Candidate SNP Markers of Atherogenesis Significantly Shifting the Affinity of TATA-Binding Protein for Human Gene Promoters Show Stabilizing Natural Selection as a Sum of Neutral Drift Accelerating Atherogenesis and Directional Natural Selection Slowing It

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
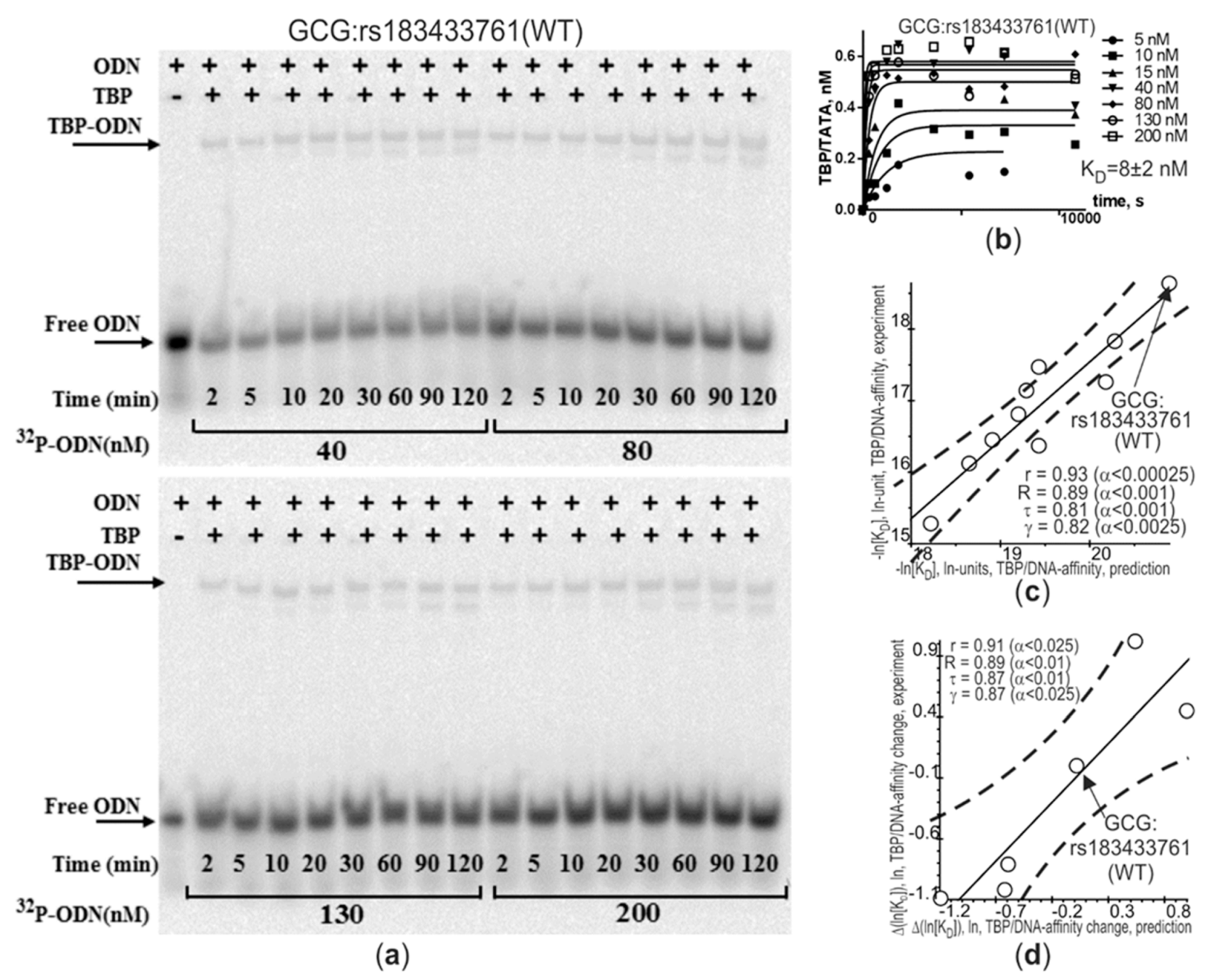
2.1. The Only Clinically Proven SNP Marker of Atherosclerosis in a TBP-Site of a Human Gene Promoter

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data: GRCh38, dbSNP Rel. 151 [12] | Result | H0: Neutral Drift [24,25] | H0: ↑ and ↓ Equivalence | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | SNPs | NGENE | NSNP | NRES | n> | n< | P(H0: n> < n<) [62] | n↑ | n↓ | P(H0: n↑ ≡ n↓) |
| 1 | Whole-genome norm for SNPs of TBP-sites [63] | 104 | 105 | 103 | 200 | 800 | >0.99 | - | - | - |
| 2 | Clinically known SNP markers of diseases at TBP-sites [45] | 33 | 203 | 51 | 14 | 37 | >0.99 | - | - | - |
| 3 | Candidate SNP markers for atherogenesis near clinical SNP markers for diseases at TBP-sites [55] | 17 | 175 | 34 | 7 | 27 | >0.99 | 19 | 15 | >0.30 |
| 4 | Candidate SNP markers for atherogenesis near clinical SNP markers for diseases at TBP-sites [this work] | 26 | 644 | 145 | 72 | 73 | 0.50 | 91 | 54 | <0.01 |
| 5 | Candidate SNP markers for atherogenesis near the TBP-sites of protein-coding genes related to the maintenance of mitochondrial genome integrity [56], this work | 4 | 464 | 77 | 48 | 29 | <0.05 | 27 | 50 | <0.01 |
| 6 | Candidate SNP markers of atherogenesis near the TBP-sites of the miRNA genes associated with instability of atherosclerotic plaque [57], this work | 4 | 81 | 16 | 11 | 5 | <0.05 | 2 | 14 | <0.01 |
| 7 | TOTAL | 34 | 1189 | 238 | - | - | - | - | - | - |
| Gene | dbSNP ID [12] or [Ref] | DNA, Genome Sequence | KD, nM | Clinical Data or Candidate SNP Markers | AS | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5′ flank | WT | min | 3′ flank | WT | min | Δ | Z | α | ρ | |||||
| CETP | rs1427119663 | cgtgggggct | 18bp | - | gggctccagg | 4 | 7 | < | 7 | 10−6 | A | hyperalpha-lipoprotei-nemia (athero-protector) | ↓ | [58] |
| rs1002690375 | gggccactta | c | t | acaccactgc | 4 | 2 | > | 11 | 10−6 | A | accelerated atherogenesis, which can be slowed down by swimming | ↑ | [61,62] | |
| rs757176551 | catatacggg | c | g | tccaggctga | 4 | 2 | > | 10 | 10−6 | A | ↑ | |||
| rs569033466 | atacatatac | g | a, t, c | ggctccaggc | 4 | 3 | > | 4 | 10−3 | B | ↑ | |||
| rs1451694749 | catacatata | c | t | gggctccagg | 4 | 2 | > | 12 | 10−6 | A | ↑ | |||
| MBL2 | rs72661131 | tctatttcta | t | c | tatagcctgc | 2 | 4 | < | 12 | 10−6 | A | stroke, pre-eclampsia, variable immune-deficiency | ↑ | [64,65,66] |
| rs1471733364 | tatttctata | t | g | agcctgcacc | 2 | 5 | < | 15 | 10−6 | A | on the Western diet, MBL2 deficit and excess quicken earlier and late atherogenesis, respectively | ↑ | [67,68,69] | |
| rs562962093 | atctatttct | a | g | tatagcctgc | 2 | 5 | < | 15 | 10−6 | A | ↑ | |||
| rs567653539 | tttctatata | g | a | cctgcaccca | 2 | 1 | > | 4 | 10−3 | B | ↑ | |||
| F3 | rs563763767 | ccctttatag | c | t * | gcgcggggca | 3 | 2 | > | 6 | 10−6 | A | myocardial infarction, thrombosis | ↑ | [70] |
| rs1439518731 | gccctttata | - | ta | gcgcgcgggg | 3 | 1 | > | 12 | 10−6 | A | accelerated atherogenesis that can be slowed down by aspirinslowed down atherogenesis | ↑ | [71,72] | |
| rs966076891:t | gccgccggcc | c | t | tttatagcgc | 3 | 2 | > | 6 | 10−6 | A | ↑ | |||
| rs966076891:g | gccgccggcc | c | g | tttatagcgc | 3 | 4 | < | 2 | 0.05 | D | ↓ | [73] | ||
| rs1190659847 | gccggccctt | t | c | atagcgcgcg | 3 | 12 | < | 19 | 10−6 | A | ↓ | |||
| TPI1 | rs1800202 | gcgctctata | t | g | aagtgggcag | 1 | 4 | < | 17 | 10−6 | A | hemolytic anemia, neu-romuscular diseases | ↑ | [74,75] |
| rs1386262216 | gcggcgctct | a | c, g | tataagtggg | 1 | 2 | < | 8 | 10−6 | A | traumatic and ischemic co-mplications of atherogenesis | ↑ | [76,77] | |
| rs781835924 | cgcggcgctc | t | c | atataagtgg | 1 | 2 | < | 10 | 10−6 | A | ↑ | |||
2.2. Human Genes Associated with Cardiovascular Diseases
2.3. Human Genes Associated with Blood Disorders
| Gene | dbSNP ID [12] or [Ref] | DNA, Genome Sequence | KD, nM | Clinical Data or Prediction | AS | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5′ flank | WT | min | 3′ flank | WT | min | Δ | Z | α | ρ | |||||
| HBB | rs33981098 | agggctgggc | a | g, c | taaaagtcag | 5 | 9 | < | 10 | 10−6 | A | thalassemia, RM | ↓ | [78] |
| rs33980857 | gggctgggca | t | a, t, g | aaaagtcagt | 5 | 21 | < | 27 | 10−6 | A | ↓ | |||
| rs397509430 | gggctgggca | t | - | aaaagtcagt | 5 | 29 | < | 34 | 10−6 | A | ↓ | |||
| rs34598529 | ggctgggcat | a | g | aaagtcaggg | 5 | 18 | < | 24 | 10−6 | A | ↓ | |||
| rs33931746 | gctgggcata | a | g, c | aagtcagggc | 5 | 11 | < | 14 | 10−6 | A | ↓ | |||
| rs281864525 | tgggcataaa | a | c * | gtcagggcag | 5 | 7 | < | 7 | 10−6 | A | thalassemia is athero-protective | ↓ | [79] | |
| rs63750953 | ctgggcataa | aa | - | gtcagggcag | 5 | 8 | < | 9 | 10−6 | A | ↓ | |||
| rs1160543272:t | ccagggctgg | g | t | cataaaagtc | 4.6 | 5.3 | < | 2 | 0.05 | D | ↓ | |||
| rs1160543272:a | ccagggctgg | g | a | cataaaagtc | 4.6 | 3.8 | > | 3 | 10−2 | C | norm (silent SNP), hemo-lytically extra-cellular HBB releases athe-rogenic heme | ↑ | [80,81] | |
| rs34500389 | cagggctggg | c | t* | ataaaagtca | 5 | 2 | > | 14 | 10−6 | A | ↑ | |||
| [80] | gctgggcata | a | t | aagtcagggc | 5 | 3 | > | 8 | 10−6 | A | ↑ | |||
| HBD | rs996092254 | aggacaggac | c | t | agcataaaag | 4 | 3 | > | 4 | 10−6 | A | ↑ | ||
| rs1473693473 | ataaaaggca | a | g | ggcagagtcg | 4 | 5 | < | 3 | 10−2 | C | thalassemia athero-protective | ↓ | [79] | |
| rs34166473 | aggaccagca | t | c | aaaaggcagg | 4 | 12 | < | 18 | 10−6 | A | ↓ | |||
| rs35518301 | caggaccagc | a | g | taaaaggcag | 4 | 8 | < | 11 | 10−6 | A | thalassemia, RM | ↓ | [78] | |
| ACKR1 | rs2814778 | ttggctctta | t | c | cttggaagca | 10 | 12 | < | 4 | 10−3 | B | leukopenia, RM | ↓ | [82,83] |
| slowed down atherogenesis | [84] | |||||||||||||
| rs1185314734 | tggctcttat | c | g | ttggaagcac | 10 | 6 | > | 8 | 10−6 | A | accelerated atherogenesis | ↑ | [85] | |
| NOS2 | [30] | gtataaatac | t | c | tcttggctgc | 1.7 | 1.4 | > | 3 | 10−2 | C | RM | ↑ | [86,87] |
| accelerated atherogenesis | [88,89] | |||||||||||||
| rs1339255364 | gcatggggtg | a | g * | gtataaatac | 1.7 | 2.0 | < | 2 | 0.05 | D | slowed down atherogenesis | ↓ | [90] | |
| F7 | [91] | ccttggaggc | a | c | gagaactttg | 53 | 62 | < | 3 | 10−2 | C | moderate bleeding | ↑ | [91] |
| atherogenesis slowed by dill | [92,93] | |||||||||||||
| rs749691733 | agaactttgc | c | t | cgtcagtccc | 53 | 66 | < | 4 | 10−3 | B | F7 excess is a biomarker of hypercholes-terolemia, which accelerates atherogenesis, which can be slowed down by olive oil | ↑ | [94,95] | |
| rs997515289 | ttggaggcag | a | c | gaactttgcc | 53 | 68 | < | 5 | 10−3 | B | ↑ | |||
| rs549591993 | gcccgtcagt | c | a | ccatggggaa | 53 | 25 | > | 13 | 10−6 | A | ↑ | |||
| rs367732974 | aactttgccc | g | a | tcagtcccat | 53 | 47 | > | 2 | 0.05 | D | ↑ | |||
| rs777947114 | agagaacttt | g | a | cccgtcagtc | 53 | 19 | > | 19 | 10−6 | A | ↑ | |||
| rs1187329967 | tggaggcaga | g | c | aactttgccc | 53 | 32 | > | 9 | 10−6 | A | ↑ | |||
| rs770113559 | gtcacccttg | g | a | aggcagagaa | 53 | 41 | > | 5 | 10−6 | A | ↑ | |||
| rs781338265 | cctctgtcac | c | a * | cttggaggca | 53 | 30 | > | 11 | 10−6 | A | ↑ | |||
| rs754814507 | cctcccccat | c | t | cctctgtcac | 53 | 45 | > | 3 | 10−3 | B | ↑ | |||
| rs1296764751 | tcccctcccc | c | t | atccctctgt | 53 | 46 | > | 2 | 0.05 | A | ↑ | |||
2.4. Human Genes Associated with Autoimmunity-Associated Diseases
| Gene | dbSNP ID [12] or [Ref] | DNA, Genome Sequence | KD, nM | Clinical Data or Candidate SNP Markers | AS | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5′ flank | WT | min | 3′ flank | WT | min | Δ | Z | α | ρ | |||||
| SOD1 | rs7277748 | ggtctggcct | a | g | taaagtagtc | 2 | 7 | < | 17 | 10−6 | A | familial amyotrophic lateral sclerosis | ↑ | [96,97,98,99] |
| rs1438766715 | agagtgggcg | 26bp | - | agtcgcggag | 2 | 79 | < | 59 | 10−6 | A | accelerated atherogenesis that can be slowed down by short-term exercise or onion extract | ↑ | [100,101,102] | |
| rs1325052558 | tctggcctat | a | g | aagtagtcgc | 2 | 9 | < | 22 | 10−6 | A | ↑ | |||
| rs966452334 | aggtctggcc | t | c | ataaagtagt | 2 | 6 | < | 16 | 10−6 | A | ↑ | |||
| INS | rs5505 | agatcactgt | c | t | cttctgccat | 53 | 44 | > | 4 | 10−3 | B | neonatal diabetes mellitus, hyper(pro)-insulinemia | ↑ | [15] |
| rs1389349459 | ctgtctccca | g | t | atcactgtcc | 53 | 14 | > | 25 | 10−6 | A | atherogenesis that can be slowed by dietary soy isoflavone as norminsuli-nemic athero-protector | ↑ | [103,104,106] | |
| rs563207167 | tcagccctgc | c | t | tgtctcccag | 53 | 44 | > | 4 | 10−3 | B | ↑ | |||
| rs1367101897 | cctcagccct | g | a | cctgtctccc | 53 | 32 | > | 9 | 10−6 | A | ↑ | |||
| rs11557611 | gatcactgtc | c | t | ttctgccatg | 53 | 60 | < | 2 | 0.05 | D | ↑ | [105] | ||
| MMP12 | rs2276109 | gatatcaact | a | g | tgagtcactc | 11 | 14 | < | 3 | 10−2 | C | lower risks of psoriasis, systemic scleroderma, and asthma | ↑ | [107,108,109,110] |
| rs572527200 | gatgatatca | a | g | ctatgagtca | 11 | 14 | < | 3 | 10−2 | C | accelerated atherogenesis that can be slowed by milk fermented with lacto-bacilli and dietary sup-plements akin to estrogens | ↑ | [111,112] | |
| rs1401366377 | tatcaactat | g | a | agtcactcat | 11 | 3 | > | 20 | 10−6 | A | ↑ | [113,114] | ||
2.5. Human Genes Associated with Obesity
2.6. Human Genes Associated with Carcinogenesis
2.7. Human Genes Associated with Developmental Disorders
2.8. Selective In Vitro Validation
2.9. In Silico Validation of Our Predictions Based on Clinical SNP Markers as a Whole
2.10. Human Genes Related to Mitochondrial Genome Integrity and Instability of the Atherosclerotic Plaque
3. Materials and Methods
3.1. DNA Sequences
3.2. Analysis of DNA Sequences
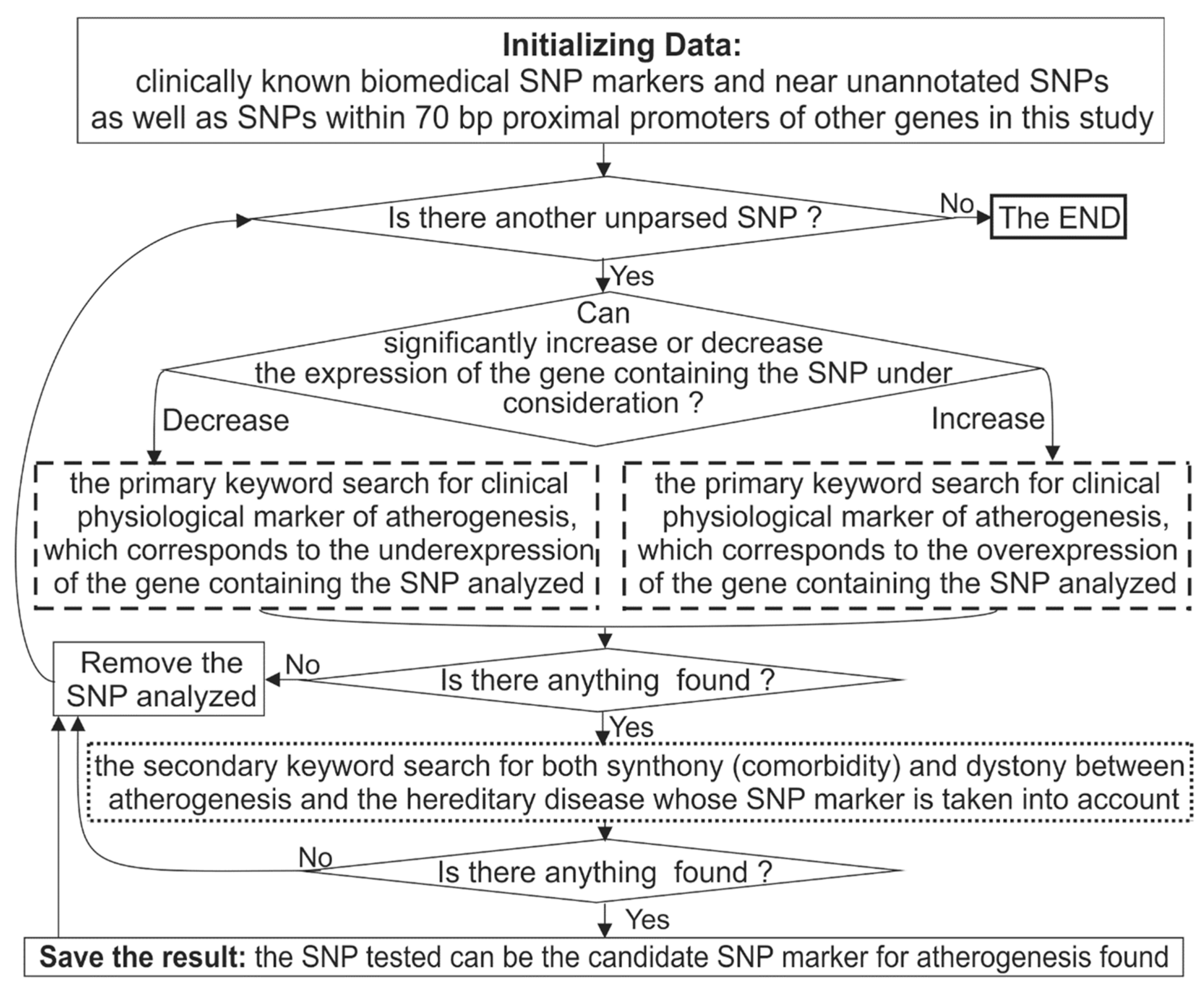
3.3. Keyword Searches in the PubMed Database
3.4. In Vitro Measurements
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EMSA | electrophoretic mobility shift assay |
| TBP | TATA-binding protein |
| SNP | single-nucleotide polymorphism |
| WHO | World Health Organization |
References
- Barquera, S.; Pedroza-Tobias, A.; Medina, C.; Hernandez-Barrera, L.; Bibbins-Domingo, K.; Lozano, R.; Moran, A.E. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch. Med. Res. 2015, 46, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.; Badimon, L.; Hansson, G.; Deanfield, J.; Bittencourt, M.; Tokgozoglu, L.; Lewis, E. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Li, A.C.; Glass, C.K. The macrophage foam cell as a target for therapeutic intervention. Nat. Med. 2002, 8, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, S.; Soda, S.; Ito, Y.; Matsui, H.; Ueno, T.; Fukushima, Y.; Ohmura, H.; Hanyu, O.; Aizawa, Y.; Miida, T. Circadian change of serum concentration of small dense LDL-cholesterol in type 2 diabetic patients. Clin. Chim. Acta 2010, 411, 253–257. [Google Scholar] [CrossRef]
- Lathe, R.; Sapronova, A.; Kotelevtsev, Y. Atherosclerosis and Alzheimer--diseases with a common cause? Inflammation, oxysterols, vasculature. BMC Geriatr. 2014, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Napoli, C.; D’Armiento, F.; Mancini, F.; Postiglione, A.; Witztum, J.; Palumbo, G.; Palinski, W. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J. Clin. Investig. 1997, 100, 2680–2690. [Google Scholar] [CrossRef] [PubMed]
- Trovato, G.M. Sustainable medical research by effective and comprehensive medical skills: Overcoming the frontiers by predictive, preventive and personalized medicine. EPMA J. 2014, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Telenti, A.; Pierce, L.; Biggs, W.; di Iulio, J.; Wong, E.; Fabani, M.; Kirkness, E.; Moustafa, A.; Shah, N.; Xie, C.; et al. Deep sequencing of 10,000 human genomes. Proc. Natl. Acad. Sci. USA 2016, 113, 11901–11906. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.; Wilder, S.; Johnson, N.; Juettemann, T.; Flicek, P. The Ensembl regulatory build. Genome Biol. 2015, 16, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.; Ward, M.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeussler, M.; Raney, B.; Hinrichs, A.; Clawson, H.; Zweig, A.; Karolchik, D.; Casper, J.; Speir, M.; Haussler, D.; Kent, W. Navigating protected genomics data with UCSC Genome Browser in a box. Bioinformatics 2015, 31, 764–766. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wu, M.; Li, L.; Liu, Z.; Zeng, W.; Jiang, R. dbWGFP: A database and web server of human whole-genome single nucleotide variants and their functional predictions. Database 2016, 2016, baw024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.; Lee, J.; Riley, G.; Jang, W.; Rubinstein, W.; Church, D.; Maglott, D. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amberger, J.; Bocchini, C.; Schiettecatte, F.; Scott, A.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuyasu, H.; Izuhara, K.; Mao, X.; Gao, P.; Arinobu, Y.; Enomoto, T.; Kawai, M.; Sasaki, S.; Dake, Y.; Hamasaki, N.; et al. Ile50Val variant of IL4R alpha upregulates IgE synthesis and associates with atopic asthma. Nat. Genet. 1998, 19, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Deplancke, B.; Alpern, D.; Gardeux, V. The genetics of transcription factor DNA binding variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef] [Green Version]
- Savinkova, L.; Ponomarenko, M.; Ponomarenko, P.; Drachkova, I.; Lysova, M.; Arshinova, T.; Kolchanov, N. TATA box polymorphisms in human gene promoters and associated hereditary pathologies. Biochemistry 2009, 74, 117–129. [Google Scholar] [CrossRef]
- Mogno, I.; Vallania, F.; Mitra, R.D.; Cohen, B. TATA is a modular component of synthetic promoters. Genome Res. 2010, 20, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, M.; Mironova, V.; Gunbin, K.; Savinkova, L. Hogness Box. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; Volme 3, pp. 491–494. [Google Scholar]
- Varzari, A.; Deyneko, I.; Tudor, E.; Turcan, S. Polymorphisms of glutathione S-transferase and methylenetetrahydrofolate reductase genes in Moldavian patients with ulcerative colitis: Genotype-phenotype correlation. Meta Gene 2016, 7, 76–82. [Google Scholar] [CrossRef]
- Pocai, B. The ICD-11 has been adopted by the World Health Assembly. World Psychiatry 2019, 18, 371–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M. Evolutionary rate at the molecular level. Nature 1968, 217, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Haldane, J.B.S. The cost of natural selection. J. Genet. 1957, 55, 511–524. [Google Scholar] [CrossRef]
- Yoo, S.; Jin, C.; Jung, D.; Choi, Y.; Choi, J.; Lee, W.; Lee, S.; Lee, J.; Cha, S.; Kim, C.; et al. Putative functional variants of XRCC1 identified by RegulomeDB were not associated with lung cancer risk in a Korean population. Cancer Genet. 2015, 208, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Deyneko, I.V.; Kalybaeva, Y.; Kel, A.E.; Blocker, H. Human-chimpanzee promoter comparisons: Property-conserved evolution? Genomics 2010, 96, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Boyle, A.; Hong, E.; Hariharan, M.; Cheng, Y.; Schaub, M.; Kasowski, M.; Karczewski, K.; Park, J.; Hitz, B.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [Green Version]
- Mathelier, A.; Fornes, O.; Arenillas, D.; Chen, C.; Denay, G.; Lee, J.; Shi, W.; Shyr, C.; Tan, G.; Worsley-Hunt, R.; et al. JASPAR 2016: A major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016, 44, D110–D115. [Google Scholar] [CrossRef] [Green Version]
- Yevshin, I.; Sharipov, R.; Valeev, T.; Kel, A.; Kolpakov, F. GTRD: A database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res. 2017, 45, D61–D67. [Google Scholar] [CrossRef]
- Kulakovskiy, I.; Vorontsov, I.; Yevshin, I.; Sharipov, R.; Fedorova, A.; Rumynskiy, E.; Medvedeva, Y.; Magana-Mora, A.; Bajic, V.; Papatsenko, D.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018, 46, D252–D259. [Google Scholar] [CrossRef]
- Levitsky, V.; Zemlyanskaya, E.; Oshchepkov, D.; Podkolodnaya, O.; Ignatieva, E.; Grosse, I.; Mironova, V.; Merkulova, T. A single ChIP-seq dataset is sufficient for comprehensive analysis of motifs co-occurrence with MCOT package. Nucleic Acids Res. 2019, 47, e139. [Google Scholar] [CrossRef] [PubMed]
- Sokolenko, A.; Sandomirskii, I.; Savinkova, L. Interaction of yeast TATA-binding protein with short promotor segments. Mol. Biol. 1996, 30, 279–285. [Google Scholar]
- Ponomarenko, M.; Ponomarenko, J.; Frolov, A.; Podkolodny, N.; Savinkova, L.; Kolchanov, N.; Overton, G. Identification of sequence-dependent features correlating to activity of DNA sites interacting with proteins. Bioinformatics 1999, 15, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Savinkova, L.; Drachkova, I.; Ponomarenko, M.; Lysova, M.; Arshinova, T.; Kolchanov, N. Interaction of recombinant TATA-binding protein with TATA-boxes of mammalian gene promoters. Ecol. Genet. 2007, 2, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, P.; Savinkova, L.; Drachkova, I.; Lysova, M.; Arshinova, T.; Ponomarenko, M.; Kolchanov, N. A step-by-step model of TBP/TATA box binding allows predicting human hereditary diseases by single nucleotide polymorphism. Dokl. Biochem. Biophys. 2008, 419, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Delgadillo, R.; Whittington, J.; Parkhurst, L.; Parkhurst, L. The TBP core domain in solution variably bends TATA sequences via a three-step binding mechanism. Biochemistry 2009, 48, 1801–1809. [Google Scholar] [CrossRef]
- Lu, Z. PubMed and beyond: A survey of web tools for searching biomedical literature. Database 2011, 2011, baq036. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, P.; Suslov, V.; Savinkova, L.; Ponomarenko, M.; Kolchanov, N. A precise equilibrium equation for four steps of binding between TBP and TATA-box allows for the prediction of phenotypical expression upon mutation. Biofizika 2010, 55, 358–369. [Google Scholar]
- Savinkova, L.; Drachkova, I.; Arshinova, T.; Ponomarenko, P.; Ponomarenko, M.; Kolchanov, N. An experimental verification of the predicted effects of promoter TATA-box polymorphisms associated with human diseases on interactions between the TATA boxes and TATA-binding protein. PLoS ONE 2013, 8, e54626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drachkova, I.; Savinkova, L.; Arshinova, T.; Ponomarenko, M.; Peltek, S.; Kolchanov, N. The mechanism by which TATA-box polymorphisms associated with human hereditary diseases influence interactions with the TATA-binding protein. Hum. Mutat. 2014, 35, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Drachkova, I.; Shekhovtsov, S.; Peltek, S.; Ponomarenko, P.; Arshinova, T.; Ponomarenko, M.; Merkulova, T.; Savinkova, L.; Kolchanov, N. Study of interaction of human TATA-binding protein with TATA-element of NOS2A gene promoter using surface plasmon resonance method. Vavilov. J. Genet. Breed. 2012, 16, 391–396. [Google Scholar]
- Arkova, O.; Kuznetsov, N.; Fedorova, O.; Kolchanov, N.; Savinkova, L. Realtime interaction between TBP and the TATA box of the human triosephosphate isomerase gene promoter in the norm and pathology. Acta Nat. 2014, 6, 36–40. [Google Scholar] [CrossRef]
- Arkova, O.; Kuznetsov, N.; Fedorova, O.; Savinkova, L. A real-time study of the interaction of TBP with a TATA box-containing duplex identical to an ancestral or minor allele of human gene LEP or TPI. J. Biomol. Struct. Dyn. 2017, 35, 3070–3081. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, M.; Rasskazov, D.; Arkova, O.; Ponomarenko, P.; Suslov, V.; Savinkova, L.; Kolchanov, N. How to use SNP_TATA_Comparator to find a significant change in gene expression caused by the regulatory SNP of this gene’s promoter via a change in affinity of the TATA-binding protein for this promoter. Biomed. Res. Int. 2015, 2015, 359835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomarenko, M.; Rasskazov, D.; Chadaeva, I.; Sharypova, E.; Ponomarenko, P.; Arkova, O.; Kashina, E.; Ivanisenko, N.; Zhechev, D.; Savinkova, L.; et al. SNP_TATA_Comparator: Genomewide landmarks for preventive personalized medicine. Front. Biosci. 2017, 9, 276–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnaev, I.; Rasskazov, D.; Arkova, O.; Ponomarenko, M.; Ponomarenko, P.; Savinkova, L.; Kolchanov, N. Hypothetical SNP markers that significantly affect the affinity of the TATA-binding protein to VEGFA, ERBB2, IGF1R, FLT1, KDR, and MET oncogene promoters as chemotherapy targets. Mol. Biol. 2016, 50, 161–173. [Google Scholar] [CrossRef]
- Arkova, O.; Ponomarenko, M.; Rasskazov, D.; Drachkova, I.; Arshinova, T.; Ponomarenko, P.; Savinkova, L.; Kolchanov, N. Obesity-related known and candidate SNP markers can significantly change affinity of TATA-binding protein for human gene promoters. BMC Genom. 2015, 16, S5. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, M.; Arkova, O.; Rasskazov, D.; Ponomarenko, P.; Savinkova, L.; Kolchanov, N. Candidate SNP markers of gender-biased autoimmune complications of monogenic diseases are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. Front. Immunol. 2016, 7, 130. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, P.; Chadaeva, I.; Rasskazov, D.; Sharypova, E.; Kashina, E.; Drachkova, I.; Zhechev, D.; Ponomarenko, M.; Savinkova, L.; Kolchanov, N. Candidate SNP markers of familial and sporadic Alzheimer’s diseases are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. Front. Aging Neurosci. 2017, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, P.; Rasskazov, D.; Suslov, V.; Sharypova, E.; Savinkova, L.; Podkolodnaya, O.; Podkolodny, N.; Tverdokhleb, N.; Chadaeva, I.; Ponomarenko, M.; et al. Candidate SNP markers of chronopathologies are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. BioMed Res. Int. 2016, 2016, 8642703. [Google Scholar] [CrossRef] [Green Version]
- Chadaeva, I.; Ponomarenko, M.; Rasskazov, D.; Sharypova, E.; Kashina, E.; Matveeva, M.; Arshinova, T.; Ponomarenko, P.; Arkova, O.; Bondar, N.; et al. Candidate SNP markers of aggressiveness-related complications and comorbidities of genetic diseases are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. BMC Genom. 2016, 17, 995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadaeva, I.; Ponomarenko, P.; Rasskazov, D.; Sharypova, E.; Kashina, E.; Zhechev, D.; Drachkova, I.; Arkova, O.; Savinkova, L.; Ponomarenko, M.; et al. Candidate SNP markers of reproductive potential are predicted by a significant change in the affinity of TATA-binding protein for human gene promoters. BMC Genom. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadaeva, I.; Ponomarenko, P.; Rasskazov, D.; Sharypova, E.; Kashina, E.; Kleshchev, M.; Ponomarenko, M.; Naumenko, V.; Savinkova, L.; Kolchanov, N.; et al. Natural selection equally supports the human tendencies in subordination and domination: A genome-wide study with in silico confirmation and in vivo validation in mice. Front. Genet. 2019, 10, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomarenko, M.; Rasskazov, D.; Chadaeva, I.; Sharypova, E.; Drachkova, I.; Ponomarenko, P.; Oshchepkova, E.; Savinkova, L.; Kolchanov, N. Candidate SNP-markers of atherosclerosis, which may significantly change the affinity of the TATA-binding protein for the human gene promoters. Russ. J. Genet. 2019, 55, 1137–1151. [Google Scholar] [CrossRef]
- Vasileiou, P.; Mourouzis, I.; Pantos, C. Principal aspects regarding the maintenance of mammalian mitochondrial genome integrity. Int. J. Mol. Sci. 2017, 18, 1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koroleva, I.; Nazarenko, M.; Kucher, A. Role of microRNA in development of instability of atherosclerotic plaque. Biochemistry 2017, 82, 1380–1390. [Google Scholar] [CrossRef]
- Plengpanich, W.; Le Goff, W.; Poolsuk, S.; Julia, Z.; Guerin, M.; Khovidhunkit, W. CETP deficiency due to a novel mutation in the CETP gene promoter and its effect on cholesterol efflux and selective uptake into hepatocytes. Atherosclerosis 2011, 216, 370–373. [Google Scholar] [CrossRef]
- Waardenberg, A.; Basset, S.; Bouveret, R.; Harvey, R. CompGO: An R package for comparing and visualizing Gene Ontology enrichment differences between DNA binding experiments. BMC Bioinform. 2015, 16, 275. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Sun, M.; Wang, J.; Cui, J.; Liu, Z.; Yu, B. A novel swine model for evaluation of dyslipidemia and atherosclerosis induced by human CETP overexpression. Lipids Health Dis. 2017, 16, 169. [Google Scholar] [CrossRef] [Green Version]
- Casquero, A.; Berti, J.; Teixeira, L.; de Oliveira, H. Chronic exercise reduces CETP and mesterolone treatment counteracts exercise benefits on plasma lipoproteins profile: Studies in transgenic mice. Lipids 2017, 52, 981–990. [Google Scholar] [CrossRef]
- Kasowski, M.; Grubert, F.; Heffelfinger, C.; Hariharan, M.; Asabere, A.; Waszak, S.; Habegger, L.; Rozowsky, J.; Shi, M.; Urban, A.; et al. Variation in transcription factor binding among humans. Science 2010, 328, 232–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 1000 Genomes Project Consortium; Abecasis, G.; Auton, A.; Brooks, L.; DePristo, M.; Durbin, R.; Handsaker, R.; Kang, H.; Marth, G.; McVean, G.; et al. An integrated map of genetic variation from 1.092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervera, A.; Planas, A.M.; Justicia, C.; Urra, X.; Jensenius, J.; Torres, F.; Lozano, F.; Chamorro, A. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS ONE 2010, 5, e8433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sziller, I.; Babula, O.; Hupuczi, P.; Nagy, B.; Rigo, B.; Szabo, G.; Papp, Z.; Linhares, I.; Witkin, S. Mannose-binding lectin (MBL) codon 54 gene polymorphism protects against development of pre-eclampsia, HELLP syndrome and pre-eclampsia-associated intrauterine growth restriction. Mol. Hum. Reprod. 2007, 13, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Boldt, A.; Culpi, L.; Tsuneto, L.; de Souza, I.; Kun, J.; Petzl-Erler, M. Diversity of the MBL2 gene in various Brazilian populations and the case of selection at the mannose-binding lectin locus. Hum. Immunol. 2006, 67, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Losin, I.; Shakhnovich, R.; Zykov, K.; Ruda, M. Cardiovascular diseases and mannose-binding lectin. Kardiologiia 2014, 54, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Troelsen, L.; Garred, P.; Christiansen, B.; Torp-Pedersen, C.; Christensen, I.; Narvestad, E.; Jacobsen, S. Double role of mannose-binding lectin in relation to carotid intima-media thickness in patients with rheumatoid arthritis. Mol. Immunol. 2010, 47, 713–718. [Google Scholar] [CrossRef]
- Matthijsen, R.; de Winther, M.; Kuipers, D.; van der Made, I.; Weber, C.; Herias, M.; Gijbels, M.; Buurman, W. Macrophage-specific expression of mannose-binding lectin controls atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2009, 119, 2188–2195. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, E.; Barbalat, V.; Nicaud, V.; Cambien, F.; Evans, A.; Morrison, C.; Arveiler, D.; Luc, G.; Ruidavets, J.; Emmerich, J.; et al. Polymorphisms in the 5′ regulatory region of the tissue factor gene and the risk of myocardial infarction and venous thromboembolism: The ECTIM and PATHROS studies. Etude Cas-Temoins de l’Infarctus du Myocarde. Paris Thrombosis case-control Study. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 892–898. [Google Scholar] [CrossRef] [Green Version]
- Hasenstab, D.; Lea, H.; Hart, C.; Lok, S.; Clowes, A. Tissue factor overexpression in rat arterial neointima models thrombosis and progression of advanced atherosclerosis. Circulation 2000, 101, 2651–2657. [Google Scholar] [CrossRef]
- Lin, H.; Yen, H.; Hsieh, S.; An, L.; Shen, K. Low-dose aspirin ameliorated hyperlipidemia, adhesion molecule, and chemokine production induced by high-fat diet in Sprague-Dawley rats. Drug Dev. Res. 2014, 75, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Ebert, J.; Wilgenbus, P.; Teiber, J.; Jurk, K.; Schwierczek, K.; Dohrmann, M.; Xia, N.; Li, H.; Spiecker, L.; Ruf, W.; et al. Paraoxonase-2 regulates coagulation activation through endothelial tissue factor. Blood 2018, 131, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Zingg, B.; Mohrenweiser, H. Molecular analysis of a series of alleles in humans with reduced activity at the triosephosphate isomerase locus. Am. J. Hum. Genet. 1996, 58, 308–316. [Google Scholar]
- Vives-Corrons, J.; Rubinson-Skala, H.; Mateo, M.; Estella, J.; Feliu, E.; Dreyfus, J. Triosephosphate isomerase deficiency with hemolytic anemia and severe neuromuscular disease: Familial and biochemical studies of a case found in Spain. Hum. Genet. 1978, 42, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Vercellotti, G.; Eaton, J.; Jacob, H. Heme uptake by endothelium synergizes polymorphonuclear granulocyte-mediated damage. Trans. Assoc. Am. Physicians 1990, 103, 17417–17419. [Google Scholar]
- Kioumourtzoglou, M.; Seals, R.; Gredal, O.; Mittleman, M.; Hansen, J.; Weisskopf, M. Cardiovascular disease and diagnosis of amyotrophic lateral sclerosis: A population based study. Amyotroph. Lateral. Scler. Frontotemporal. Degener. 2016, 17, 548–554. [Google Scholar] [CrossRef]
- Martiney, J.; Cerami, A.; Slater, A. Inhibition of hemozoin formation in Plasmodium falciparum trophozoite extracts by heme analogs: Possible implication in the resistance to malaria conferred by the beta-thalassemia trait. Mol. Med. 1996, 2, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Luo, W.; Wang, J.; Guo, C.; Wolffe, S.; Wang, J.; Sun, E.; Bradley, K.; Campbell, A.; Eitzman, D. Paradoxical protection from atherosclerosis and thrombosis in a mouse model of sickle cell disease. Br. J. Haematol. 2013, 162, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Collins, F.; Weissman, S. The molecular genetics of human hemoglobin. Prog. Nucleic Acid Res. Mol. Biol. 1984, 31, 315–462. [Google Scholar]
- Gall, T.; Petho, D.; Nagy, A.; Hendrik, Z.; Mehes, G.; Potor, L.; Gram, M.; Åkerstrom, B.; Smith, A.; Nagy, P.; et al. Heme induces endoplasmic reticulum stress (HIER stress) in human aortic smooth muscle cells. Front. Physiol. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Nalls, M.; Wilson, J.; Patterson, N.; Tandon, A.; Zmuda, J.; Huntsman, S.; Garcia, M.; Hu, D.; Li, R.; Beamer, B.; et al. Admixture mapping of white cell count: Genetic locus responsible for lower white blood cell count in the Health ABC and Jackson Heart studies. Am. J. Hum. Genet. 2008, 82, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Michon, P.; Woolley, I.; Wood, E.M.; Kastens, W.; Zimmerman, P.; Adams, J. Duffy-null promoter heterozygosity reduces DARC expression and abrogates adhesion of the P. vivax ligand required for blood-stage infection. FEBS Lett. 2001, 495, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; Liu, Q.; Lionakis, M.; Marino, A.; Anderson, S.; Swamydas, M.; Murphy, P. Atypical chemokine receptor 1 deficiency reduces atherogenesis in ApoE-knockout mice. Cardiovasc. Res. 2015, 106, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Gencer, S.; van der Vorst, E.P.C.; Aslani, M.; Weber, C.; Doring, Y.; Duchene, J. Atypical chemokine receptors in cardiovascular disease. Thromb. Haemost. 2019, 119, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, M.; Udhayakumar, V.; Levesque, M.; Booth, J.; Roberts, J.; Tkachuk, A.; Pole, A.; Coon, H.; Kariuki, S.; Nahlen, B.; et al. A new NOS2 promoter polymorphism associated with increased nitric oxide production and protection from severe malaria in Tanzanian and Kenyan children. Lancet 2002, 360, 1468–1475. [Google Scholar] [CrossRef]
- Clark, I.; Rockett, K.; Burgner, D. Genes, nitric oxide and malaria in African children. Trends Parasitol. 2003, 19, 335–337. [Google Scholar] [CrossRef]
- Zhao, J.; Shyue, S.; Lin, S.; Wei, J.; Lee, T. Excess nitric oxide impairs LXR(α)-ABCA1-dependent cholesterol efflux in macrophage foam cells. J. Cell Physiol. 2014, 229, 117–125. [Google Scholar]
- Jovanovic, A.; Sudar-Milovanovic, E.; Obradovic, M.; Pitt, S.; Stewart, A.; Zafirovic, S.; Stanimirovic, J.; Radak, D.; Isenovic, E. Influence of a high-fat diet on cardiac iNOS in female rats. Curr. Vasc. Pharmacol. 2017, 15, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Ponnuswamy, P.; Ostermeier, E.; Schrottle, A.; Chen, J.; Huang, P.; Ertl, G.; Nieswandt, B.; Kuhlencordt, P. Oxidative stress and compartment of gene expression determine proatherosclerotic effects of inducible nitric oxide synthase. Am. J. Pathol. 2009, 174, 2400–2410. [Google Scholar] [CrossRef] [Green Version]
- Kavlie, A.; Hiltunen, L.; Rasi, V.; Prydz, H. Two novel mutations in the human coagulation factor VII promoter. Thromb. Haemost. 2003, 90, 194–205. [Google Scholar] [CrossRef]
- Zacharski, L.; Delprete, S.; Kisiel, W.; Hunt, J.; Cornell, C.; Marrin, C. Atherosclerosis and coronary bypass surgery in hereditary factor VII deficiency. Am. J. Med. 1988, 84, 955–959. [Google Scholar] [CrossRef]
- Roche, H.; Zampelas, A.; Knapper, J.; Webb, D.; Brooks, C.; Jackson, K.; Wright, J.; Gould, B.; Kafatos, A.; Gibney, M.; et al. Effect of long-term olive oil dietary intervention on postprandial triacylglycerol and factor VII metabolism. Am. J. Clin. Nutr. 1998, 68, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Mitropoulos, K.; Esnouf, M.; Meade, T. Increased factor VII coagulant activity in the rabbit following diet-induced hypercholesterolaemia. Evidence for increased conversion of VII to alpha VIIa and higher flux within the coagulation pathway. Atherosclerosis 1987, 63, 43–52. [Google Scholar] [CrossRef]
- Setorki, M.; Rafieian-Kopaei, M.; Merikhi, A.; Heidarian, E.; Shahinfard, N.; Ansari, R.; Nasri, H.; Esmael, N.; Baradaran, A. Suppressive impact of anethum graveolens consumption on biochemical risk factors of atherosclerosis in hypercholesterolemic rabbits. Int. J. Prev. Med. 2013, 4, 889–895. [Google Scholar] [PubMed]
- Niemann, S.; Broom, W.; Brown, R.H., Jr. Analysis of a genetic defect in the TATA box of the SOD1 gene in a patient with familial amyotrophic lateral sclerosis. Muscle Nerve 2007, 36, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Goldacre, R.; Ramagopalan, S.; Talbot, K.; Goldacre, M. Autoimmune disease preceding amyotrophic lateral sclerosis: An epidemiologic study. Neurology 2013, 81, 1222–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ende, N.; Weinstein, F.; Chen, R.; Ende, M. Human umbilical cord blood effect on sod mice (amyotrophic lateral sclerosis). Life Sci. 2000, 67, 53–59. [Google Scholar] [CrossRef]
- Charles, B.; Hsieh, M.; Adeyemo, A.; Shriner, D.; Ramos, E.; Chin, K.; Srivastava, K.; Zakai, N.; Cushman, M.; McClure, L.; et al. Analyses of genome wide association data, cytokines, and gene expression in African-Americans with benign ethnic neutropenia. PLoS ONE 2018, 13, e0194400. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Padilla, J.; Jenkins, N.; Laughlin, M. Exercise training and vascular cell phenotype in a swine model of familial hypercholesterolaemia: Conduit arteries and veins. Exp. Physiol. 2014, 99, 454–465. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Tang, C.; Jin, H.; Du, J. Effects of onion extract on endogenous vascular H2S and adrenomedulin in rat atherosclerosis. Curr. Pharm. Biotechnol. 2011, 12, 1427–1439. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, L.; Wang, Z.; Roberts, L.J., 2nd; Lin, X.; Zhao, Y.; Guo, Z. Overexpression of antioxidant enzymes in ApoE-deficient mice suppresses benzo(a)pyrene-accelerated atherosclerosis. Atherosclerosis 2009, 207, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Ning, J.; Yang, X.; Liu, Z. Excess exposure to insulin is the primary cause of insulin resistance and its associated atherosclerosis. Curr. Mol. Pharmacol. 2011, 4, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, Z.; Huang, G.; Xiao, Y.; Li, Z.; Liu, C.; Na, R. Long-term effects intensive medical therapy on the development and progression of subclinical atherosclerosis and the metabolic syndrome in Chinese patients with type 2 diabetes mellitus. Medicine 2016, 95, 5201. [Google Scholar] [CrossRef] [PubMed]
- Badin, J.; Kole, A.; Stivers, B.; Progar, V.; Pareddy, A.; Alloosh, M.; Sturek, M. Alloxan-induced diabetes exacerbates coronary atherosclerosis and calcification in Ossabaw miniature swine with metabolic syndrome. J. Transl. Med. 2018, 16, 58. [Google Scholar] [CrossRef]
- Ferguson, J.; Ryan, M.; Gibney, E.; Brennan, L.; Roche, H.; Reilly, M. Dietary isoflavone intake is associated with evoked responses to inflammatory cardiometabolic stimuli and improved glucose homeostasis in healthy volunteers. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 996–1003. [Google Scholar] [CrossRef] [Green Version]
- Starodubtseva, N.; Sobolev, V.; Soboleva, A.; Nikolaev, A.; Bruskin, S. Genes expression of metalloproteinases (MMP-1, MMP-2, MMP-9, and MMP-12) associated with psoriasis. Russ. J. Genet. 2011, 47, 1117–1123. [Google Scholar] [CrossRef]
- Manetti, M.; Ibba-Manneschi, L.; Fatini, C.; Guiducci, S.; Cuomo, G.; Bonino, C.; Bazzichi, L.; Liakouli, V.; Giacomelli, R.; Abbate, R.; et al. Association of a functional polymorphism in the matrix metalloproteinase-12 promoter region with systemic sclerosis in an Italian population. J. Rheumatol. 2010, 37, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Hunninghake, G.; Cho, M.; Tesfaigzi, Y.; Soto-Quiros, M.; Avila, L.; Lasky-Su, J.; Stidley, C.; Melen, E.; Soderhall, C.; Hallberg, J.; et al. MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med. 2009, 361, 2599–2608. [Google Scholar] [CrossRef] [Green Version]
- Hemminki, K.; Li, X.; Sundquist, J.; Sundquist, K. Subsequent autoimmune or related disease in asthma patients: Clustering of diseases or medical care? Ann. Epidemiol. 2010, 20, 217–222. [Google Scholar] [CrossRef]
- Motterle, A.; Xiao, Q.; Kiechl, S.; Pender, S.; Morris, G.; Willeit, J.; Caulfield, M.; Ye, S. Influence of matrix metalloproteinase-12 on fibrinogen level. Atherosclerosis 2012, 220, 351–354. [Google Scholar] [CrossRef]
- Bukowska, H.; Pieczul-Mroz, J.; Jastrzebska, M.; Chelstowski, K.; Naruszewicz, M. Decrease in fibrinogen and LDL-cholesterol levels upon supplementation of diet with Lactobacillus plantarum in subjects with moderately elevated cholesterol. Atherosclerosis 1998, 137, 437–438. [Google Scholar] [PubMed]
- Liang, J.; Liu, E.; Yu, Y.; Kitajima, S.; Koike, T.; Jin, Y.; Morimoto, M.; Hatakeyama, K.; Asada, Y.; Watanabe, T.; et al. Macrophage metalloelastase accelerates the progression of atherosclerosis in transgenic rabbits. Circulation 2006, 113, 1993–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Bajpai, A.; Hawthorne, E.; Bae, Y.; Castagnino, P.; Monslow, J.; Pure, E.; Spiller, K.; Assoian, R. Cardiovascular protection in females linked to estrogen-dependent inhibition of arterial stiffening and macrophage MMP12. JCI Insight 2019, 4, 122742. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Kawai, K.; Inoue, S.; Murayama, Y.; Fujieda, K.; Kuzuya, N.; Fujita, T.; Koide, Y.; Yamashita, K. Hyperglucagonemia of insulin autoimmune syndrome induced by methimazole in a patient with Graves’ disease. Endocrinol. Jpn. 1989, 36, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.; Saunders, G. Structure of the human glucagon gene. Nucleic Acids Res. 1986, 14, 4719–4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, R.; Kosch, C.; Sanchez, A.; Sabate, J.; Berk, L.; Shavlik, G. Effect of dietary protein on serum insulin and glucagon levels in hyper- and normocholesterolemic men. Atherosclerosis 1989, 76, 55–61. [Google Scholar] [CrossRef]
- Galassetti, P.; Larson, J.; Iwanaga, K.; Salsberg, S.; Eliakim, A.; Pontello, A. Effect of a high-fat meal on the growth hormone response to exercise in children. J. Pediatr. Endocrinol. Metab. 2006, 19, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Skrypnik, K.; Suliburska, J.; Skrypnik, D.; Pilarski, L.; Regula, J.; Bogdanski, P. The genetic basis of obesity complications. Acta Sci. Pol. Technol. Aliment. 2017, 16, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.; Ma, Z.; Pyla, R.; Segar, L. Leptin treatment inhibits the progression of atherosclerosis by attenuating hypercholesterolemia in type 1 diabetic Ins2(+/Akita):apoE(-/-) mice. Atherosclerosis 2012, 225, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, A.; Sasaki, J.; Han, H.; Huang, W.; Kugi, M.; Koga, T.; Ichiki, S.; Shinkawa, T.; Arakawa, K. Compound heterozygosity for an apolipoprotein A1 gene promoter mutation and a structural nonsense mutation with apolipoprotein A1 deficiency. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 348–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimers, G.; Jackson, C.; Rickards, J.; Chan, P.; Cohn, J.; Rye, K.; Barter, P.; Rodgers, K. Inhibition of rupture of established atherosclerotic plaques by treatment with apolipoprotein A-I. Cardiovasc. Res. 2011, 91, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, A. Plasma lipid and lipoprotein levels in obese post-menopausal women: Effects of a short-term low-protein diet and exercise. Maturitas 1990, 12, 121–126. [Google Scholar] [CrossRef]
- Priyadarshini, S.; Pradhan, B.; Griebel, P.; Aich, P. Cortisol regulates immune and metabolic processes in murine adipocytes and macrophages through HTR2c and HTR5a serotonin receptors. Eur. J. Cell Biol. 2018, 97, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Wyler, S.; Wan, R.; Castorena, C.; Ahmed, N.; Mathew, D.; Lee, S.; Liu, C.; Elmquist, J. The atypical antipsychotic olanzapine causes weight gain by targeting serotonin receptor 2C. J. Clin. Investig. 2017, 127, 3402–3406. [Google Scholar] [CrossRef] [Green Version]
- Strandberg, L.; Mellstrom, D.; Ljunggren, O.; Grundberg, E.; Karlsson, M.; Holmberg, A.; Orwoll, E.; Eriksson, A.; Svedberg, J.; Bengtsson, M.; et al. IL6 and IL1B polymorphisms are associated with fat mass in older men: The MrOS Study Sweden. Obesity 2008, 16, 710–713. [Google Scholar] [CrossRef]
- Hayashi, F.; Watanabe, M.; Nanba, T.; Inoue, N.; Akamizu, T.; Iwatani, Y. Association of the -31C/T functional polymorphism in the interleukin-1beta gene with the intractability of Graves’ disease and the proportion of T helper type 17 cells. Clin. Exp. Immunol. 2009, 158, 281–286. [Google Scholar] [CrossRef]
- Borkowska, P.; Kucia, K.; Rzezniczek, S.; Paul-Samojedny, M.; Kowalczyk, M.; Owczarek, A.; Suchanek, R.; Medrala, T.; Kowalski, J. Interleukin-1beta promoter (-31T/C and -511C/T) polymorphisms in major recurrent depression. J. Mol. Neurosci. 2011, 44, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Zhou, X.; Zheng, F.; Xu, X.; Lin, Y.; Yang, J. Influence of interleukin-1 beta genetic polymorphism, smoking and alcohol drinking on the risk of non-small cell lung cancer. Clin. Chim. Acta 2010, 411, 1441–1446. [Google Scholar] [CrossRef]
- Wang, Y.; Kato, N.; Hoshida, Y.; Yoshida, H.; Taniguchi, H.; Goto, T.; Moriyama, M.; Otsuka, M.; Shiina, S.; Shiratori, Y.; et al. Interleukin-1beta gene polymorphisms associated with hepatocellular carcinoma in hepatitis C virus infection. Hepatology 2003, 37, 65–71. [Google Scholar] [CrossRef]
- El-Omar, E.; Carrington, M.; Chow, W.; McColl, K.; Bream, J.; Young, H.; Herrera, J.; Lissowska, J.; Yuan, C.; Rothman, N.; et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000, 404, 398–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Carrillo, D.; Garza-Gonzalez, E.; Betancourt-Linares, R.; Monico-Manzano, T.; Antunez-Rivera, C.; Roman-Roman, A.; Flores-Alfaro, E.; Illades-Aguiar, B.; Fernandez-Tilapa, G. Association of IL1B -511C/-31T haplotype and Helicobacter pylori vacA genotypes with gastric ulcer and chronic gastritis. BMC Gastroenterol. 2010, 10, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.; Zernecke, A. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Han, X.; Li, R.; Zhao, W.; Bai, B.; Yan, C.; Dong, X. Anti-atherosclerosis of oligomeric proanthocyanidins from Rhodiola rosea on rat model via hypolipemic, antioxidant, anti-inflammatory activities together with regulation of endothelial function. Phytomedicine 2018, 51, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Giunzioni, I.; Bonomo, A.; Bishop, E.; Castiglioni, S.; Corsini, A.; Bellosta, S. Cigarette smoke condensate affects monocyte interaction with endothelium. Atherosclerosis 2014, 234, 383–390. [Google Scholar] [CrossRef]
- Abbas, A.; Lechevrel, M.; Sichel, F. Identification of new single nucleotid polymorphisms (SNP) in alcohol dehydrogenase class IV ADH7 gene within a French population. Arch. Toxicol. 2006, 80, 201–205. [Google Scholar] [CrossRef]
- Lainas, P.; Fuks, D.; Gaujoux, S.; Machroub, Z.; Fregeville, A.; Perniceni, T.; Mal, F.; Dousset, B.; Gayet, B. Preoperative imaging and prediction of oesophageal conduit necrosis after oesophagectomy for cancer. Br. J. Surg. 2017, 104, 1346–1354. [Google Scholar] [CrossRef]
- Peltoketo, H.; Piao, Y.; Mannermaa, A.; Ponder, B.; Isomaa, V.; Poutanen, M.; Winqvist, R.; Vihko, R. A point mutation in the putative TATA box, detected in nondiseased individuals and patients with hereditary breast cancer, decreases promoter activity of the 17 beta-hydroxysteroid dehydrogenase type 1 gene 2 (EDH17B2) in vitro. Genomics 1994, 23, 250–252. [Google Scholar] [CrossRef]
- Ma, A.; Wang, D.; An, Y.; Fang, W.; Zhu, H. Comparative transcriptomic analysis of mice liver treated with different AMPK activators in a mice model of atherosclerosis. Oncotarget 2017, 8, 6594–6604. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Gauri, M.; Li, T.; Wang, R.; Lin, S. Current knowledge of the multifunctional 17β-hydroxysteroid dehydrogenase type 1 (HSD17B1). Gene 2016, 588, 54–61. [Google Scholar] [CrossRef]
- Chistiakov, D.; Sobenin, I.; Bobryshev, Y.; Orekhov, A. Mitochondrial dysfunction and mitochondrial DNA mutations in atherosclerotic complications in diabetes. World J. Cardiol. 2012, 4, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Cervelli, T.; Borghini, A.; Galli, A.; Andreassi, M. DNA damage and repair in atherosclerosis: Current insights and future perspectives. Int. J. Mol. Sci. 2012, 13, 16929–16944. [Google Scholar] [CrossRef] [PubMed]
- Coxhead, J.; Williams, E.; Mathers, J. DNA mismatch repair status may influence anti-neoplastic effects of butyrate. Biochem. Soc. Trans. 2005, 33, 728–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Qu, X.; Yuan, F.; Zhang, M.; Fang, W. A RTK-based functional RNAi screen reveals determinants of PTX-3 expression. Int. J. Clin. Exp. Pathol. 2013, 6, 660–668. [Google Scholar] [PubMed]
- Philips, S.; Richter, A.; Oesterreich, S.; Rae, J.; Flockhart, D.; Perumal, N.; Skaar, T. Functional characterization of a genetic polymorphism in the promoter of the ESR2 gene. Horm. Cancer 2012, 3, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McRobb, L.S.; McGrath, K.C.Y.; Tsatralis, T.; Liong, E.C.; Tan, J.T.M.; Hughes, G.; Handelsman, D.J.; Heather, A.K. Estrogen receptor control of atherosclerotic calcification and smooth muscle cell osteogenic differentiation. Arterioscler. Thromb.Vasc. Biol. 2017, 37, 1127–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.J.; Kruszka, B.; Delaney, J.A.; He, K.; Burke, G.L.; Alonso, A.; Bild, D.E.; Budoff, M.; Michos, E.D. Calcium intake from diet and supplements and the risk of coronary artery calcification and its progression among older adults: 10-year follow-up of the Multi-Ethnic Study of Atherosclerosis (MESA). J. Am. Heart Assoc. 2016, 5, e003815. [Google Scholar] [CrossRef]
- Al-Shakfa, F.; Dulucq, S.; Brukner, I.; Milacic, I.; Ansari, M.; Beaulieu, P.; Moghrabi, A.; Laverdiere, C.; Sallan, S.; Silverman, L.; et al. DNA variants in region for noncoding interfering transcript of dihydrofolate reductase gene and outcome in childhood acute lymphoblastic leukemia. Clin. Cancer Res. 2009, 15, 6931–6938. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.; Chen, W.; Dikalov, S.; Li, L. Regulation of endothelial cell tetrahydrobiopterin pathophysiological and therapeutic implications. Adv. Pharmacol. 2010, 60, 107–132. [Google Scholar]
- Okumura, K.; Tsukamoto, H. Folate in smokers. Clin. Chim. Acta 2011, 412, 521–526. [Google Scholar] [CrossRef]
- Wilhelmson, A.; Bourghardt-Fagman, J.; Gogos, J.; Fogelstrand, P.; Tivesten, A. Catechol-O-methyltransferase is dispensable for vascular protection by estradiol in mouse models of atherosclerosis and neointima formation. Endocrinology 2011, 152, 4683–4690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lievens, D.; Habets, K.; Robertson, A.; Laouar, Y.; Winkels, H.; Rademakers, T.; Beckers, L.; Wijnands, E.; Boon, L.; Mosaheb, M.; et al. Abrogated transforming growth factor beta receptor II (TGFβRII) signalling in dendritic cells promotes immune reactivity of T cells resulting in enhanced atherosclerosis. Eur. Heart J. 2013, 34, 3717–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, J.; Zhao, D.; Zhang, L.; Song, S.; Li, Y.; Sun, F.; Ding, X.; Yu, C.; Li, Y.; Liu, M.; et al. Type III transforming growth factor-β receptor drives cardiac hypertrophy through β-arrestin2-dependent activation of calmodulin-dependent protein kinase II. Hypertension 2016, 68, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dol-Gleizes, F.; Delesque-Touchard, N.; Mares, A.; Nestor, A.; Schaeffer, P.; Bono, F. A new synthetic FGF receptor antagonist inhibits arteriosclerosis in a mouse vein graft model and atherosclerosis in apolipoprotein E-deficient mice. PLoS ONE 2013, 8, e80027. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Rui, X.; He, S. Effect of heparin-derived oligosaccharide on bFGFR1 and bFGFR2 in vascular smooth muscle cells. Vasc. Endovasc. Surg. 2014, 48, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Hartgerink, J.; Cramm, J.; de Vos, A.; Bakker, T.; Steyerberg, E.; Mackenbach, J.; Nieboer, A. Situational awareness, relational coordination and integrated care delivery to hospitalized elderly in the Netherlands: A comparison between hospitals. BMC Geriatr. 2014, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.; Calvert, P.; Mercer, J.; Harrison, J.; Baker, L.; Figg, N.; Kumar, S.; Wang, J.; Hurst, L.; Obaid, D.; et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Ren, J.; Li, G.; Wu, J.; Wu, X.; Wang, G.; Gu, G.; Ren, H.; Hong, Z.; Li, J. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018, 9, 403. [Google Scholar] [CrossRef]
- Cioffi, F.; Senese, R.; Lasala, P.; Ziello, A.; Mazzoli, A.; Crescenzo, R.; Liverini, G.; Lanni, A.; Goglia, F.; Iossa, S. Fructose-rich diet affects mitochondrial DNA damage and repair in rats. Nutrients 2017, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Reddyvari, H.; Govatati, S.; Matha, S.; Korla, S.; Malempati, S.; Pasupuleti, S.; Bhanoori, M.; Nallanchakravarthula, V. Therapeutic effect of green tea extract on alcohol induced hepatic mitochondrial DNA damage in albino wistar rats. J. Adv. Res. 2017, 8, 289–295. [Google Scholar] [CrossRef]
- Stein, S.; Lohmann, C.; Handschin, C.; Stenfeldt, E.; Boren, J.; Luscher, T.; Matter, C. ApoE-/- PGC-1α-/- mice display reduced IL-18 levels and do not develop enhanced atherosclerosis. PLoS ONE 2010, 5, e13539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Zhang, J.; Wang, H. PGC-1α limits angiotensin II-induced rat vascular smooth muscle cells proliferation via attenuating NOX1-mediated generation of reactive oxygen species. Biosci. Rep. 2015, 35, e00252. [Google Scholar] [CrossRef]
- Vernochet, C.; Mourier, A.; Bezy, O.; Macotela, Y.; Boucher, J.; Rardin, M.; An, D.; Lee, K.; Ilkayeva, O.; Zingaretti, C.; et al. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab. 2012, 16, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Azuma, H.; Aihara, K.; Fujimura, M.; Akaike, M.; Mitsui, T.; Matsumoto, T. Vascular smooth muscle cell proliferation is dependent upon upregulation of mitochondrial transcription factor A (mtTFA) expression in injured rat carotid artery. Atherosclerosis 2005, 178, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Johnson, H.; Rao, M.; Martin, G.; Sancheti, H.; Silkwood, K.; Decker, C.; Nguyen, K.; Casian, J.; Cadenas, E.; et al. Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free Radic. Biol. Med. 2017, 102, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, J.; Cheng, K.; Figg, N.; Gorenne, I.; Mahmoudi, M.; Griffin, J.; Vidal-Puig, A.; Logan, A.; Murphy, M.; Bennett, M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ. Res. 2010, 107, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Yu, E.; Figg, N.; Cheng, K.; Prime, T.; Griffin, J.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.; Bennett, M. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef]
- Liu, L.; Nagai, I.; Gao, Y.; Matsushima, Y.; Kawai, Y.; Sayama, K. Effects of catechins and caffeine on the development of atherosclerosis in mice. Biosci. Biotechnol. Biochem. 2017, 81, 1948–1955. [Google Scholar] [CrossRef] [Green Version]
- Razani, B.; Feng, C.; Semenkovich, C. p53 is required for chloroquine-induced atheroprotection but not insulin sensitization. J. Lipid Res. 2010, 51, 1738–1746. [Google Scholar] [CrossRef] [Green Version]
- Nair, V.D. Activation of p53 signaling initiates apoptotic death in a cellular model of Parkinson’s disease. Apoptosis 2006, 11, 955–966. [Google Scholar] [CrossRef]
- Stamatelopoulos, K.; Karatzi, K.; Sidossis, L. Noninvasive methods for assessing early markers of atherosclerosis: The role of body composition and nutrition. Curr. Opin. Clin. Nutr. Metab. Care. 2009, 12, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Teng, I.; Tsai, M.; Shih, S.; Tsuei, B.; Chang, H.; Chuang, Y.; Lin, C.; Chern, C.; Chen, S. Chalcone derivatives enhance ATP-binding cassette transporters A1 in human THP-1 macrophages. Molecules 2018, 23, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Wang, Y. Berberine alleviates hepatic lipid accumulation by increasing ABCA1 through the protein kinase C δ pathway. Biochem. Biophys. Res. Commun. 2018, 498, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, D.; Zhang, H. Investigation of the underlying genes and mechanism of macrophage-enriched ruptured atherosclerotic plaques using bioinformatics method. J. Atheroscler. Thromb. 2019, 26, 636–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Kim, C.; Simmons, R.; Jo, H. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: Mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2206–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergenreider, E.; Heydt, S.; Treguer, K.; Boettger, T.; Horrevoets, A.; Zeiher, A.; Scheffer, M.; Frangakis, A.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012, 14, 249–256. [Google Scholar] [CrossRef]
- Castiglioni, S.; Monti, M.; Arnaboldi, L.; Canavesi, M.; Ainis Buscherini, G.; Calabresi, L.; Corsini, A.; Bellosta, S. ABCA1 and HDL3 are required to modulate smooth muscle cells phenotypic switch after cholesterol loading. Atherosclerosis 2017, 266, 8–15. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Cao, C.; Yang, X.; Ma, S.; Han, X.; Yang, X.; Yang, A.; Tian, J.; Xu, H.; et al. A regulatory circuit involving miR-143 and DNMT3a mediates vascular smooth muscle cell proliferation induced by homocysteine. Mol. Med. Rep. 2016, 13, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Xuekelati, S.; Zhang, Y.; Yin, Y.; Li, Y.; Chai, R.; Li, X.; Peng, Y.; Wu, J.; Guo, X. Expression levels of atherosclerosis-associated miR-143 and miR-145 in the plasma of patients with hyperhomocysteinaemia. BMC Cardiovasc. Disord. 2017, 17, 163. [Google Scholar] [CrossRef]
- Hahn, S.; Buratowski, S.; Sharp, P.; Guarente, L. Yeast TATA-binding protein TFIID binds to TATA elements with both consensus and nonconsensus DNA sequences. Proc. Natl. Acad. Sci. USA 1989, 86, 5718–5722. [Google Scholar] [CrossRef] [Green Version]
- Bucher, P. Weight matrix descriptions of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. J. Mol. Biol. 1990, 212, 563–578. [Google Scholar] [CrossRef]
- Karas, H.; Knuppel, R.; Schulz, W.; Sklenar, H.; Wingender, E. Combining structural analysis of DNA with search routines for the detection of transcription regulatory elements. Comput. Applic. Biosci. 1996, 12, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, B. Purification of the human TATA-binding protein, TBP. Methods Mol. Biol. 1995, 37, 359–367. [Google Scholar] [PubMed]




| Gene | dbSNP ID [12] or [Ref] | DNA, Genome Sequence | KD, nM | Clinical Data or Candidate SNP Markers | AS | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5′ flank | WT | min | 3′ flank | WT | min | Δ | Z | α | ρ | |||||
| GCG | rs183433761 | gctggagagt | a | g | tataaaagca | 1 | 2 | < | 8 | 10−6 | A | hypogluca-gonemia and postprandial increased ratio [INS]:[GCG] accelerates atherogenesis | ↑ | [115,116,117,118] |
| rs1342326265 | tataaaagca | g | a | tgcgccttgg | 0.9 | 1.2 | < | 5 | 10−6 | A | ↑ | |||
| rs757035851 | tatataaaag | cag | - | tgcgccttgg | 0.9 | 1.1 | < | 3 | 10−3 | B | ↑ | |||
| rs773542506 | tggagagtat | a | g | taaaagcagt | 1 | 2 | < | 14 | 10−6 | A | ↑ | |||
| rs984859897 | agctggagag | t | c | atataaaagc | 1 | 2 | < | 11 | 10−6 | A | ↑ | |||
| LEP | rs201381696 | tcgggccgct | a | g | taagaggggc | 4 | 12 | < | 18 | 10−6 | A | hypoleptine-mia, obesity | ↑ | [119] |
| rs1458202641 | ggggcgggca | a | a | gaggggcggg | 4 | 6 | < | 8 | 10−6 | A | accelerate atherogenesis | ↑ | [120] | |
| rs34104384 | ccgctataag | a | t | ggggcgggca | 4 | 3 | > | 4 | 10−3 | B | slowed down atherogenesis (athero-protector) | ↓ | [121] | |
| rs200487063 | tgatcgggcc | g | a | ctataagagg | 4 | 2 | > | 6 | 10−6 | A | ↓ | |||
| rs1249322424 | gtgatcgggc | c | a | gctataagag | 4 | 3 | > | 3 | 10−2 | C | ↓ | |||
| APOA1 | [122] | tgcagacata | a | c | ataggccctg | 3 | 4 | < | 5 | 10−6 | A | hematuria, fatty liver, obesity | ↑ | [122] |
| rs1428975217 | acataaatag | g | t | ccctgcaaga | 2.6 | 3.0 | < | 2 | 0.05 | D | accelerated atherogenesis (exogenous APOA1 is atheropro-tector) | ↑ | [123,124] | |
| rs1017922094 | cagacataaa | t | c | aggccctgca | 3 | 6 | < | 9 | 10−6 | A | ↑ | |||
| rs1297144980 | cctggctgca | 6bp | - | aataggccct | 3 | 21 | < | 30 | 10−6 | A | ↑ | |||
| HTR2C | rs3813929 | cccctcatcc | c | t | gcttttggcc | 73 | 56 | > | 6 | 10−6 | A | obesity com-plication of olanzapine antipsycho-tic therapy | ↑ | [15] |
| rs1348095721 | agagcgtggt | g | a | cagattcacc | 73 | 56 | > | 5 | 10−3 | B | hypercor-tisolemia accelerates atherogenesis that can be slowed down by lorcaserin | ↑ | [125,126] | |
| rs1444133212 | caagagcgtg | c | t | caagagcgtg | 73 | 39 | > | 13 | 10−6 | A | ↑ | |||
| rs886838672 | ccgcttttgg | c | t | ccaagagcgt | 73 | 34 | > | 15 | 10−6 | A | ↑ | |||
| rs1376972872 | tcccgctttt | g | t | gcccaagagc | 73 | 43 | > | 11 | 10−6 | A | ↑ | |||
| rs1222709869 | tggctcctcc | c | t | ctcatcccgc | 73 | 50 | > | 8 | 10−6 | A | ↑ | |||
| IL1B | rs1143627 | ttttgaaagc | c | t | ataaaaacag | 5 | 2 | > | 15 | 10−6 | A | obesity, Gra-ves’ disease, depression; lung, liver, breast, and gastric cancer | ↑ | [127,128,129,130,131,132,133] |
| accelerated atherogenesis, | [134] | |||||||||||||
| rs549858786 | tgaaagccat | a | t | aaaacagcga | 5 | 7 | < | 8 | 10−6 | A | slowed down atherogenesis | ↓ | [135,136] | |
| Gene | dbSNP ID [12] or [Ref] | DNA, Genome Sequence | KD, nM | Clinical Data or Candidate SNP Markers | AS | [Ref] | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5′ flank | WT | min | 3′ flank | WT | min | Δ | Z | α | ρ | |||||
| ADH7 | rs17537595 | gctgctgtta | t | c | atacaacaga | 1 | 3 | < | 13 | 10−6 | A | esophageal cancer | ↑ | [137] |
| rs372329931 | agctgctgtt | a | g | tatacaacag | 1 | 3 | < | 13 | 10−6 | A | post-esopha-gectomy necrosis comorbid with atherogenesis | ↑ | [138] | |
| rs755152695 | caagctgctg | t | c | tatatacaac | 1.0 | 1.4 | < | 4 | 10−3 | B | ↑ | |||
| rs1238877951 | gcacaagctg | c | a | tgttatatac | 1.0 | 1.2 | < | 3 | 10−2 | C | ↑ | |||
| HSD17B1 | rs201739205 | aggtgatatc | a | c | agcccagagc | 13 | 18 | < | 5 | 10−3 | B | breast cancer | ↓ | [139] |
| rs748743528 | gcaggtgata | t | c | caagcccaga | 13 | 28 | < | 13 | 10−6 | A | biomarker of the atherogenesis slowed down by drug IMM-H007 | ↓ | [140] | |
| rs779674159 | agcaggtgat | a | t | tcaagcccag | 13 | 35 | < | 18 | 10−6 | A | ↓ | |||
| rs1282820277 | cgaagcaggt | g | a | atatcaagcc | 13 | 7 | > | 9 | 10−6 | A | accelerated atherogenesis | ↑ | [141] | |
| rs755636251 | ggcgaagcag | g | t | tgatatcaag | 13 | 11 | > | 2 | 0.05 | D | ↑ | |||
| rs1332869256 | gggcgaagca | g | c | gtgatatcaa | 13 | 11 | > | 4 | 10−3 | B | ↑ | |||
| MLH1 | rs63750527 | tacaacaaag | g | c | ggacttcaga | 11 | 9 | > | 4 | 10−3 | B | nonpolyposis colon cancer | ↓ | [15] |
| rs756099600 | atacaacaaa | g | a, c | gggacttcag | 11 | 10 | > | 3 | 0.05 | D | ↓ | |||
| rs753671152 | caaaggggac | t | a | tcagaaatgt | 11 | 9 | > | 4 | 10−3 | B | improved DNA mismatch repair can slow down atherogenesis | ↓ | [142,143,144] | |
| rs1424963586 | gatacaacaa | a | c | ggggacttca | 11 | 9 | > | 3 | 10−2 | C | ↓ | |||
| rs752622244 | cttggagggg | g | t | atacaacaaa | 11 | 3 | > | 17 | 10−6 | A | ↓ | |||
| rs587778905 | gcttggaggg | 21bp | taaa | agaaatgtca | 11 | 19 | < | 9 | 10−6 | A | accelerated atherogenesis, which can be slowed down by restricting alcohol and red meat intake | ↑ | ||
| rs34285587 | gggggataca | a | g* | caaaggggac | 11 | 20 | < | 9 | 10−6 | A | ↑ | |||
| rs864622145 | ggagggggat | a | g | caacaaaggg | 11 | 27 | < | 17 | 10−6 | A | ↑ | |||
| RET | rs10900296 | gccggcgctt | a | g, c | cctcgcttca | 30 | 90 | < | 20 | 10−6 | A | pheochro-mocytoma | ↑ | [15] |
| rs551321384 | agccggcgct | t | c | acctcgcttc | 30 | 75 | < | 17 | 10−6 | A | dysregulated atheroprotector pentraxin-3 | ↑ | [145] | |
| rs1191017949 | cagccggcgc | t | c | tacctcgctt | 30 | 48 | < | 7 | 10−6 | A | ↑ | |||
| rs1237152255 | tacctcgctt | c | t | ttacctcgct | 30 | 20 | > | 6 | 10−6 | A | slowed down atherogenesis | ↓ | ||
| rs1372293149 | cgcagccggc | g | a | cttacctcgc | 30 | 16 | > | 10 | 10−6 | A | ↓ | |||
| rs10900297 | gcgcttacct | c | a | gcttcagtcc | 30 | 9 | > | 17 | 10−6 | A | pheochro-mocytoma | ↓ | [15] | |
| ESR2 | rs35036378 | cctctcggtc | t | g | ttaaaaggaa | 6 | 8 | < | 5 | 10−3 | B | ESR2-deficient pT1 tumor | ↑ | [146] |
| rs766797386 | ttaaaaggaa | g | t | aaggggctta | 6 | 7 | < | 3 | 10−2 | C | accelerated calcification | ↑ | [147,148] | |
| DHFR | rs10168 | ctgcacaaat | g | a | gggacgaggg | 15 | 9 | > | 9 | 10−6 | A | resistance to methotrexate | ↓ | [149] |
| rs750793297 | tgcacaaatg | g | t | ggacgagggg | 15 | 13 | > | 3 | 10−2 | C | DHFR deficit is a biomarker of accelerated atherogenesis, which can be slowed down by smoking restriction | ↓ | [150,151] | |
| rs1464445339 | ggggcggggc | 35bp | - | ggccacaatt | 15 | 170 | < | 44 | 10−6 | A | ↑ | |||
| rs766799008 | gcctgcacaa | a | g | tggggacgag | 15 | 19 | < | 3 | 10−3 | B | ↑ | |||
| rs764508464 | gcctgcacaa | a | - | tggggacgag | 15 | 37 | < | 17 | 10−6 | A | ↑ | |||
| rs754122321 | ctcgcctgca | c | g | aaatggggac | 15 | 25 | < | 9 | 10−6 | A | ↑ | |||
| rs1328822484 | cctcgcctgc | a | g | caaatgggga | 15 | 58 | < | 25 | 10−6 | A | ↑ | |||
| Gene | dbSNP ID [12] or [Ref] | DNA, Genome Sequence | KD, nM | Clinical Data or Candidate SNP Markers | AS | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5′ flank | WT | min | 3′ flank | WT | min | Δ | Z | α | ρ | |||||
| COMT | rs370819229 | ccgcccgcca | c | a* | ggcctgcgtc | 187 | 211 | < | 2 | 0.05 | D | dilated car-diomyopathy | ↓ | [15] |
| rs901020754 | acggcctgcg | t | c | ccgccaccgg | 187 | 243 | < | 5 | 10−3 | B | slowed atherogenesis due to both estradiol (substrate) and 2-metho-xy estradiol (metabolite), which are athero-protective | ↓ | [152] | |
| rs779542396 | cgccacggcc | t | c | gcgtccgcca | 187 | 243 | < | 5 | 10−3 | B | ↓ | |||
| rs45593642 | ccaccggaag | c | a | gccctcctaa | 187 | 115 | > | 9 | 10−6 | A | ↓ | |||
| rs45581136 | gccaccggaa | g | a | cgccctccta | 187 | 160 | > | 3 | 10−2 | C | ↓ | |||
| rs868447575 | cgtccgccac | c | a, t | ggaagcgccc | 187 | 74 | > | 17 | 10−6 | A | ↓ | |||
| rs1369731401 | gcgtccgcca | c | a | cggaagcgcc | 187 | 118 | > | 8 | 10−6 | A | ↓ | |||
| rs928358205 | ctgcgtccgc | c | t | accggaagcg | 187 | 82 | > | 13 | 10−6 | A | ↓ | |||
| rs1060501404 | gcctgcgtcc | g | a | ccaccggaag | 187 | 137 | > | 5 | 10−6 | A | ↓ | |||
| rs1296549321 | ggcctgcgtc | c | t | gccaccggaa | 187 | 152 | > | 4 | 10−3 | B | ↓ | |||
| rs1333679310 | cacggcctgc | g | a | tccgccaccg | 187 | 110 | > | 10 | 10−6 | A | ↓ | |||
| rs981175339 | ccacggcctg | c | a, t | cgtccgccac | 187 | 113 | > | 9 | 10−6 | A | ↓ | |||
| rs748298389 | gccacggcct | g | t | cgtccgccac | 187 | 70 | > | 18 | 10−6 | A | ↓ | |||
| rs1428300695 | ccgccacggc | c | t* | tgcgtccgcc | 187 | 117 | > | 9 | 10−6 | A | ↓ | |||
| rs562298402 | cgcccgccac | g | a | gcctgcgtcc | 187 | 150 | > | 4 | 10−3 | B | ↓ | |||
| rs1249101844 | cgcaccccgc | 15bp | - | ccgccaccgg | 187 | 83 | > | 15 | 10−6 | A | ↓ | |||
| rs777650793 | gtccgccacc | g | a | gaagcgccct | 187 | 82 | > | 15 | 10−6 | A | cardiovascu-lar disease | ↓ | [15] | |
| TGFBR2 | rs138010137 | cgcagcgctg | a | g | gttgaagttg | 29 | 39 | < | 6 | 10−6 | A | aortic thora-cic aneurysm & dissection | ↑ | [15] |
| rs1300366819 | cgctgagttg | a | g | agttgagtga | 29 | 53 | < | 16 | 10−6 | A | unchecked T-cell activation accelerates atherogenesis | ↑ | [153] | |
| rs1310294304 | agcgctgagt | t | a | gaagttgagt | 29 | 11 | > | 17 | 10−6 | A | hypertension accelerates atherogenesis | ↑ | [154] | |
| FGFR2 | rs886046768 | agagcgcggt | g | a | gagagccgag | 115 | 31 | > | 22 | 10−6 | A | cranio-synostosis | ↑ | [15] |
| rs1212347974 | tggaggagag | c | t | gcggtggaga | 115 | 99 | > | 3 | 10−2 | C | accelerated atherogenesis that can be slowed down by heparin-derived oligo-saccharide | ↑ | [155,156] | |
| rs1027484343 | ctggaggaga | g | t* | cgcggtggag | 115 | 101 | > | 3 | 10−2 | C | ↑ | |||
| rs1226640384 | ggctggagga | g | c | agcgcggtgg | 115 | 99 | > | 3 | 10−2 | C | ↑ | |||
| rs1189849606 | gcggctggag | g | a | agagcgcggt | 115 | 48 | > | 17 | 10−6 | A | ↑ | |||
| rs971411400 | atggtggtaa | c | t* | agtcatcctg | 13 | 11 | > | 3 | 10−2 | C | ↑ | |||
| rs778187292 | cctgtatggt | g | a | gtaacagtca | 13 | 7 | > | 9 | 10−6 | A | ↑ | |||
| rs1377663539 | gggcggcggc | t | c | ggaggagagc | 115 | 139 | < | 3 | 10−2 | C | slowed atherogenesis | ↑ | ||
| rs751951199 | aatcgcctgt | a | g | tggtggtaac | 13 | 30 | < | 13 | 10−6 | A | ↑ | |||
| rs757648006 | tcttaatcgc | c | g | tgtatggtgg | 13 | 17 | < | 3 | 10−2 | C | ↑ | |||
| rs387906677 | atcgcctgta | t | g | ggtggtaaca | 13 | 27 | < | 11 | 10−6 | A | bent bone dysplasia | ↑ | [15] | |
| ↑ | ||||||||||||||
| Gene | dbSNP ID [12] or [Ref] | DNA, Genome Sequence | KD, nM | Clinical Data or Candidate SNP Markers | AS | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5′ flank | WT | min | 3′ flank | WT | min | Δ | Z | α | ρ | |||||
| POLG | rs776506626 | gaagcgctgg | a | g | tatttaccag | 5 | 8 | < | 6 | 10−6 | A | accelerated atherogenesis, which can be slowed down by antioxidant MitoQ | ↑ | [158,159,160,161] |
| rs142347031 | gagcgcttac | t | g | aatgcagttt | 6 | 13 | < | 13 | 10−6 | A | ↑ | |||
| rs888362291 | ggacaccatg | a | c | gcatgcacat | 41 | 53 | < | 5 | 10−3 | B | ↑ | |||
| rs1460331646 | cctggacacc | a | g | tgagcatgca | 41 | 53 | < | 5 | 10−3 | B | ↑ | |||
| rs760935752 | cgtttcctgg | a | g | caccatgagc | 41 | 53 | < | 5 | 10−3 | B | ↑ | |||
| rs1266453407:g | ttggcggccc | t | g | cctattggtc | 24 | 30 | < | 3 | 10−3 | B | ↑ | |||
| rs1266453407:a | ttggcggccc | t | a | cctattggtc | 24 | 20 | > | 3 | 10−2 | C | slowed atherogenesis, while fructose-rich diet can accelerate mitochondrial DNA damage as atherogenic nutrition, as can alcohol, the effect of which could be reversed by green tea | ↓ | ||
| rs1257127556 | ccctcctatt | g | a | gtcgcgctgg | 24 | 5 | > | 22 | 10−6 | A | ↓ | |||
| rs1210506140 | gccctcctat | t | a | ggtcgcgctg | 24 | 8 | > | 16 | 10−6 | A | ↓ | |||
| rs3176154 | tggcggccct | c | t | ctattggtcg | 24 | 14 | > | 9 | 10−6 | A | ↓ | |||
| rs1024469208 | cctcacagac | c | t | tcggcccctg | 35 | 29 | > | 3 | 10−2 | C | ↓ | |||
| rs747270023 | cccccaccct | c | t * | acagacctcg | 35 | 15 | > | 13 | 10−6 | A | ↓ | |||
| rs2307434 | cccccaccct | c | - | acagacctcg | 35 | 21 | > | 8 | 10−6 | A | ↓ | |||
| rs983016336 | cttcccccac | c | a | ctcacagacc | 35 | 25 | > | 6 | 10−6 | A | ↓ | |||
| rs1318374817 | tgattgtctt | c | a | ccccaccctc | 35 | 22 | > | 9 | 10−6 | A | ↓ | |||
| rs768707883 | gctcctgatt | g | a | tcttccccca | 35 | 20 | > | 9 | 10−6 | A | ↓ | |||
| rs974500502 | tctcctgctc | c | t | tgattgtctt | 35 | 23 | > | 8 | 10−6 | A | ↓ | |||
| rs142460560 | tgaagcgctg | g | t | atatttacca | 5 | 3 | > | 7 | 10−6 | A | ↓ | |||
| rs769410130 | cagggctaag | c | t * | agcttccagc | 41 | 9 | > | 23 | 10−6 | A | ↓ | |||
| rs749799663 | ggccatctca | g | a | ggctaagcag | 41 | 33 | > | 4 | 10−3 | B | ↓ | |||
| rs769104909 | acatggccat | c | a | tcagggctaa | 41 | 16 | > | 16 | 10−6 | A | ↓ | |||
| rs1309549222 | gcatgcacat | g | a | gccatctcag | 41 | 17 | > | 15 | 10−6 | A | ↓ | |||
| rs767895645 | ccatgagcat | g | a * | cacatggcca | 41 | 10 | > | 20 | 10−6 | A | ↓ | |||
| rs144068243 | acaccatgag | c | t | atgcacatgg | 41 | 17 | > | 18 | 10−6 | A | ↓ | |||
| rs1206132846 | gtttcctgga | c | t | accatgagca | 41 | 28 | > | 7 | 10−6 | A | ↓ | |||
| rs766414821 | gcatgcgttt | c | t | ctggacacca | 41 | 35 | > | 3 | 10−2 | C | ↓ | |||
| rs1333954036 | tggtgttcga | c | t | gtggaggtct | 62 | 36 | > | 7 | 10−6 | A | ↓ | |||
| rs953495957 | gggggaggcc | g | a, t | tacccgtggc | 62 | 43 | > | 5 | 10−3 | B | ↓ | |||
| rs961105910 | tccagccagt | a | t | aaagaagcca | 16 | 7 | > | 11 | 10−6 | A | ↓ | |||
| PGC1A | rs1254748756 | ggactgtagt | a | g | agacaggtgc | 5 | 18 | < | 18 | 10−6 | A | slowed atherogenesis | ↓ | [162] |
| rs1206245736 | tgtttggatg | t | c | gtaaatgcag | 5 | 10 | < | 10 | 10−6 | A | ↓ | |||
| rs772816414 | gtttggatgt | g | a | taaatgcagg | 5 | 2 | > | 17 | 10−6 | A | ↓ | [163] | ||
| rs1334636034 | gtgtttggat | g | a | tgtaaatgca | 5 | 4 | > | 3 | 10−2 | B | ↓ | |||
| TFAM | rs1349790536 | gcccccatct | a | t | ccgaccggat | 18 | 25 | < | 5 | 10−6 | A | slowed atherogenesis, while chronic alcohol intake upregulates TFAM as pro-atherogenic risk factor | ↓ | [164,166] |
| rs1442727766 | cccgccccca | t | g | ctaccgaccg | 18 | 25 | < | 5 | 10−6 | A | ↓ | |||
| rs756889032 | catgtggggc | g | t * | tgctgagtgc | 39 | 44 | < | 2 | 0.05 | D | ↓ | |||
| rs200473819 | ctccgaagca | t | c | gtggggcgtg | 39 | 50 | < | 5 | 10−3 | B | ↓ | |||
| rs1247582808 | tctccgaagc | a | g | tgtggggcgt | 39 | 50 | < | 5 | 10−3 | B | ↓ | |||
| rs1468597986 | tcccgttact | a | g | tttctgaact | 5 | 11 | < | 9 | 10−6 | A | ↓ | |||
| rs1036018716 | cctctcccgt | t | c | actatttctg | 5 | 11 | < | 10 | 10−6 | A | ↓ | |||
| rs943871999 | cccatctacc | g | a | accggatgtt | 18 | 14 | > | 4 | 10−3 | B | propensity to intimal thickening of an artery or vein related to accelerated atherogenesis | ↑ | [165] | |
| rs571704530 | ccccatctac | c | t | gaccggatgt | 18 | 13 | > | 5 | 10−3 | B | ↑ | |||
| rs911015016 | cccccatcta | c | t | cgaccggatg | 18 | 15 | > | 3 | 10−2 | C | ↑ | |||
| rs1355238441 | tccgaagcat | g | a | tggggcgtgc | 39 | 13 | > | 18 | 10−6 | A | ↑ | |||
| rs1350851808 | tttctccgaa | g | c | catgtggggc | 39 | 34 | > | 3 | 0.05 | D | ↑ | |||
| rs1448492458 | gatggcgttt | c | g | tccgaagcat | 39 | 30 | > | 4 | 10−3 | B | ↑ | |||
| rs1337389336 | gagcgatggc | g | a * | tttctccgaa | 39 | 32 | > | 4 | 10−3 | B | ↑ | |||
| ATM | rs540204119 | cgggagtagg | t | c | agctgcgtgg | 12 | 15 | < | 3 | 10−2 | C | accelerated atherogenesis, which can be slowed down by anti-malarial drug chloroquine when p53 expression is normal, as can mitochondria-targeted antioxidant MitoQ regardless of p53 level | ↑ | [167,168,169,170,171,172] |
| rs960185644 | aagcgggagt | a | c | ggtagctgcg | 12 | 15 | < | 3 | 10−2 | C | ↑ | |||
| rs758371056 | gtatttagta | c | t | ttttagtcag | 3 | 5 | < | 7 | 10−6 | A | ↑ | |||
| rs1424898053 | tctctcgtat | t | c | tagtactttt | 3 | 5 | < | 8 | 10−6 | A | ↑ | |||
| rs1418169685 | ctctctcgta | t | c | ttagtacttt | 3 | 4 | < | 4 | 10−3 | B | ↑ | |||
| rs1439694580 | tgatctctct | c | g | tcgtatttag | 3 | 5 | < | 4 | 10−3 | B | ↑ | |||
| rs1461551448 | cgggtccaat | a | c | accctccatc | 23 | 45 | < | 12 | 10−6 | A | ↑ | |||
| rs1479937619 | agccgggtcc | a | g | ataaccctcc | 23 | 29 | < | 4 | 10−3 | B | ↑ | |||
| rs1179356361 | ccagcatagc | c | t | gggtccaata | 23 | 26 | < | 2 | 0.05 | D | ↑ | |||
| rs1290688759 | ttcacagata | t | c | aaaatattaa | 3 | 5 | < | 12 | 10−6 | A | ↑ | |||
| rs778072373 | gttcacagat | a | g | taaaatatta | 3 | 5 | < | 10 | 10−6 | A | ↑ | |||
| rs1162474448 | aacggaagtt | a | g | atatgatcat | 6 | 14 | < | 12 | 10−6 | A | ↑ | |||
| rs951064054 | gccgcggttg | a | c | tactactttg | 6 | 17 | < | 13 | 10−6 | A | ↑ | |||
| rs773550815:g | gccgggtcca | a | g | taaccctcca | 23 | 33 | < | 7 | 10−6 | A | ↑ | |||
| rs773550815:t | gccgggtcca | a | t | taaccctcca | 23 | 21 | > | 2 | 0.05 | D | slowed atherogenesis, while caffeine (ATM inhibi-tor) can post-prandially in-crease arterial stiffness for up to 3 h per cup of coffee as pro-atherogenic factor unlike mixtures of caffeine & catechins in tea as athero-protectors | ↓ | ||
| rs976385260 | acgacgaggg | c | a * | gaagagggtg | 69 | 50 | > | 6 | 10−6 | A | ↓ | |||
| rs1440912817 | gacgacgagg | g | t | cgaagagggt | 69 | 51 | > | 6 | 10−6 | A | ↓ | |||
| rs1382260245 | ggaggacgac | g | a | agggcgaaga | 69 | 36 | > | 12 | 10−6 | A | ↓ | |||
| rs942361620 | gcggggagga | c | t | gacgagggcg | 69 | 49 | > | 6 | 10−6 | A | ↓ | |||
| rs1457610144 | gggcggggag | g | a | acgacgaggg | 69 | 61 | > | 2 | 0.05 | D | ↓ | |||
| rs973002497 | agacttggag | 15bp | - | gggcggggag | 69 | 56 | > | 4 | 10−3 | B | ↓ | |||
| rs1222755392 | ggcggggatg | a | t | ggagggcggg | 69 | 53 | > | 5 | 10−3 | B | ↓ | |||
| rs1242858005 | ggaggggcgg | g | a | gatgaggagg | 69 | 57 | > | 4 | 10−3 | B | ↓ | |||
| rs1235571571 | gcgggagtag | g | a | tagctgcgtg | 12 | 7 | > | 7 | 10−6 | A | ↓ | |||
| rs1254449321 | agcgggagta | g | a | gtagctgcgt | 12 | 8 | > | 6 | 10−6 | A | ↓ | |||
| rs763320013 | ggtccaataa | c | t | cctccatccc | 23 | 19 | > | 4 | 10−3 | B | ↓ | |||
| rs373553992 | tccttctgtc | c | t | agcatagccg | 23 | 16 | > | 4 | 10−3 | B | ↓ | |||
| rs1397272250 | gacgttcaca | g | t | atataaaata | 3 | 1 | > | 10 | 10−6 | A | ↓ | |||
| rs1401830174 | tcgaaacagt | c | t | ataacggaag | 6 | 4 | > | 3 | 10−2 | C | ↓ | |||
| rs1310604038 | ccaccgccgc | g | c, t | gttgatacta | 6 | 5 | > | 2 | 0.05 | D | ↓ | |||
| Gene | dbSNP ID [12] or [Ref] | DNA, Genome Sequence | KD, nM | Clinical Data or Candidate SNP Markers | AS | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5′ flank | WT | min | 3′ flank | WT | min | Δ | Z | α | ρ | |||||
| MIR10B | rs1388274194 | cagcgaccta | g | t | cagcgaccta | 14 | 4 | > | 14 | 10−6 | A | ABCA1 over-inhibition that accelerates atherogenesis, which can be slowed down by dietary supplements of berberine | ↑ | [173,174] |
| rs564940769 | cggcagcgac | c | g * | taggtacctc | 14 | 25 | < | 8 | 10−6 | A | slowed atherogenesis | ↓ | ||
| MIR21 | rs752908264 | gtgacatctc | c | t | atggctgtac | 33 | 17 | > | 11 | 10−6 | A | reduced calcification of atherosclerotic plaque | ↓ | [175] |
| rs781007656 | atcgtgacat | c | a | tccatggctg | 33 | 10 | > | 20 | 10−6 | A | ↓ | |||
| rs368618785 | ctaccatcgt | g | t | acatctccat | 33 | 10 | > | 20 | 10−6 | A | ↓ | |||
| rs772840282 | actgtctgct | t | a | gttttgccta | 33 | 22 | > | 7 | 10−6 | A | ↓ | |||
| MIR143 | rs369969688 | cctctaacac | c | t | ccttctcctg | 17 | 13 | > | 5 | 10−3 | B | prevented transformation of cells over-loaded with cholesterol in-to foam cells slows down atherogenesis | ↓ | [176,177,178] |
| rs142037609 | tcccctctaa | c | t | accccttctc | 17 | 5 | > | 17 | 10−6 | A | ↓ | |||
| rs1035277319 | caggtcccct | c | a | taacacccct | 17 | 5 | > | 16 | 10−6 | A | ↓ | |||
| rs999653534 | ccaggtcccc | t | a | ctaacacccc | 17 | 14 | > | 3 | 10−2 | C | ↓ | |||
| rs1369382070 | caccccttct | - | c | cctggccagg | 17 | 22 | < | 5 | 10−3 | B | accelerated atherogenesis | ↑ | [179] | |
| rs568314295 | ccctctaaca | c | g, t | cccttctcct | 17 | 21 | < | 3 | 10−3 | B | ↑ | |||
| rs1033081876 | cccctctaac | a | c | ccccttctcc | 17 | 44 | < | 16 | 10−6 | A | ↑ | |||
| MIR145 | rs909856793 | cagctggtcc | t | c | tagggacacg | 45 | 56 | < | 3 | 10−3 | B | accelerated atherogenesis | ↑ | [177,178,180] |
| rs746241408 | cttagggaca | c | a, t | ggcggccttg | 45 | 38 | > | 3 | 0.05 | D | prevented transformation of cells over-loaded with cholesterol in-to foam cells slows down atherogenesis | ↓ | ||
| rs778670319 | tccttaggga | c | t | acggcggcct | 45 | 32 | > | 5 | 10−6 | D | ↓ | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponomarenko, M.; Rasskazov, D.; Chadaeva, I.; Sharypova, E.; Drachkova, I.; Oshchepkov, D.; Ponomarenko, P.; Savinkova, L.; Oshchepkova, E.; Nazarenko, M.; et al. Candidate SNP Markers of Atherogenesis Significantly Shifting the Affinity of TATA-Binding Protein for Human Gene Promoters Show Stabilizing Natural Selection as a Sum of Neutral Drift Accelerating Atherogenesis and Directional Natural Selection Slowing It. Int. J. Mol. Sci. 2020, 21, 1045. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031045
Ponomarenko M, Rasskazov D, Chadaeva I, Sharypova E, Drachkova I, Oshchepkov D, Ponomarenko P, Savinkova L, Oshchepkova E, Nazarenko M, et al. Candidate SNP Markers of Atherogenesis Significantly Shifting the Affinity of TATA-Binding Protein for Human Gene Promoters Show Stabilizing Natural Selection as a Sum of Neutral Drift Accelerating Atherogenesis and Directional Natural Selection Slowing It. International Journal of Molecular Sciences. 2020; 21(3):1045. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031045
Chicago/Turabian StylePonomarenko, Mikhail, Dmitry Rasskazov, Irina Chadaeva, Ekaterina Sharypova, Irina Drachkova, Dmitry Oshchepkov, Petr Ponomarenko, Ludmila Savinkova, Evgeniya Oshchepkova, Maria Nazarenko, and et al. 2020. "Candidate SNP Markers of Atherogenesis Significantly Shifting the Affinity of TATA-Binding Protein for Human Gene Promoters Show Stabilizing Natural Selection as a Sum of Neutral Drift Accelerating Atherogenesis and Directional Natural Selection Slowing It" International Journal of Molecular Sciences 21, no. 3: 1045. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031045