N were reported as stable and major QTLs, identified in more than one individual environment, with
R2 > 10%. Besides, by comparing the physical genomic regions of QTLs identified in both populations (ZY and K3N) and mapping methods (CIM and MCIM), two major and novel QTLs, viz.,
qSW-1-3ZY and
qSLT-20-3K3N, were characterized commonly in both mapping methods. These above seven unique and stable QTLs significantly represent potential loci for the improvement of seed sizes and shapes in soybeans. Hence, identification of many new and unique QTLs in the present study suggests distinct genetic architecture in the population derived from the diverse Chinese cultivated soybean genotypes and the need to use more germplasm for revealing the complex genetic basis of soybeans. The favorable alleles for seed size and shape traits were contributed by both parents of two RIL populations, viz., ZY and K3N. Therefore, it is critical to note that not only the higher phenotype parent contributes beneficial alleles but also the contribution of favorable alleles by lower phenotype parents cannot be disregarded; similar results are also described earlier [
30].
The stability of the QTL is essential for use in a breeding program. In addition to novel stable QTLs identified for both seed size and shape traits, this study also identified 37 and 21 QTLs through the CIM and MCIM methods, which have been previously colocalized in the same physical interval by earlier studies (see references in
Table 1,
Table 2 and
Table 3). Out of these colocalized QTLs, 12 and 3 QTLs detected by the CIM and MCIM methods were major (
R2 > 10%). Therefore, our results showed the reliability of QTL mapping. Furthermore, these QTLs can be utilized as principal targets to identify the candidate genes and MAS in future studies.
It has been demonstrated that epistatic and QTL by environment interaction effects are the two crucial genetic factors that make an enormous contribution to the phenotypic variation observed in complex traits, and the knowledge of those interaction effects is vital for understanding the genetic mechanism of complex traits [
39,
40]. Previous studies revealed that the seed sizes and shapes of soybeans is significantly affected by the environment [
36]. Moreover, knowledge of specific QTL by environment interactions can guide the search of varieties adapted to particular environments. The QTLs with more significant additive effects are often considered more stable [
41,
42]. For example, the
qSW-1-1ZY and
qST-18-2ZY (additive effect: 0.14) identified in both CIM and MCIM methods; though,
qSLT-6-5ZY (additive effect: 0.001) was detected only in the MCIM method (ZY only) (
Table 3). The genetic architecture of seed size and shape also includes epistatic interactions between QTLs [
11,
43]. Hence, ignoring intergenic interactions will lead to the overestimation of individual QTL eff
ects, and the underestimation of genetic variance [
44], consequently, could result in a substantial drop in the genetic response to MAS, particularly at late generations [
45]. In the present study, 16 pairs of digenetic epistatic QTLs pairs were identified for seed size and shape in both populations and expressed phenotypic variations that varied from 1.71 to 9.70% (
Table 4). Except for
qSL-13-3ZY,
qSL-13-4ZY, and
qST-13-4ZY, all the remaining epistatic QTLs do not possess additive effects alone, suggesting that these loci might serve as modifying genes that interact with other genes to affect the phenotypes of seed sizes and shapes (
Table 4). All 16 pairs have significant AA, but only four QTL pairs, viz.,
qSL-2-1K3N and
qSL-2-2K3N,
qST-9-1K3N and
qST-12-4K3N,
qSLT-2-3K3N and
qSLT-7-1K3N, and
qSWT-6-1K3N and
qSWT-8-4K3N, hold significant AAE interaction effects. However, the total AAE phenotypic variations expressed by these four epistatic pairs was 19.14%. These results show that epistatic and environmental interactions are fundamental for understanding the genetic basis of seed sizes and shapes in soybeans, demonstrating that these eff
ects should be considered in a QTL mapping program and could increase the accuracy of phenotypic value predictions in MAS.
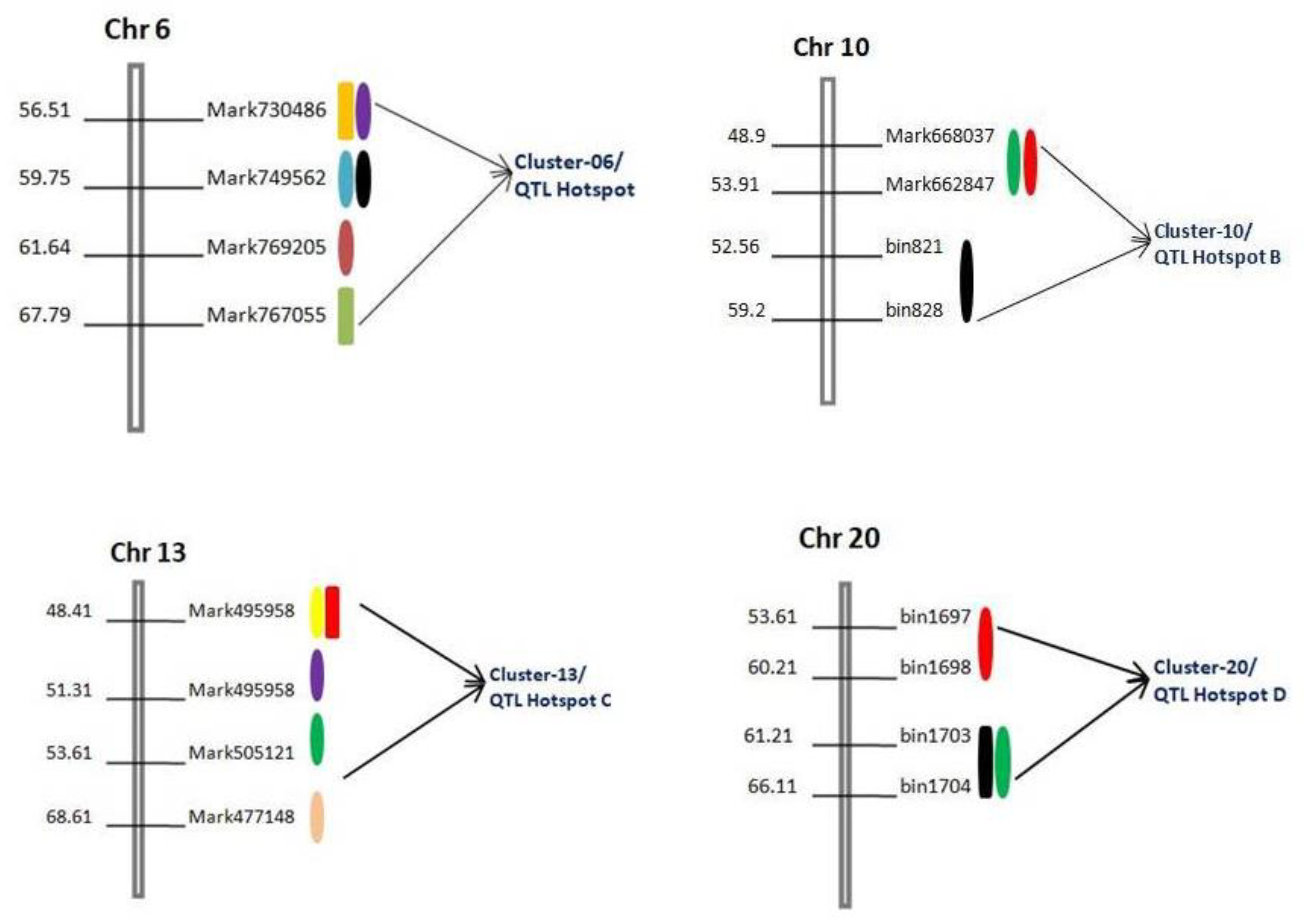
Colocalization of QTLs on chromosomes for different traits related to seed size and shape were also observed in this study. This colocalization of QTLs linked to related traits on chromosomes was reported earlier in soybeans and referred to as “QTL cluster/hotspots” [
46]. In this study, we scrutinized a few genomic regions containing QTL clusters and found four QTL clusters/hotspots on four different chromosomes, viz., Chr06, Chr10, Chr13, and Chr20 (
Figure 3 and
Table 5). The QTLs within each cluster/hotspot are associated with three or more traits related to seed sizes and shapes in soybeans. The highest number of six and five QTLs were observed in “QTL Hotspot A” and “QTL Hotspot C”, respectively, harboring QTLs for more than three traits related to seed size and shape (
Figure 3 and
Table 5). The other two hotspots, viz., “QTL Hotspot B” and “QTL Hotspot D”, contain three QTLs, each for three different traits related to seed size and shape (
Table 5). These QTLs clusters/hotspots have not reported and added to the growing knowledge of the genetic control of these traits. The phenomenon of the QTL clustering might represent a linkage of genes/QTLs or result from the pleiotropic effects of a single QTL in the same genomic region [
47]. This colocalization of QTLs for different seed size and shape traits was following the fact that they were highly significantly correlated with each other (
Table 1). These “QTL hotspot” regions showed that the QTLs linkage/pleiotropy could facilitate the enhancement of seed size and shape. Previously, some of the QTLs for other traits have also been identified in the same region of “QTL Hotspot A” on chromosome 06, which are related to seed oil and protein content [
48,
49] and days to flowering [
50]. In the case of “QTL Hotspot B”, QTLs related to seed weight and seed yield [
51], length of the reproductive stage [
33], days to flowering, and maturity [
33] were reported in the same physical interval.
Similarly, earlier studies have also reported QTLs for seed weight [
7] and seed volume [
33] in the “QTL Hotspot C” region on Chr13. In “QTL Hotspot D”, QTLs related to seed maturity [
33] and seed oil content [
52] have formerly reported. Seed oil and protein content in soybeans have reported a significant correlation with seed size and shape [
53], as seed oil and protein content represents a major component of soybean seeds, representing 38–42% and 18–22%, respectively; hence, these traits are directly related to seed sizes and shapes in soybeans [
13]. Both seed size and shape are important yield component traits [
54] and it has been reported that days to flowering and maturity is directly correlated to yield in soybeans [
55,
56], signifying the potential probability of common genic factors for these traits and also showing the necessity to promote further study for these regions. These QTL clusters have provided some valuable information to define genome regions associated with different traits. Based on the comprehensive analysis of clusters in this study, breeding programs targeting an increase of seed sizes and shapes with high yield and superior quality can focus on hotspot clustering and select QTLs around the region. Finally, the existence of QTL clusters/hotspots has provided proof that genes related to some crop traits are more densely concentrated in certain genomic regions of crop genomes than others [
33,
51].
Identification of candidate genes underlying the QTL region is of great interest for practical plant breeding. Earlier studies based on QTL mapping of seed size and shape did not practice mining for candidate genes [
22,
54], and, to date, only a few seed size/shape-related genes have been isolated from the soybean. For example, the
Ln gene has a large effect on the number of seeds per pod and seed size/shape [
57], and, recently, the
PP2C-1 (protein phosphatase type-2 C) allele from wild soybean accession ZYD7 were found to contribute toward the increase in seed size/shape [
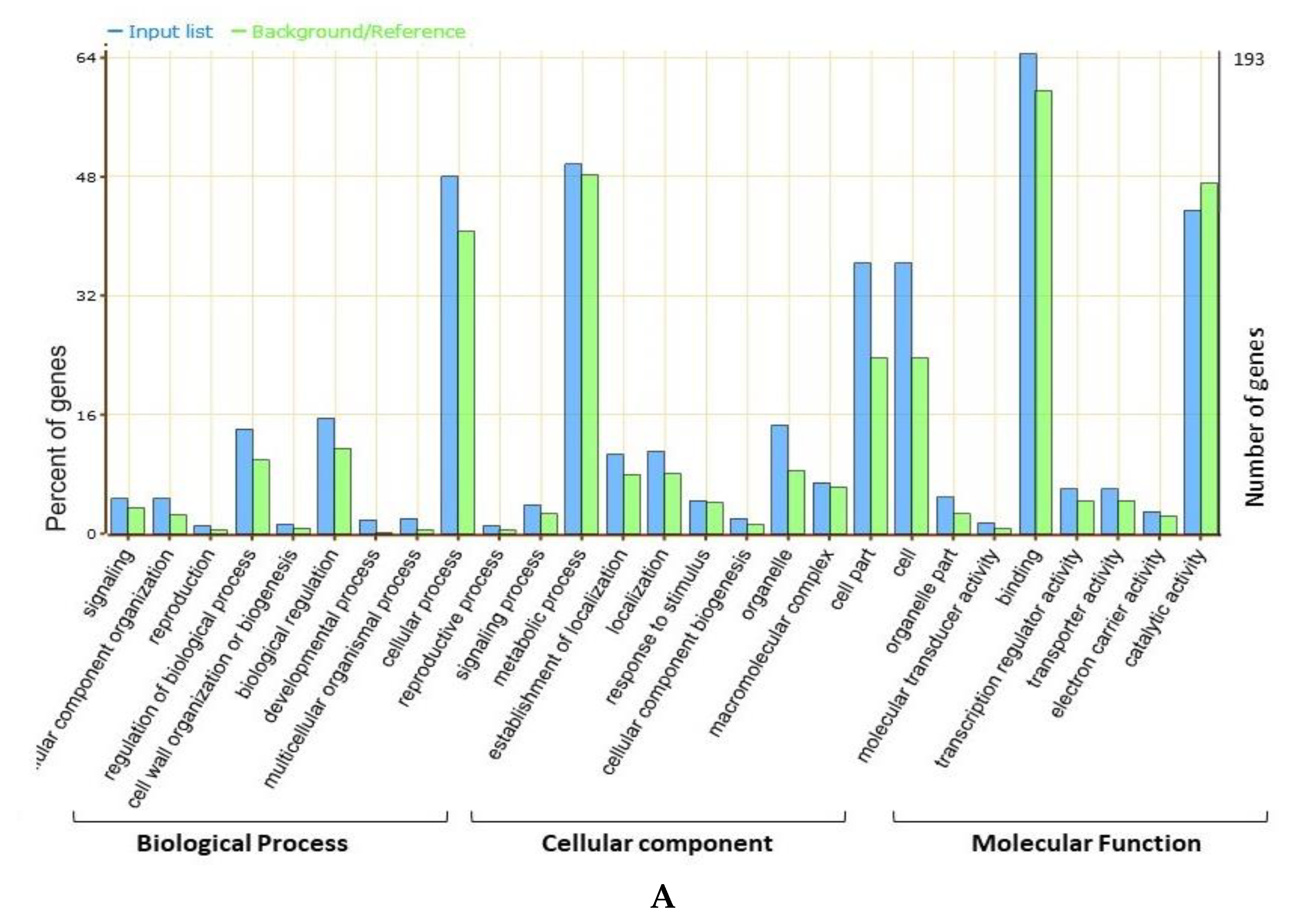
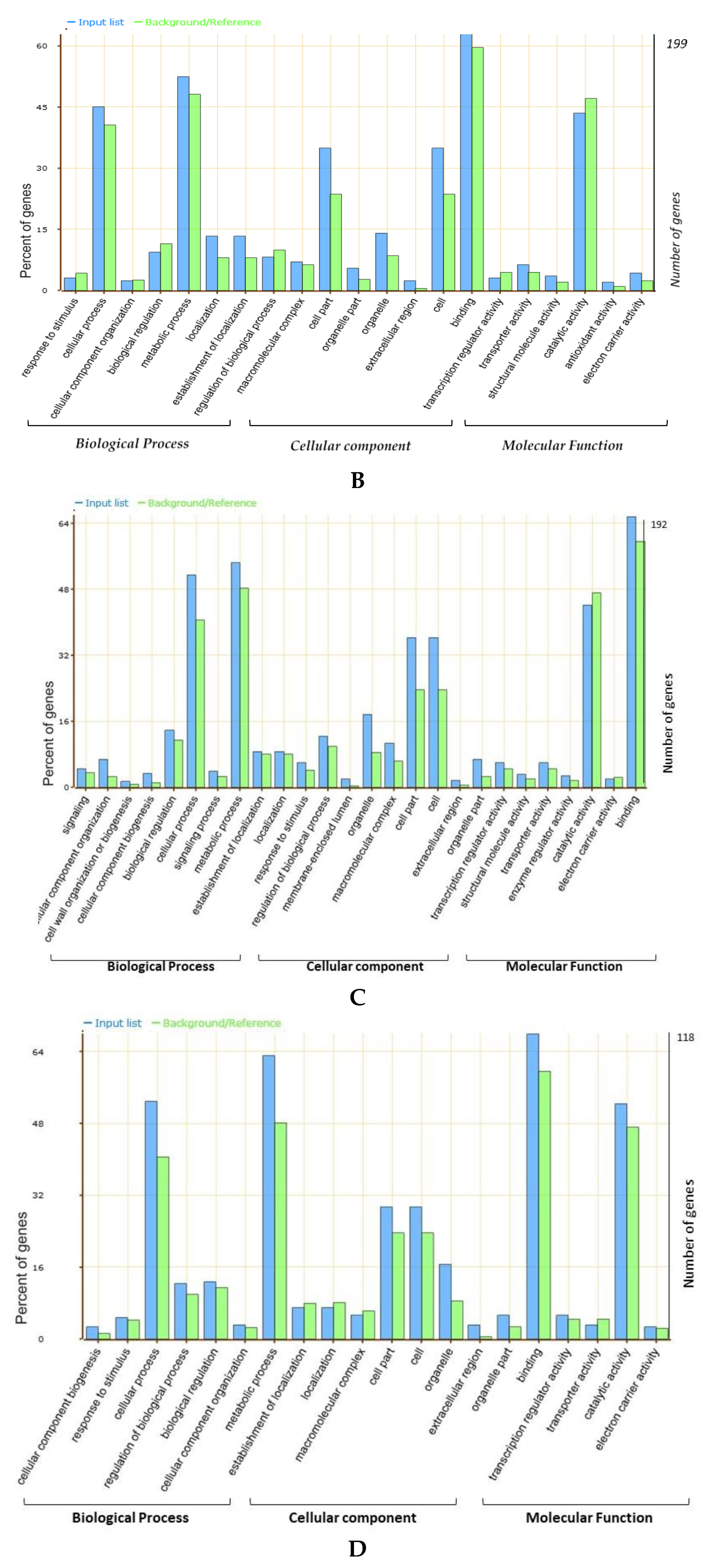
58]. Based on the gene annotations, available literature, and GO enrichment analysis, the present study identified the possible candidate genes regulating the seed sizes and shapes in soybeans that underlies the four categorized “QTL hotspots”. Gene ontology (GO) analysis revealed that most of the genes underlying the above four “QTL hotspots” belong to the terms cellular process, metabolic process, cell part, cell, catalytic activity, and binding, and these elements are reported as being vital in seed development [
59,
60,
61]. A total of 2406 gene models were mined within the physical interval of the four “QTL hotspots.” Out of them, 88 were considered as possible candidate genes, based on the GO enrichment analysis, gene function, and available literature. These 88 predicted candidate genes have functions that are directly or indirectly involved in seed development, influencing the shape and size of seeds, such as lipid storage, transport and metabolic processes, signal transduction of plant hormones, degradation of the ubiquitin-proteasome pathway, fatty acid beta-oxidation, the brassinosteroid-mediated signaling pathway, and the auxin biosynthetic process (
Table S5). From the available gene expression data (RNA-seq), 23 of the 88 predicted candidate genes expressed significantly higher gene expression, particularly in seed development stages, root nodules, leaf, and pod shell (
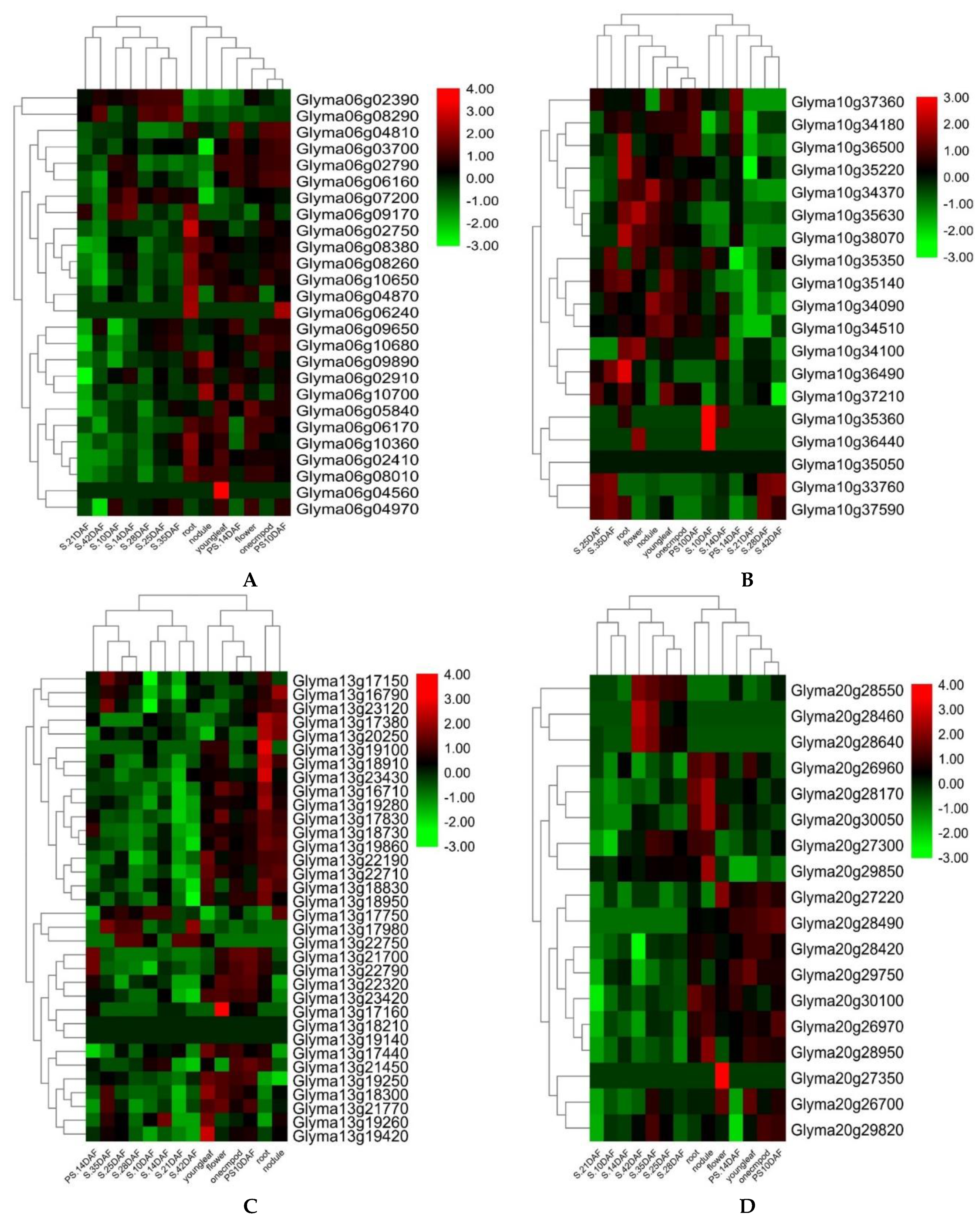
Figure 5 and
Table S5). Out of these 23 genes, five genes, viz.,
Glyma06g04810,
Glyma06g03700,
Glyma13g17980,
Glyma13g21770, and
Glyma20g30100 have functions that are related to seed development, ovule development, endosperm, and embryo development, which have been reported to directly contribute to seed sizes and shapes in crop plants, including soybeans [
62,
63]. Likewise,
Glyma06g02390,
Glyma06g06160,
Glyma06g07200, and
Glyma13g18730 encode RING/U-box superfamily proteins/protein ubiquitination. The ubiquitin pathway has recently been known to play an essential part in seed size determination [
60]. Several factors involved in ubiquitin-related activities have been revealed to determine seed sizes in
Arabidopsis and rice [
60]. Genes, viz.,
Glyma06g08290,
Glyma20g28460,
Glyma20g28640,
Glyma20g29750,
Glyma20g28550,
Glyma13g22790,
Glyma20g29750, and
Glyma20g27300, function in lipid storage, seed maturation, and cell growth, which have formerly been reported to determine seed size and shape in oilseeds, including soybeans [
64]. For example, overexpression of
GmMYB73 promotes lipid accumulation in soybean seeds, which leads to increased seed sizes in soybeans [
65]. Genes, viz.,
Glyma06g09650,
Glyma10g35360,
Glyma10g36440,
Glyma13g17750, and
Glyma13g21700, are involved in auxin biosynthesis, responses to auxin stimulus, and responses to ethylene stimulus. The auxin regulates seed weights and sizes in
Arabidopsis [
22,
66].
Glyma06g10700 functions to regulate the brassinosteroid stimulus, which positively governs seed size [
62]. Hence, based on the gene function, GO, and RNA-Seq analysis, the above 23 genes were considered as the most potentially possible candidate genes for regulating the seed sizes and shapes in soybeans. However, it requires further validation and verification to confirm their actual roles in seed sizes/shapes in soybeans, as well as their future uses for the improvement of seed quality traits. Some of these genes were already included in our ongoing project for functional validation to ascertain their effects on the seed sizes and shapes. Hence, the precise identification of QTLs in a specific physical interval through the use of a high-density map would make it easy to identify candidate genes.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}