Innate Immunity: A Common Denominator between Neurodegenerative and Neuropsychiatric Diseases

 ,
,  , , ,
, , ,  , and
, and
Abstract
:
1. Introduction
2. Neurodegenerative Diseases
2.1. Alzheimer’s Disease
2.2. Parkinson’s Disease
- (i)
- Post-mortem pathological studies. Examinations of histopathological samples from human Parkinson’s diseased brains have revealed a wide distribution of activated microglia (positive for the MHC class II) in several brain regions, including the striatum, often in combination with α-synuclein-positive Lewy neurites [52,53,54]. In addition, inflammatory mediators which are released by, or which promote the activation of, microglia have been identified in the brain tissue of PD patients. Indeed, a high level of the CXC-family chemokine ligand 12 (CXCL12) and its receptor CXC-family chemokine receptor 4 (CXCR4) [55], as well as TNFα, IL-1β, interferon γ (IFNγ), nitric oxide synthase (NOS), and reactive oxygen species (ROS) have been found in the nigral tissue. Higher concentrations of the proinflammatory interleukins (IL-1β, IL-2, IL-6) and TNFα have also been found in the striatum [56,57,58,59].
- (ii)
- In vivo studies on biological samples (cerebrospinal fluid and blood). The same panel of proinflammatory mediators have been found both in cerebrospinal fluid (IL-1β, IL-6, and TNFα) and blood (serum and plasma) samples [58,59,60,61,62,63,64], thus confirming the role of these microglial-related inflammatory mediators.
- (iii)
- In vivo PET imaging studies. Studies that used the PK11195 PET ligand, a selective ligand for the peripheral benzodiazepine binding site (PBBS), which is considered a selective marker of in vivo microglial activation, demonstrated a widespread microglial activation in early stages of PD disease [65], but not in the late course of the disease. The new highly specific DPA714 (N,N-diethyl-2-[4-(2-fluoroethoxy)phenyl]-5,7-dimethylpyrazolo[1,5-a]-pyrimidine-3-acetamide) PET ligand has also been used to measure the regional distribution of activated microglia in PD patients, showing neuroinflammation within the substantia nigra of the most affected hemisphere [66]. In addition, the P2X7 (P2X purinoceptor 7 receptor) PET ligand revealed microglial P2X7 availability in acute but not chronic rodent models of PD [67]. Taken together, all these findings demonstrate the key role of activated microglia in the regions critically involved in PD, which takes place early in PD development.
2.3. Huntington’s Disease
2.4. Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
3. Psychiatric Diseases
3.1. Depression
3.2. Schizophrenia
3.3. Post-Traumatic Stress Disorder
3.4. Autism Spectrum Disorder
4. Differences and Similarities between Neurological and Psychiatric Disorders
- (i)
- Response mechanisms of innate immunity. The overall response is similar for both disorders and is characterized by inflammation mediated by resident glial cells, which produce molecules which, in turn, facilitate the recruitment of other immune cells, including the peripheral innate immunity cells (Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8 and Figure 9). These soluble inflammatory molecules include cytokines, chemokines, and complement proteins, with a profile that presents subtle differences among these disorders. Table 1 reports the main cytokines and chemokines involved in neurological and psychiatric diseases.
- (ii)
- Triggering of response and toxicity. Neurons are characterized by a notable susceptibility to inflammatory thrust, like that produced by cytokines, inducible nitric oxide synthase (iNOS), and phagocytic NADPH oxidase in the brain. If uncontrolled, these stimuli cause neuronal death via oxidative damage. The neurotoxicity is clearly more evident in neurological diseases, especially in neurodegenerative ones. Indeed, unlike psychiatric disorders, neurodegenerative diseases are characterized by severe, inexorable neuronal loss, and lead to the atrophy of specific brain regions. A particular characteristic of neurodegenerative diseases is the accumulation of misfolded proteins that contribute to neurotoxicity and in themselves are an inflammatory stimulus that can trigger and amplify the immune response.
- (iii)
- Gene/environment interaction and age of onset. Recent evidence has revealed that both neurological and psychiatric disorders share mechanisms of individual predisposition to the development of the disease. Despite this, there are differences in the weight of the genetic component between these disorders. Some neurodegenerative diseases are directly caused by a genetic mutation (such as HD, or some forms of AD, PD, FTD, and ALS). In other cases, the genetic component represents a risk factor that interacts with environmental factors, but the brain compensatory abilities are such that, despite the degenerative process beginning many years earlier, the disease becomes evident in senile or pre-senile age. In psychiatric diseases, the genetic susceptibility component always needs to fit together with environmental factors, sometimes represented by intercurrent infections, toxic environmental substances, or other insults that stimulate the inflammatory response in a period in which the correct brain architecture is being built (childhood or even intrauterine life), giving rise to neurodevelopmental disorders. In addition, there is a widespread genetic overlap across psychiatric disorders. In contrast, neurological disorders seem to be genetically distinct from each another and from psychiatric disorders, thus suggesting that genetic influence is not similar among these disorders. Differences in the timing and means of gene–environment interactions, as well as differences in interactions between genes, can produce clinical peculiarities of these [290]. The lack of large-scale genetic variants shared between neurological and psychiatric diseases could underpin the main etiological and pathogenetic differences, and be combined with neuroinflammatory mechanisms in these disorders.
- (iv)
- Gut microbiota–brain axis. The complex and multifaceted crosstalk between the microbiota, immune system, and CNS is a fundamental emerging pathophysiological aspect shared by both psychiatric and neurodegenerative disorders. Growing evidence suggests that brain-resident and peripheral immune cells play a pivotal role in managing gut microbiota–brain interaction. The gut microbiota is a critical factor in modulating the activities of glial cells resident in the brain, which are essential for several key processes, such as neurogenesis, neural growth, synapse homeostasis, neurotransmission, CNS immune response, and BBB integrity. The microbiota also activates peripheral immune response, with critical effects on brain inflammation. Accordingly, the interaction between gut microbiota and the immune system is a key factor in the pathogenetic cascade, leading to neurodevelopmental, psychiatric, and neurodegenerative diseases.
- (v)
- Therapeutic perspective. The therapeutic strategies currently adopted in neurological and psychiatric disorders are symptomatic and aim to restore the altered neurotransmitter balance or to provide depleted mediators. In psychiatry, they have led to the improvement of some disorders, and have made severe conditions, such as schizophrenia, more manageable. In the field of degenerative diseases, although therapeutic strategies are useful for counteracting the main symptoms, they do not lead to effective action on the course of the disease. Further knowledge on the immunological mechanisms underlying these pathologies may lead to common therapeutic strategies, aimed at modulating the inflammatory response underlying these disorders.
5. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| BBB | Blood-brain barrier |
| CNS | Central nervous system |
| CRP | C-reactive protein |
| CD | Crohn’s disease |
| CXCL | “C-X-C” motif chemokine ligand |
| CXCR | “C-X-C” motif chemokine receptor |
| DAMP | Damage associated molecular pattern |
| DC | Dendritic cell |
| DPR | Dipeptide repeats |
| FTD | Frontotemporal dementia |
| GWA | Genome-wide association studies |
| HD | Huntington’s disease |
| HLA-DR | Human leukocyte antigen – DR isotype |
| IL | Interleukin |
| IL-1RN | Interleukin 1 receptor antagonist protein |
| IFN | Interferon |
| iPSC | Induced pluripotent stem cell |
| IBD | Inflammatory bowel disease |
| LE | Limbic encephalitis |
| MBL | Mannan-binding lectin |
| MND | Motor neuron disease |
| NK | Natural killer |
| PAMP | Pathogen associated molecular pattern |
| PD | Parkinson’s disease |
| PRRs | Pattern-recognition receptors |
| ROS | Reactive oxygen species |
| TNF | Tumor necrosis factor |
| TGF | Transforming growth factor |
| UC | Ulcerative colitis |
References
- Arts, R.J.W.; Joosten, L.A.B.; Netea, M.G. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front. Immunol. 2018, 9, 298. [Google Scholar] [CrossRef]
- Salam, A.P.; Borsini, A.; Zunszain, P.A. Trained innate immunity: A salient factor in the pathogenesis of neuroimmune psychiatric disorders. Mol. Psychiatry 2018, 23, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Guadagno, E.; Presta, I.; Maisano, D.; Donato, A.; Pirrone, C.K.; Cardillo, G.; Corrado, S.D.; Mignogna, C.; Mancuso, T.; Donato, G.; et al. Role of macrophages in brain tumor growth and progression. Int. J. Mol. Sci. 2018, 19, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presta, I.; Vismara, M.; Novellino, F.; Donato, A.; Zaffino, P.; Scali, E.; Pirrone, K.C.; Spadea, M.F.; Malara, N.; Donato, G. Innate immunity cells and the neurovascular unit. Int. J. Mol. Sci. 2018, 19, 3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of Protein Aggregation in Mitochondrial Dysfunction and Neurodegeneration in Alzheimer’s and Parkinson’s Diseases. NeuroMolecular Med. 2003, 4, 21–35. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Novellino, F.; López, M.E.; Vaccaro, M.G.; Miguel, Y.; Delgado, M.L.; Maestu, F. Association Between Hippocampus, Thalamus, and Caudate in Mild Cognitive Impairment APOEε4 Carriers: A Structural Covariance MRI Study. Front. Neurol. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Novellino, F.; Vasta, R.; Sarica, A.; Chiriaco, C.; Salsone, M.; Morelli, M.; Arabia, G.; Saccà, V.; Nicoletti, G.; Quattrone, A. Relationship between Hippocampal Subfields and Category Cued Recall in AD and PDD: A Multimodal MRI Study. Neuroscience 2018, 371, 506–517. [Google Scholar] [CrossRef]
- Hensley, K. Neuroinflammation in Alzheimer’s disease: Mechanisms, pathologic consequences, and potential for therapeutic manipulation. J. Alzheimer’s Dis. 2010, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Thériault, P.; Elali, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467. [Google Scholar] [CrossRef]
- Edison, P.; Archer, H.A.; Gerhard, A.; Hinz, R.; Pavese, N.; Turkheimer, F.E.; Hammers, A.; Tai, Y.F.; Fox, N.; Kennedy, A.; et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol. Dis. 2008, 32, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Yokokura, M.; Mori, N.; Yagi, S.; Yoshikawa, E.; Kikuchi, M.; Yoshihara, Y.; Wakuda, T.; Sugihara, G.; Takebayashi, K.; Suda, S.; et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 343–351. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Salani, F.; Sterbini, V.; Sacchinelli, E.; Garramone, M.; Bossù, P. Is Innate Memory a Double-Edge Sword in Alzheimer’s Disease? A Reappraisal of New Concepts and Old Data. Front. Immunol. 2019, 10, 1768. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Djordjevic, J.; Sabbir, M.G.; Albensi, B.C. Traumatic Brain Injury as a Risk Factor for Alzheimer’s Disease: Is Inflammatory Signaling a Key Player? Curr. Alzheimer Res. 2016, 13, 730–738. [Google Scholar] [CrossRef]
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer’s Res. Ther. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Du Yan, S.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Invest. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Festoff, B.W.; Sajja, R.K.; van Dreden, P.; Cucullo, L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflammation 2016, 13, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, P.; Schachner, M.; Shen, Y.Q. HMGB1 in development and diseases of the central nervous system. Mol. Neurobiol. 2012, 45, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Majd, S.; Power, J.; Majd, Z. Alzheimer’s Disease and Cancer: When Two Monsters Cannot Be Together. Front. Neurosci. 2019, 13, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Valle, J.; Tejero, H.; Ibáñez, K.; Portero, J.L.; Krallinger, M.; Al-Shahrour, F.; Tabarés-Seisdedos, R.; Baudot, A.; Valencia, A. A molecular hypothesis to explain direct and inverse co-morbidities between Alzheimer’s Disease, Glioblastoma and Lung cancer. Sci. Rep. 2017, 7, 4474. [Google Scholar] [CrossRef] [Green Version]
- Lehrer, S. Glioma and Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2018, 2, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Donato, G.; Martinez Hoyos, J.; Amorosi, A.; Maltese, L.; Lavano, A.; Volpentesta, G.; Signorelli, F.; Pentimalli, F.; Pallante, P.; Ferraro, G.; et al. High mobility group A1 expression correlates with the histological grade of human glial tumors. Oncol. Rep. 2004, 11, 1209–1213. [Google Scholar] [CrossRef]
- Manabe, T.; Katayama, T.; Sato, N.; Gomi, F.; Hitomi, J.; Yanagita, T.; Kudo, T.; Honda, A.; Mori, Y.; Matsuzaki, S.; et al. Induced HMGA 1a expression causes aberrant splicing a Presenilin-2 pre-mRNA in sporadic Alzhiemer’s disease. Cell Death Differ. 2003, 10, 698–708. [Google Scholar] [CrossRef]
- Katayama, T.; Imaizumi, K.; Manabe, T.; Hitomi, J.; Kudo, T.; Tohyama, M. Induction of neuronal death by ER stress in Alzheimer’s disease. J. Chem. Neuroanat. 2004, 28, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Manabe, T.; Ohe, K.; Katayama, T.; Matsuzaki, S.; Yanagita, T.; Okuda, H.; Bando, Y.; Imaizumi, K.; Reeves, R.; Tohyama, M.; et al. HMGA1a: sequence-specific RNA-binding factor causing sporadic Alzheimer’s disease-linked exon skipping of presenilin-2 pre-mRNA. Genes Cells 2007, 12, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Sumter, T.F.; Xian, L.; Huso, T.; Koo, M.; Chang, Y.-T.; Almasri, T.N.; Chia, L.; Inglis, C.; Reid, D.; Resar, L.M.S. The High Mobility Group A1 (HMGA1) Transcriptome in Cancer and Development. Curr. Mol. Med. 2016, 16, 353–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohe, K.; Mayeda, A. HMGA1a Trapping of U1 snRNP at an Authentic 5′ Splice Site Induces Aberrant Exon Skipping in Sporadic Alzheimer’s Disease. Mol. Cell. Biol. 2010, 30, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Messineo, S.; Laria, A.E.; Arcidiacono, B.; Chiefari, E.; Luque Huertas, R.M.; Foti, D.P.; Brunetti, A. Cooperation between HMGA1 and HIF-1 Contributes to Hypoxia-Induced VEGF and Visfatin Gene Expression in 3T3-L1 Adipocytes. Front. Endocrinol. (Lausanne) 2016, 7, 73. [Google Scholar] [CrossRef]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenes by up-regulating BACE1 gene expression—PubMed result. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Z.; Esiri, M.M.; Smith, A.D. The cell division cycle and the pathophysiology of Alzheimer’s disease. Neuroscience 1998, 87, 731–739. [Google Scholar]
- Majd, S.; Zarifkar, A.; Rastegar, K.; Takhshid, M.A. Different fibrillar Abeta 1-42 concentrations induce adult hippocampal neurons to reenter various phases of the cell cycle. Brain Res. 2008, 1218, 224–229. [Google Scholar] [CrossRef]
- Weichhart, T.; Säemann, M.D. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann. Rheum. Dis. 2008, 67, iii70–iii74. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Di Domenico, F. Targeting mTOR to reduce Alzheimer-related cognitive decline: from current hits to future therapies. Expert Rev. Neurother. 2017, 17, 33–45. [Google Scholar] [CrossRef]
- Van Praag, H.; Schinder, A.F.; Christle, B.R.; Toni, N.; Palmer, T.D.; Gage, F.H. Functional neurogenesis in the adult hippocampus. Nature 2002, 415, 1030–1034. [Google Scholar] [CrossRef]
- Snyder, J.S.; Hong, N.S.; McDonald, R.J.; Wojtowicz, J.M. A role for adult neurogenesis in spatial long-term memory. Neuroscience 2005, 130, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, K.; Stathopoulos, A.; Pimplikar, S.W. APP intracellular domain impairs adult neurogenesis in transgenic mice by inducing neuroinflammation. PLoS ONE 2010, 5, e11866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Rolls, A.; Shechter, R.; London, A.; Ziv, Y.; Ronen, A.; Levy, R.; Schwartz, M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat. Cell Biol. 2007, 9, 1081–1088. [Google Scholar] [CrossRef]
- Kokaia, Z.; Martino, G.; Schwartz, M.; Lindvall, O. Cross-talk between neural stem cells and immune cells: The key to better brain repair? Nat. Neurosci. 2012, 15, 1078–1087. [Google Scholar] [CrossRef]
- Dickson, D.W.; Braak, H.; Duda, J.E.; Duyckaerts, C.; Gasser, T.; Halliday, G.M.; Hardy, J.; Leverenz, J.B.; Del Tredici, K.; Wszolek, Z.K.; et al. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 2009, 8, 1150–1157. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy bodies [8]. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Cerasa, A.; Novellino, F.; Quattrone, A. Connectivity Changes in Parkinson’s Disease. Curr. Neurol. Neurosci. Rep. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the: Substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Joers, V.; Tansey, M.G.; Mulas, G.; Carta, A.R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 2017, 155, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Shimoji, M.; Pagan, F.; Healton, E.B.; Mocchetti, I. CXCR4 and CXCL12 expression is increased in the nigro-striatal system of Parkinson’s disease. Neurotox. Res. 2009, 16, 318–328. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Narabayashi, H.; Riederer, P.; Nagatsu, T. Transforming growth factor-β1 levels are elevated in the striatum and in ventricular cerebrospinal fluid in Parkinson’s disease. Neurosci. Lett. 1995, 193, 129–132. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Hunot, S.; Dugas, N.; Faucheux, B.; Hartmann, A.; Tardieu, M.; Debré, P.; Agid, Y.; Dugas, B.; Hirsch, E.C. FcεRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-α in glial cells. J. Neurosci. 1999, 19, 3440–3447. [Google Scholar] [CrossRef] [Green Version]
- Brodacki, B.; Staszewski, J.; Toczyłowska, B.; Kozłowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFα, and INFγ concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Katsarou, Z.; Bostantjopoulou, S.; Hatzizisi, O.; Giza, E.; Soler-Cardona, A.; Kyriazis, G. ¿Factores inmunes o depresión? La fatiga relacionada con la enfermedad de Parkinson. Rev. Neurol. 2007, 45, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.R.; dos Santos, L.V.; Santos, R.M.S.; Campos, A.L.F.; Pimenta, A.L.; de Oliveira, M.S.; Bacheti, G.G.; Rocha, N.P.; Teixeira, A.L.; Christo, P.P.; et al. IL-6 serum levels are elevated in Parkinson’s disease patients with fatigue compared to patients without fatigue. J. Neurol. Sci. 2016, 370, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, A.; Fagone, P.; Donzuso, G.; Mangano, K.; Dibilio, V.; Caponnetto, S.; Bendtzen, K.; Zappia, M.; Nicoletti, F. Parkinson’s disease is associated with increased serum levels of macrophage migration inhibitory factor. Cytokine 2011, 55, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Blum-Degena, D.; Müller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Lavisse, S.; Wimberley, C.; Goutal, S.; Bottlaender, M.; Kuhnast, B.; Peyronneau, M.; Sarazin, M.; Hantraye, P.; Thiriez, C.; Remy, P. 18F-DPA714 PET reveals neuroinflammatory activity in the Substantia nigra of patients with Parkinson disease. In Proceedings of the Mov Disord; Wiley: Fontenay Aux Roses, France, 2019; p. 1913. [Google Scholar]
- Crabbé, M.; Van Der Perren, A.; Bollaerts, I.; Kounelis, S.; Baekelandt, V.; Bormans, G.; Casteels, C.; Moons, L.; Laere, K. Van Increased P2X7 receptor binding is associated with neuroinflammation in acute but not chronic rodent models for Parkinson’s disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted overexpression of human α-synuclein triggers microglial activation and an adaptive immune response in a mouse model of parkinson disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Guajardo, V.; Febbraro, F.; Kirik, D.; Romero-Ramos, M. Microglia acquire distinct activation profiles depending on the degree of α-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS ONE 2010, 5, e8784. [Google Scholar] [CrossRef]
- De Marco, E.V.; Tarantino, P.; Rocca, F.E.; Provenzano, G.; Civitelli, D.; De Luca, V.; Annesi, F.; Carrideo, S.; Cirò Candiano, I.C.; Romeo, N.; et al. Alpha-synuclein promoter haplotypes and dementia in Parkinson’s disease. Am. J. Med. Genet. Part. B Neuropsychiatr. Genet. 2008, 147, 403–407. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, S.S.; Cho, J.J.; Choi, D.H.; Hwang, O.; Shin, D.H.; Chun, H.S.; Beal, M.F.; Joh, T.H. Matrix metalloproteinase-3: A novel signaling proteinase from apoptotic neuronal cells that activates microglia. J. Neurosci. 2005, 25, 3701–3711. [Google Scholar] [CrossRef] [Green Version]
- Zecca, L.; Zucca, F.A.; Wilms, H.; Sulzer, D. Neuromelanin of the substantia nigra: A neuronal black hole with protective and toxic characteristics. Trends Neurosci. 2003, 26, 578–580. [Google Scholar] [CrossRef]
- Zecca, L.; Zucca, F.A.; Albertini, A.; Rizzio, E.; Fariello, R.G. A proposed dual role of neuromelanin in the pathogenesis of Parkinson’s disease. Neurology 2006, 67, S8–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia Nigra: An update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Punati, A.; Newman, M.B. Progressive dopamine neuron loss in Parkinson’s disease: The multiple hit hypothesis. Cell Transplant. 2006, 15, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvoisin, R.C.; Yahr, M.D.; Schweitzer, M.D.; Merritt, H.H. Parkinsonism Before and Since the Epidemic of Encephalitis Lethargica. Arch. Neurol. 1963, 9, 232–236. [Google Scholar] [CrossRef]
- Pradhan, S.; Pandey, N.; Shashank, S.; Gupta, R.K.; Mathur, A. Parkinsonism due to predominant involvement of substantia nigra in Japanese encephalitis. Neurology 1999, 53, 1781–1786. [Google Scholar] [CrossRef]
- Elbaz, A.; Levecque, C.; Clavel, J.; Vidal, J.S.; Richard, F.; Amouyel, P.; Alpérovitch, A.; Chartier-Harlin, M.C.; Tzourio, C. CYP2D6 Polymorphism, Pesticide Exposure, and Parkinson’s Disease. Ann. Neurol. 2004, 55, 430–434. [Google Scholar] [CrossRef]
- Langston, J.; Ballard, P.; Tetrud, J.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [Green Version]
- Sadek, A.H.; Rauch, R.; Schulz, P.E. Parkinsonism due to Manganism in a Welder. Int. J. Toxicol. 2003, 22, 393–401. [Google Scholar] [CrossRef]
- Hudnell, H.K. Effects from environmental Mn exposures: A review of the evidence from non-occupational exposure studies. Neurotoxicology 1999, 20, 379–398. [Google Scholar] [PubMed]
- Iregren, A. Manganese neurotoxicity in industrial exposures: Proof of effects, critical exposure level, and sensitive tests. Neurotoxicology 1999, 20, 315–324. [Google Scholar] [PubMed]
- Venkateshappa, C.; Harish, G.; Mythri, R.B.; Mahadevan, A.; Srinivas Bharath, M.M.; Shankar, S.K. Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: Implications for Parkinson’s disease. Neurochem. Res. 2012, 37, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Zhang, P.; Agid, Y.; Javoy-Agid, F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 1993, 52, 1–6. [Google Scholar] [CrossRef]
- Berbel, P.; Innocenti, G.M. The development of the corpus callosum in cats: A light- and electron- microscopic study. J. Comp. Neurol. 1988, 276, 132–156. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.P.Q.; Perreau, V.M.; Shultz, S.R.; Brady, R.D.; Lei, E.; Dixit, S.; Taylor, J.M.; Beart, P.M.; Boon, W.C. Inflammation in Traumatic Brain Injury: Roles for Toxic A1 Astrocytes and Microglial–Astrocytic Crosstalk. Neurochem. Res. 2019, 44, 1410–1424. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell. Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef]
- Söllvander, S.; Nikitidou, E.; Brolin, R.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Croisier, E.; Graeber, M.B. Glial degeneration and reactive gliosis in alpha-synucleinopathies: The emerging concept of primary gliodegeneration. Acta Neuropathol. 2006, 112, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Terada, S.; Ishizu, H.; Yokota, O.; Tsuchiya, K.; Nakashima, H.; Ishihara, T.; Fujita, D.; Uéda, K.; Ikeda, K.; Kuroda, S. Glial involvement in diffuse Lewy body disease. Acta Neuropathol. 2003, 105, 163–169. [Google Scholar] [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Rostami, J.; Holmqvist, S.; Lindström, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergström, J.; Roybon, L.; Erlandsson, A. Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef] [Green Version]
- Di Fonzo, A.; Chien, H.F.; Socal, M.; Giraudo, S.; Tassorelli, C.; Iliceto, G.; Fabbrini, G.; Marconi, R.; Fincati, E.; Abbruzzese, G.; et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 2007, 68, 1557–1562. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Gründemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Duran, R.; Lopez, G.; Kurzawa-Akanbi, M.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M.; Theuns, J.; Crosiers, D.; Cras, P.; et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013, 70, 727–735. [Google Scholar] [CrossRef] [PubMed]
- De Marco, E.V.; Annesi, G.; Tarantino, P.; Rocca, F.E.; Provenzano, G.; Civitelli, D.; Candiano, I.C.C.; Annesi, F.; Carrideo, S.; Condino, F.; et al. Glucocerebrosidase gene mutations are associated with Parkinson’s disease in southern Italy. Mov. Disord. 2008, 23, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Henry, A.G.; Aghamohammadzadeh, S.; Samaroo, H.; Chen, Y.; Mou, K.; Needle, E.; Hirst, W.D. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum. Mol. Genet. 2015, 24, 6013–6028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Bandopadhyay, R.; Lashley, T.; Renton, A.E.M.; Kingsbury, A.E.; Kumaran, R.; Kallis, C.; Vilariño-Güell, C.; O’Sullivan, S.S.; Lees, A.J.; et al. LRRK2 expression in idiopathic and G2019S positive Parkinson’s disease subjects: A morphological and quantitative study. Neuropathol. Appl. Neurobiol. 2011, 37, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef]
- Higashi, S.; Moore, D.J.; Yamamoto, R.; Minegishi, M.; Sato, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; Hino, H.; et al. Abnormal localization of leucine-rich repeat kinase 2 to the endosomal-lysosomal compartment in lewy body disease. J. Neuropathol. Exp. Neurol. 2009, 68, 994–1005. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.M.; Van Der Pol, S.M.A.; Jansen, Q.; Witte, M.E.; Van Der Valk, P.; Rozemuller, A.J.M.; Drukarch, B.; De Vries, H.E.; Van Horssen, J. Association of Parkinson disease-related protein PINK1 with Alzheimer disease and multiple sclerosis brain lesions. Free Radic. Biol. Med. 2011, 50, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Witte, M.E.; Bol, J.G.J.M.; Gerritsen, W.H.; van der Valk, P.; Drukarch, B.; van Horssen, J.; Wilhelmus, M.M.M. Parkinson’s disease-associated parkin colocalizes with Alzheimer’s disease and multiple sclerosis brain lesions. Neurobiol. Dis. 2009, 36, 445–452. [Google Scholar] [CrossRef]
- Van Horssen, J.; Drexhage, J.A.R.; Flor, T.; Gerritsen, W.; van der Valk, P.; de Vries, H.E. Nrf2 and DJ1 are consistently upregulated in inflammatory multiple sclerosis lesions. Free Radic. Biol. Med. 2010, 49, 1283–1289. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marco, E.V.; Annesi, G.; Tarantino, P.; Nicoletti, G.; Civitelli, D.; Messina, D.; Annesi, F.; Arabia, G.; Salsone, M.; Condino, F.; et al. DJ-1 is a Parkinson’s disease susceptibility gene in southern Italy. Clin. Genet. 2010, 77, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Nagakubo, D.; Taira, T.; Kitaura, H.; Ikeda, M.; Tamai, K.; Iguchi-Ariga, S.M.M.; Ariga, H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 1997, 231, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.; Scumaci, D.; Di Cello, A.; Venturella, R.; Donato, G.; Faniello, M.C.; Quaresima, B.; Cuda, G.; Zullo, F.; Costanzo, F. DJ-1 in endometrial cancer a possible biomarker to improve differential diagnosis between subtypes. Int. J. Gynecol. Cancer 2014, 24, 649–658. [Google Scholar] [CrossRef]
- Bourdenx, M.; Dehay, B. What lysosomes actually tell us about Parkinson’s disease? Ageing Res. Rev. 2016, 32, 140–149. [Google Scholar] [CrossRef]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Waak, J.; Weber, S.S.; Waldenmaier, A.; Görner, K.; Alunni-Fabbroni, M.; Schell, H.; Vogt-Weisenhorn, D.; Pham, T.T.; Reumers, V.; Baekelandt, V.; et al. Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 2009, 23, 2478–2489. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Scornaienchi, V.; Civitelli, D.; De Marco, E.V.; Annesi, G.; Tarantino, P.; Rocca, F.E.; Greco, V.; Provenzano, G.; Annesi, F.; Nicoletti, G.; et al. Mutation analysis of the PINK1 gene in Southern Italian patients with early- and late-onset parkinsonism. Park. Relat. Disord. 2012, 18, 651–653. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Shen, R.; Agnihotri, S.K.; Chen, Y.; Huang, Z.; Büeler, H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saft, C.; Zange, J.; Andrich, J.; Müller, K.; Lindenberg, K.; Landwehrmeyer, B.; Vorgerd, M.; Kraus, P.H.; Przuntek, H.; Schöls, L. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington’s disease. Mov. Disord. 2005, 20, 674–679. [Google Scholar] [CrossRef]
- Stüwe, S.H.; Goetze, O.; Lukas, C.; Klotz, P.; Hoffmann, R.; Banasch, M.; Orth, M.; Schmidt, W.E.; Gold, R.; Saft, C. Hepatic mitochondrial dysfunction in manifest and premanifest Huntington disease. Neurology 2013, 80, 743–746. [Google Scholar] [CrossRef]
- Taherzadeh-Fard, E.; Saft, C.; Akkad, D.A.; Wieczorek, S.; Haghikia, A.; Chan, A.; Epplen, J.T.; Arning, L. PGC-1alpha downstream transcription factors NRF-1 and TFAM are genetic modifiers of Huntington disease. Mol. Neurodegener. 2011, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.Y.; Fang, Z.H.; Yu, Z.X.; Wang, C.E.; Li, S.H.; Li, X.J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Silvestroni, A.; Faull, R.L.M.; Strand, A.D.; Möllera, T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport 2009, 20, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Imaging microglial activation in Huntington’s disease. Brain Res. Bull. 2007, 72, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Zhao, Z.; Chen, Y. The astrocytic GABA(A)/benzodiazepine-like receptor: the Joker receptor for benzodiazepine-mimetic drugs? Recent Pat. CNS Drug Discov. 2006, 1, 93–103. [Google Scholar] [CrossRef]
- Kwan, W.; Träger, U.; Davalos, D.; Chou, A.; Bouchard, J.; Andre, R.; Miller, A.; Weiss, A.; Giorgini, F.; Cheah, C.; et al. Mutant huntingtin impairs immune cell migration in Huntington disease. J. Clin. Invest. 2012, 122, 4737–4747. [Google Scholar] [CrossRef] [Green Version]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Phukan, J.; Pender, N.P.; Hardiman, O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007, 6, 994–1003. [Google Scholar] [CrossRef]
- Ng, A.S.L.; Rademakers, R.; Miller, B.L. Frontotemporal dementia: A bridge between dementia and neuromuscular disease. Ann. N. Y. Acad. Sci. 2015, 1338, 71–93. [Google Scholar] [CrossRef] [Green Version]
- Lomen-Hoerth, C.; Anderson, T.; Miller, B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002, 59, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Spalloni, A.; Longone, P. Cognitive impairment in amyotrophic lateral sclerosis, clues from the SOD1 mouse. Neurosci. Biobehav. Rev. 2016, 60, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Crespi, C.; Dodich, A.; Cappa, S.F.; Canessa, N.; Iannaccone, S.; Corbo, M.; Lunetta, C.; Falini, A.; Cerami, C. Multimodal MRI quantification of the common neurostructural bases within the FTD-ALS continuum. Neurobiol. Aging 2018, 62, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Kamminga, J.; Leslie, F.V.C.; Hsieh, S.; Caga, J.; Mioshi, E.; Hornberger, M.; Ballard, K.J.; Kiernan, M.C.; Hodges, J.R.; Burrell, J.R. Syntactic comprehension deficits across the FTD-ALS continuum. Neurobiol. Aging 2016, 41, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Puentes, F.; Malaspina, A.; Van Noort, J.M.; Amor, S. Non-neuronal cells in ALS: Role of glial, immune cells and blood-CNS barriers. Proc. Brain Pathol. 2016, 26, 248–257. [Google Scholar] [CrossRef]
- Bright, F.; Werry, E.L.; Dobson-Stone, C.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kassiou, M.; et al. Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol. 2019, 15, 540–555. [Google Scholar] [CrossRef]
- Clayton, E.L.; Mancuso, R.; Tolstrup Nielsen, T.; Mizielinska, S.; Holmes, H.; Powell, N.; Norona, F.; Overgaard Larsen, J.; Milioto, C.; Wilson, K.M.; et al. Early microgliosis precedes neuronal loss and behavioural impairment in mice with a frontotemporal dementia-causing CHMP2B mutation. Hum. Mol. Genet. 2017, 26, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Goldknopf, I.L.; Sheta, E.A.; Bryson, J.; Folsom, B.; Wilson, C.; Duty, J.; Yen, A.A.; Appel, S.H. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 342, 1034–1039. [Google Scholar] [CrossRef]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of Dendritic Cells, MCP-1, and Activated Microglia/Macrophages in Amyotrophic Lateral Sclerosis Spinal Cord Tissue. Ann. Neurol. 2004, 55, 221–235. [Google Scholar] [CrossRef]
- Troost, D.; Sillevis Smitt, P.A.E.; de Jong, J.M.B.B.; Swaab, D.F. Neurofilament and glial alterations in the cerebral cortex in amyotrophic lateral sclerosis. Acta Neuropathol. 1992, 84, 664–673. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle and Nerve 2002, 26, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar] [PubMed]
- Sasaki, S.; Maruyama, S. Immunocytochemical and ultrastructural studies of the motor cortex in amyotrophic lateral sclerosis. Acta Neuropathol. 1994, 87, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Jara, J.H.; Genç, B.; Stanford, M.J.; Pytel, P.; Roos, R.P.; Weintraub, S.; Mesulam, M.M.; Bigio, E.H.; Miller, R.J.; Özdinler, P.H. Evidence for an early innate immune response in the motor cortex of ALS. J. Neuroinflammation 2017, 14, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagnin, A.; Rossor, M.; Sampson, E.L.; MacKinnon, T.; Banati, R.B. In vivo detection of microglial activation in frontotemporal dementia. Ann. Neurol. 2004, 56, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kwong, L.K.; Truax, A.C.; Vanmassenhove, B.; Kretzschmar, H.A.; Van Deerlin, V.M.; Clark, C.M.; Grossman, M.; Miller, B.L.; Trojanowski, J.Q.; et al. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J. Neuropathol. Exp. Neurol. 2007, 66, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Beers, D.R.; Zhao, W.; Liao, B.; Kano, O.; Wang, J.; Huang, A.; Appel, S.H.; Henkel, J.S. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain. Behav. Immun. 2011, 25, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Pesaresi, M.G.; Gerbino, V.; Grosskreutz, J.; Carrì, M.T. Amyotrophic lateral sclerosis: New insights into underlying molecular mechanisms and opportunities for therapeutic intervention. Antioxidants Redox Signal. 2012, 17, 1277–1330. [Google Scholar] [CrossRef]
- Marchetti, B.; L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C. Vulnerability to Parkinson’s Disease: Towards an Unifying Theory of Disease Etiology. In Encyclopedia of Environmental Health; Elsevier: Amsterdam, The Netherlands, 2011; pp. 690–704. [Google Scholar] [CrossRef]
- Andersen, P.M. Genetics of sporadic ALS. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2001, 2, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kjældgaard, A.L.; Pilely, K.; Olsen, K.S.; Pedersen, S.W.; Lauritsen, A.Ø.; Møller, K.; Garred, P. Amyotrophic lateral sclerosis: The complement and inflammatory hypothesis. Mol. Immunol. 2018, 102, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Xie, W.; Le, W.; Beers, D.R.; He, Y.; Henkel, J.S.; Simpson, E.P.; Yen, A.A.; Xiao, Q.; Appel, S.H. Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J. Neuropathol. Exp. Neurol. 2004, 63, 964–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Invest. 2017, 127, 3250–3258. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.A.; Frick, P.; Grässer, F.A.; Gendron, T.F.; Petrucelli, L.; Cashman, N.R.; Edbauer, D.; Kremmer, E.; Prudlo, J.; Troost, D.; et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015, 130, 845–861. [Google Scholar] [CrossRef] [Green Version]
- Saberi, S.; Stauffer, J.E.; Jiang, J.; Garcia, S.D.; Taylor, A.E.; Schulte, D.; Ohkubo, T.; Schloffman, C.L.; Maldonado, M.; Baughn, M.; et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 135, 459–474. [Google Scholar] [CrossRef]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate invitro and invivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.H.; Nichol, K.E.; Sitch, T.; Sheu, P.; Chubb, C.; Miller, B.L.; Tomaselli, K.J.; Kim, R.C.; Cotman, C.W. DNA damage and activated caspase-3 expression in neurons and astrocytes: Evidence for apoptosis in frontotemporal dementia. Exp. Neurol. 2000, 163, 9–19. [Google Scholar] [CrossRef]
- Kersaitis, C.; Halliday, G.M.; Kril, J.J. Regional and cellular pathology in frontotemporal dementia: Relationship to stage of disease in cases with and without Pick bodies. Acta Neuropathol. 2004, 108, 515–523. [Google Scholar] [CrossRef]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Lai, L.; Butchbach, M.E.; Stockinger, M.P.; Shan, X.; Bishop, G.A.; Lin, C.L.G. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum. Mol. Genet. 2003, 12, 2519–2532. [Google Scholar] [CrossRef]
- Pardo, A.C.; Wong, V.; Benson, L.M.; Dykes, M.; Tanaka, K.; Rothstein, J.D.; Maragakis, N.J. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1G93A mice. Exp. Neurol. 2006, 201, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Mendonça, D.M.F.; Chimelli, L.; Martinez, A.M.B. Expression of ubiquitin and proteasome in motorneurons and astrocytes of spinal cords from patients with amyotrophic lateral sclerosis. Neurosci. Lett. 2006, 404, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Pasinelli, P.; Houseweart, M.K.; Brown, R.H.; Cleveland, D.W. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu, Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 13901–13906. [Google Scholar] [CrossRef] [Green Version]
- Ferraiuolo, L.; Heath, P.R.; Holden, H.; Kasher, P.; Kirby, J.; Shaw, P.J. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J. Neurosci. 2007, 27, 9201–9219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sta, M.; Sylva-Steenland, R.M.R.; Casula, M.; de Jong, J.M.B.V.; Troost, D.; Aronica, E.; Baas, F. Innate and adaptive immunity in amyotrophic lateral sclerosis: Evidence of complement activation. Neurobiol. Dis. 2011, 42, 211–220. [Google Scholar] [CrossRef]
- Annunziata, P.; Volpi, N. High levels of C3c in the cerebrospinal fluid from amyotrophic lateral sclerosis patients. Acta Neurol. Scand. 1985, 72, 61–64. [Google Scholar] [CrossRef]
- Ganesalingam, J.; An, J.; Shaw, C.E.; Shaw, G.; Lacomis, D.; Bowser, R. Combination of neurofilament heavy chain and complement C3 as CSF biomarkers for ALS. J. Neurochem. 2011, 117, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, Y.; Yamada, T. Increased concentration of C4d complement protein in CSF in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 1994, 57, 859–861. [Google Scholar] [CrossRef] [Green Version]
- Bahia El Idrissi, N.; Bosch, S.; Ramaglia, V.; Aronica, E.; Baas, F.; Troost, D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 72. [Google Scholar] [CrossRef] [Green Version]
- Heurich, B.; el Idrissi, N.B.; Donev, R.M.; Petri, S.; Claus, P.; Neal, J.; Morgan, B.P.; Ramaglia, V. Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J. Neuroimmunol. 2011, 235, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Bento-Abreu, A.; Nonneman, A.; Haeck, W.; Staats, K.; Geelen, V.; Hersmus, N.; Küsters, B.; Van Den Bosch, L.; Van Damme, P.; et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 2013, 136, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pape, K.; Tamouza, R.; Leboyer, M.; Zipp, F. Immunoneuropsychiatry—Novel perspectives on brain disorders. Nat. Rev. Neurol. 2019, 15, 317–328. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; Suy, E.; Vandervorst, C.; De Jonckheere, C.; Raus, J. Immune disturbances during major depression: Upregulated expression of interleukin-2 receptors. Neuropsychobiology 1990, 24, 115–120. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; Suy, E.; Vandervorst, C.; DeJonckheere, C.; Raus, J. Depression-related disturbances in mitogen-induced lymphocyte responses and interleukin-1β and soluble interleukin-2 receptor production. Acta Psychiatr. Scand. 1991, 84, 379–386. [Google Scholar] [CrossRef]
- Maes, M. Evidence for an immune response in major depression: A review and hypothesis. Prog. Neuropsychopharmacol. Biol. Psychiatry 1995, 19, 11–38. [Google Scholar] [CrossRef]
- Smith, R.S. The macrophage theory of depression. Med. Hypotheses 1991, 35, 298–306. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Howren, M.B.; Lamkin, D.M.; Suls, J. Associations of depression with c-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med. 2009, 71, 171–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichenberg, A.; Yirmiya, R.; Schuld, A.; Kraus, T.; Haack, M.; Morag, A.; Pollmächer, T. Cytokine-associated emotional and cognitive disturbances in humans. Arch. Gen. Psychiatry 2001, 58, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Harrison, N.A.; Brydon, L.; Walker, C.; Gray, M.A.; Steptoe, A.; Critchley, H.D. Inflammation Causes Mood Changes Through Alterations in Subgenual Cingulate Activity and Mesolimbic Connectivity. Biol. Psychiatry 2009, 66, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 2018, 37, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Hannestad, J.; DellaGioia, N.; Ortiz, N.; Pittman, B.; Bhagwagar, Z. Citalopram reduces endotoxin-induced fatigue. Brain. Behav. Immun. 2011, 25, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Raison, C.L.; Miller, A.H. When not enough is too much: The role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am. J. Psychiatry 2003, 160, 1554–1565. [Google Scholar] [CrossRef]
- Wichers, M.C.; Kenis, G.; Koek, G.H.; Robaeys, G.; Nicolson, N.A.; Maes, M. Interferon-α-induced depressive symptoms are related to changes in the cytokine network but not to cortisol. J. Psychosom. Res. 2007, 62, 207–214. [Google Scholar] [CrossRef]
- Capuron, L.; Gumnick, J.F.; Musselman, D.L.; Lawson, D.H.; Reemsnyder, A.; Nemeroff, C.B.; Miller, A.H. Neurobehavioral effects of interferon-α in cancer patients: Phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 2002, 26, 643–652. [Google Scholar] [CrossRef]
- Musselman, D.L.; Lawson, D.H.; Gumnick, J.F.; Manatunga, A.K.; Penna, S.; Goodkin, R.S.; Greiner, K.; Nemeroff, C.B.; Miller, A.H. Paroxetine for the prevention of depression induced by high-dose interferon alfa. N. Engl. J. Med. 2001, 344, 961–966. [Google Scholar] [CrossRef]
- Ho, P.S.; Yeh, Y.W.; Huang, S.Y.; Liang, C.S. A shift toward T helper 2 responses and an increase in modulators of innate immunity in depressed patients treated with escitalopram. Psychoneuroendocrinology 2015, 53, 246–255. [Google Scholar] [CrossRef] [PubMed]
- APA Depressive Disorders. In Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association (Ed.) American Psychiatric Association Press: Washington, DC, USA, 2013; pp. 180–182. [Google Scholar]
- Rooney, A.G.; Brown, P.D.; Reijneveld, J.C.; Grant, R. Depression in glioma: A primer for clinicians and researchers. J. Neurol. Neurosurg. Psychiatry 2014, 85, 230–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamels, M.; Mariman, A.; Kalala, O.; Van Den Broecke, C.; Delesie, L.; Tobback, E.; Van Roost, D.; Vogelaers, D. Chordoid meningioma in an adult patient presenting with chronic fatigue and systemic inflammation. Acta Clin. Belg. 2013, 68, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Donato, G.; Ferraro, G.; Signorelli, F.; Iofrida, G.; Lavano, A.; Amorosi, A.; Maltese, L.; Perrotta, I.; Tripepi, S.; Pardatscher, K.; et al. Chordoid meningioma: Case report and literature review. Ultrastruct. Pathol. 2006, 30, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Presta, I.; Guadagno, E.; Di Vito, A.; Malara, N.; Mignogna, C.; Maisano, D.; Donato, A.; Cardillo, G.; Del Basso De Caro, M.L.; Donato, G. Innate immunity may play a role in growth and relapse of chordoid meningioma. Int. J. Immunopathol. Pharmacol. 2017, 30, 429–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalat, R.; Munshi, N.C. Diagnosis of Castleman Disease. Hematol. Oncol. Clin. North. Am. 2018, 32, 53–64. [Google Scholar] [CrossRef]
- Lakhdar, R.; Siala, F.; Khouadja, A.; Ben Romdhane, S.; Ouechtati, W.; Thameur, H.; Belhani, A. Right atrial myxoma in a patient with mood disturbances. Tunis. Med. 2003, 81 (Suppl. 8) (Suppl. 8), 666–669. [Google Scholar]
- Di Vito, A.; Mignogna, C.; Donato, G. The mysterious pathways of cardiac myxomas: A review of histogenesis, pathogenesis and pathology. Histopathology 2015, 66, 321–332. [Google Scholar] [CrossRef]
- Di Vito, A.; Santise, G.; Mignogna, C.; Chiefari, E.; Cardillo, G.; Presta, I.; Arturi, F.; Malara, N.; Brunetti, F.; Donato, A.; et al. Innate immunity in cardiac myxomas and its pathological and clinical correlations. Innate Immun. 2018, 24, 47–53. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, D.; Salvadore, G.; Hsu, B.; Curran, M.; Casper, C.; Vermeulen, J.; Kent, J.M.; Singh, J.; Drevets, W.C.; et al. The effects of interleukin-6 neutralizing antibodies on symptoms of depressed mood and anhedonia in patients with rheumatoid arthritis and multicentric Castleman’s disease. Brain. Behav. Immun. 2017, 66, 156–164. [Google Scholar] [CrossRef]
- Perrotta, I.; Bruno, L.; Maltese, L.; Russo, E.; Donato, A.; Donato, G. Immunohistochemical Analysis of the Ubiquitin-conjugating Enzyme UbcH10 in Lung Cancer: A Useful Tool for Diagnosis and Therapy. J. Histochem. Cytochem. 2012, 60, 359–365. [Google Scholar] [CrossRef]
- Kayser, M.S.; Kohler, C.G.; Dalmau, J. Psychiatric manifestations of paraneoplastic disorders. Am. J. Psychiatry 2010, 167, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexopoulos, H.; Dalakas, M.C. The immunobiology of autoimmune encephalitides. J. Autoimmun. 2019, 104. [Google Scholar] [CrossRef] [PubMed]
- Wesselingh, R.; Butzkueven, H.; Buzzard, K.; Tarlinton, D.; O’Brien, T.J.; Monif, M. Innate Immunity in the Central Nervous System: A Missing Piece of the Autoimmune Encephalitis Puzzle? Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Vogelzangs, N.; de Jonge, P.; Smit, J.H.; Bahn, S.; Penninx, B.W. Cytokine production capacity in depression and anxiety. Transl. Psychiatry 2016, 6, e825. [Google Scholar] [CrossRef]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage polarization and meta-inflammation. Transl. Res. 2018, 191, 29–44. [Google Scholar] [CrossRef]
- Lamers, F.; Van Oppen, P.; Comijs, H.C.; Smit, J.H.; Spinhoven, P.; Van Balkom, A.J.L.M.; Nolen, W.A.; Zitman, F.G.; Beekman, A.T.F.; Penninx, B.W.J.H. Comorbidity patterns of anxiety and depressive disorders in a large cohort study: The Netherlands Study of Depression and Anxiety (NESDA). J. Clin. Psychiatry 2011, 72, 342–348. [Google Scholar] [CrossRef]
- Liukkonen, T.; Räsänen, P.; Jokelainen, J.; Leinonen, M.; Järvelin, M.R.; Meyer-Rochow, V.B.; Timonen, M. The association between anxiety and C-reactive protein (CRP) levels: Results from the Northern Finland 1966 Birth Cohort Study. Eur. Psychiatry 2011, 26, 363–369. [Google Scholar] [CrossRef]
- Vogelzangs, N.; Beekman, A.T.F.; De Jonge, P.; Penninx, B.W.J.H. Anxiety disorders and inflammation in a large adult cohort. Transl. Psychiatry 2013, 3, e249. [Google Scholar] [CrossRef] [Green Version]
- Rosenblat, J.D.; McIntyre, R.S. Bipolar Disorder and Inflammation. Psychiatr. Clin. North. Am. 2016, 39, 125–137. [Google Scholar] [CrossRef]
- De Baumont, A.; Maschietto, M.; Lima, L.; Carraro, D.M.; Olivieri, E.H.; Fiorini, A.; Barreta, L.A.N.; Palha, J.A.; Belmonte-de-Abreu, P.; Moreira Filho, C.A.; et al. Innate immune response is differentially dysregulated between bipolar disease and schizophrenia. Schizophr. Res. 2015, 161, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, K. Integrated theory to unify status among schizophrenia and manic depressive illness. Med. Hypotheses 2015, 85, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Foldager, L.; Köhler, O.; Steffensen, R.; Thiel, S.; Kristensen, A.S.; Jensenius, J.C.; Mors, O. Bipolar and panic disorders may be associated with hereditary defects in the innate immune system. J. Affect. Disord. 2014, 164, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hao, Y.; Fan, F.; Zhang, B. The Role of Microbiome in Insomnia, Circadian Disturbance and Depression. Front. Psychiatry 2018, 9, 669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, M.; Daniels, J.K.; Spitzer, C.; Lampe, A.; El Aidy, S. Depressed gut? the microbiota-diet-inflammation trialogue in depression. Curr. Opin. Psychiatry 2017, 30, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Ritter, A.; Rothenberg, K. Advances in Management of Neuropsychiatric Syndromes in Neurodegenerative Diseases. Curr. Psychiatry Rep. 2019, 21, 79. [Google Scholar] [CrossRef] [Green Version]
- Holmquist, S.; Nordström, A.; Nordström, P. The association of depression with subsequent dementia diagnosis: A Swedish nationwide cohort study from 1964 to 2016. PLoS Med. 2020, 17, e1003016. [Google Scholar] [CrossRef] [Green Version]
- Santos, L.E.; Beckman, D.; Ferreira, S.T. Microglial dysfunction connects depression and Alzheimer’s disease. Brain. Behav. Immun. 2016, 55, 151–165. [Google Scholar] [CrossRef]
- Mechawar, N.; Savitz, J. Neuropathology of mood disorders: do we see the stigmata of inflammation? Transl. Psychiatry 2016, 6, e946. [Google Scholar] [CrossRef] [Green Version]
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- Veselý, B.; Dufek, M.; Thon, V.; Brozman, M.; Királová, S.; Halászová, T.; Koriťáková, E.; Rektor, I. Interleukin 6 and complement serum level study in Parkinson’s disease. J. Neural Transm. 2018, 125, 875–881. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatic Association. Schizophrenia Spectrum and Other Psychotic Disorders. In Diagnostic and Statistical Manual of Mental Disorders, 1st ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Haller, C.S.; Padmanabhan, J.L.; Lizano, P.; Torous, J.; Keshavan, M. Recent advances in understanding schizophrenia. F1000Prime Rep. 2014, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.G.; Han, B.; Wu, Y.; Mignot, E.; Ollila, H.M.; Barker, J.; Spain, S.; Dand, N.; Trembath, R.; Martin, J.; et al. Cross-disorder analysis of schizophrenia and 19 immune-mediated diseases identifies shared genetic risk. Hum. Mol. Genet. 2019, 28, 3498–3513. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Zare-Shahabadi, A.; Rezaei, N. Cytokine Alterations in Schizophrenia: An Updated Review. Front. Psychiatry 2019, 10, 892. [Google Scholar] [CrossRef] [Green Version]
- Long-Smith, C.; O’Riordan, K.J.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota-Gut-Brain Axis: New Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 477–502. [Google Scholar] [CrossRef] [Green Version]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, L.; Gage, F.H. Microglia, complement and schizophrenia. Nat. Neurosci. 2019, 22, 333–334. [Google Scholar] [CrossRef]
- Shalev, H.; Serlin, Y.; Friedman, A. Breaching the Blood-Brain Barrier as a Gate to Psychiatric Disorder. Cardiovasc. Psychiatry Neurol. 2009, 2009, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pollak, T.A.; Drndarski, S.; Stone, J.M.; David, A.S.; McGuire, P.; Abbott, N.J. The blood–brain barrier in psychosis. The Lancet Psychiatry 2018, 5, 79–92. [Google Scholar] [CrossRef]
- Najjar, S.; Pahlajani, S.; De Sanctis, V.; Stern, J.N.H.; Najjar, A.; Chong, D. Neurovascular Unit Dysfunction and Blood–Brain Barrier Hyperpermeability Contribute to Schizophrenia Neurobiology: A Theoretical Integration of Clinical and Experimental Evidence. Front. Psychiatry 2017, 8, 83. [Google Scholar] [CrossRef]
- Rački, V.; Petrić, D.; Kučić, N.; Gržeta, N.; Jurdana, K.; Rončević-Gržeta, I. Cortical gray matter loss in schizophrenia: Could microglia be the culprit? Med. Hypotheses 2016, 88, 18–21. [Google Scholar] [CrossRef]
- Patel, J.P.; Frey, B.N. Disruption in the blood-brain barrier: The missing link between brain and body inflammation in bipolar disorder? Neural Plast. 2015, 2015. [Google Scholar] [CrossRef]
- Schiavone, S.; Mhillaj, E.; Neri, M.; Morgese, M.G.; Tucci, P.; Bove, M.; Valentino, M.; Di Giovanni, G.; Pomara, C.; Turillazzi, E.; et al. Early Loss of Blood-Brain Barrier Integrity Precedes NOX2 Elevation in the Prefrontal Cortex of an Animal Model of Psychosis. Mol. Neurobiol. 2017, 54, 2031–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayernia, Z.; Jaquet, V.; Krause, K.H. New insights on NOX enzymes in the central nervous system. Antioxidants Redox Signal. 2014, 20, 2815–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilhardt, F.; Haslund-Vinding, J.; Jaquet, V.; McBean, G. Microglia antioxidant systems and redox signalling. Br. J. Pharmacol. 2017, 174, 1719–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakilian, A.; Razavi-Nasab, S.M.; Ravari, A.; Mirzaei, T.; Moghadam-Ahmadi, A.; Jalali, N.; Bahramabadi, R.; Rezayati, M.; Yazdanpanah-Ravari, A.; Bahmaniar, F.; et al. Vitamin B12 in Association with Antipsychotic Drugs Can Modulate the Expression of Pro-/Anti-Inflammatory Cytokines in Alzheimer Disease Patients. Neuroimmunomodulation 2017, 24, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Fredericks, D.; Norton, J.C.; Atchison, C.; Schoenhaus, R.; Pill, M.W. Parkinson’s disease and Parkinson’s disease psychosis: a perspective on the challenges, treatments, and economic burden. Am. J. Manag. Care 2017, 23, S83–S92. [Google Scholar]
- Winograd-Gurvich, C.; Fitzgerald, P.B.; Georgiou-Karistianis, N.; Bradshaw, J.L.; White, O.B. Negative symptoms: A review of schizophrenia, melancholic depression and Parkinson’s disease. Brain Res. Bull. 2006, 70, 312–321. [Google Scholar] [CrossRef]
- Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. The relation between α-synuclein and microglia in Parkinson’s disease: Recent developments. Neuroscience 2015, 302, 47–58. [Google Scholar] [CrossRef]
- Mendoza, C.; Barreto, G.E.; Ávila-Rodriguez, M.; Echeverria, V. Role of neuroinflammation and sex hormones in war-related PTSD. Mol. Cell. Endocrinol. 2016, 434, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.N.; Pearce, B.D.; Biron, C.A.; Miller, A.H. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005, 18, 41–78. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.M.; Saligan, L.; Woods, S.; Page, G. PTSD is associated with an excess of inflammatory immune activities. Perspect. Psychiatr. Care 2009, 45, 262–277. [Google Scholar] [CrossRef] [PubMed]
- Vidović, A.; Vilibić, M.; Sabioncello, A.; Gotovac, K.; Rabatić, S.; Folnegović-Šmalc, V.; Dekaris, D. Circulating lymphocyte subsets, natural killer cell cytotoxicity, and components of hypothalamic-pituitary-adrenal axis in croatian war veterans with posttraumatic stress disorder: Cross-sectional study. Croat. Med. J. 2007, 48, 198–206. [Google Scholar] [PubMed]
- Breen, M.S.; Maihofer, A.X.; Glatt, S.J.; Tylee, D.S.; Chandler, S.D.; Tsuang, M.T.; Risbrough, V.B.; Baker, D.G.; O’Connor, D.T.; Nievergelt, C.M.; et al. Gene networks specific for innate immunity define post-traumatic stress disorder. Mol. Psychiatry 2015, 20, 1538–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torshizi, A.D.; Wang, K. Deconvolution of Transcriptional Networks in Post-Traumatic Stress Disorder Uncovers Master Regulators Driving Innate Immune System Function. Sci. Rep. 2017, 7, 1–11. [Google Scholar]
- Pennington, M.L.; Cullinan, D.; Southern, L.B. Defining Autism: Variability in State Education Agency Definitions of and Evaluations for Autism Spectrum Disorders. Autism Res. Treat. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klei, L.; Sanders, S.J.; Murtha, M.T.; Hus, V.; Lowe, J.K.; Willsey, A.J.; Moreno-De-Luca, D.; Yu, T.W.; Fombonne, E.; Geschwind, D.; et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 2012, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Estes, M.L.; McAllister, A.K. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 469–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, T.C.; Lien, Y.T.; Wang, S.; Huang, S.L.; Chen, C.Y. Comorbidity of Atopic Disorders with Autism Spectrum Disorder and Attention Deficit/Hyperactivity Disorder. J. Pediatr. 2016, 171, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Lyall, K.; Van de Water, J.; Ashwood, P.; Hertz-Picciotto, I. Asthma and allergies in children with autism spectrum disorders: Results from the CHARGE study. Autism Res. 2015, 8, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta - Mol. Basis Dis. 2012, 1822, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, Y.; Dong, H.; Xu, Y.; Zhang, S. Induction of microglial activation by mediators released from mast cells. Cell. Physiol. Biochem. 2016, 38, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Patterson, P.H. Cytokines and CNS Development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef] [Green Version]
- Hornig, M.; Bresnahan, M.A.; Che, X.; Schultz, A.F.; Ukaigwe, J.E.; Eddy, M.L.; Hirtz, D.; Gunnes, N.; Lie, K.K.; Magnus, P.; et al. Prenatal fever and autism risk. Mol. Psychiatry 2018, 23, 759–766. [Google Scholar] [CrossRef]
- McCarthy, M.M.; Wright, C.L. Convergence of Sex Differences and the Neuroimmune System in Autism Spectrum Disorder. Biol. Psychiatry 2017, 81, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Huang, L.; Li, X.; Li, H.; Zhou, Y.; Zhu, H.; Pan, T.; Kendrick, K.M.; Xu, W. Immunological cytokine profiling identifies TNF-α as a key molecule dysregulated in autistic children. Oncotarget 2017, 8, 82390–82398. [Google Scholar] [CrossRef] [Green Version]
- Custódio, C.S.; Mello, B.S.F.; Filho, A.J.M.C.; de Carvalho Lima, C.N.; Cordeiro, R.C.; Miyajima, F.; Réus, G.Z.; Vasconcelos, S.M.M.; Barichello, T.; Quevedo, J.; et al. Neonatal Immune Challenge with Lipopolysaccharide Triggers Long-lasting Sex- and Age-related Behavioral and Immune/Neurotrophic Alterations in Mice: Relevance to Autism Spectrum Disorders. Mol. Neurobiol. 2018, 55, 3775–3788. [Google Scholar] [CrossRef]
- Jyonouchi, H.; Geng, L.; Davidow, A.L. Cytokine profiles by peripheral blood monocytes are associated with changes in behavioral symptoms following immune insults in a subset of ASD subjects: An inflammatory subtype? J. Neuroinflammation 2014, 11, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarsland, D.; Påhlhagen, S.; Ballard, C.G.; Ehrt, U.; Svenningsson, P. Depression in Parkinson disease--epidemiology, mechanisms and management. Nat. Rev. Neurol. 2011, 8, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, S.; Arnedo, V.; Devinsky, O. Psychosis in epilepsy patients. Epilepsia 2007, 48, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Hinson, V.K.; Haren, W.B. Psychogenic movement disorders. Lancet Neurol. 2006, 5, 695–700. [Google Scholar] [CrossRef]
- Crossley, N.A.; Scott, J.; Ellison-Wright, I.; Mechelli, A. Neuroimaging distinction between neurological and psychiatric disorders. Br. J. Psychiatry 2015, 207, 429–434. [Google Scholar] [CrossRef] [Green Version]
- Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P.; Malik, R.; et al. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar]
- Northrup, N.A.; Yamamoto, B.K. Neuroimmune pharmacology from a neuroscience perspective. J. Neuroimmune Pharmacol. 2011, 6, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Bielekova, B.; Martin, R. Development of biomarkers in multiple sclerosis. Brain 2004, 127, 1463–1478. [Google Scholar] [CrossRef] [Green Version]











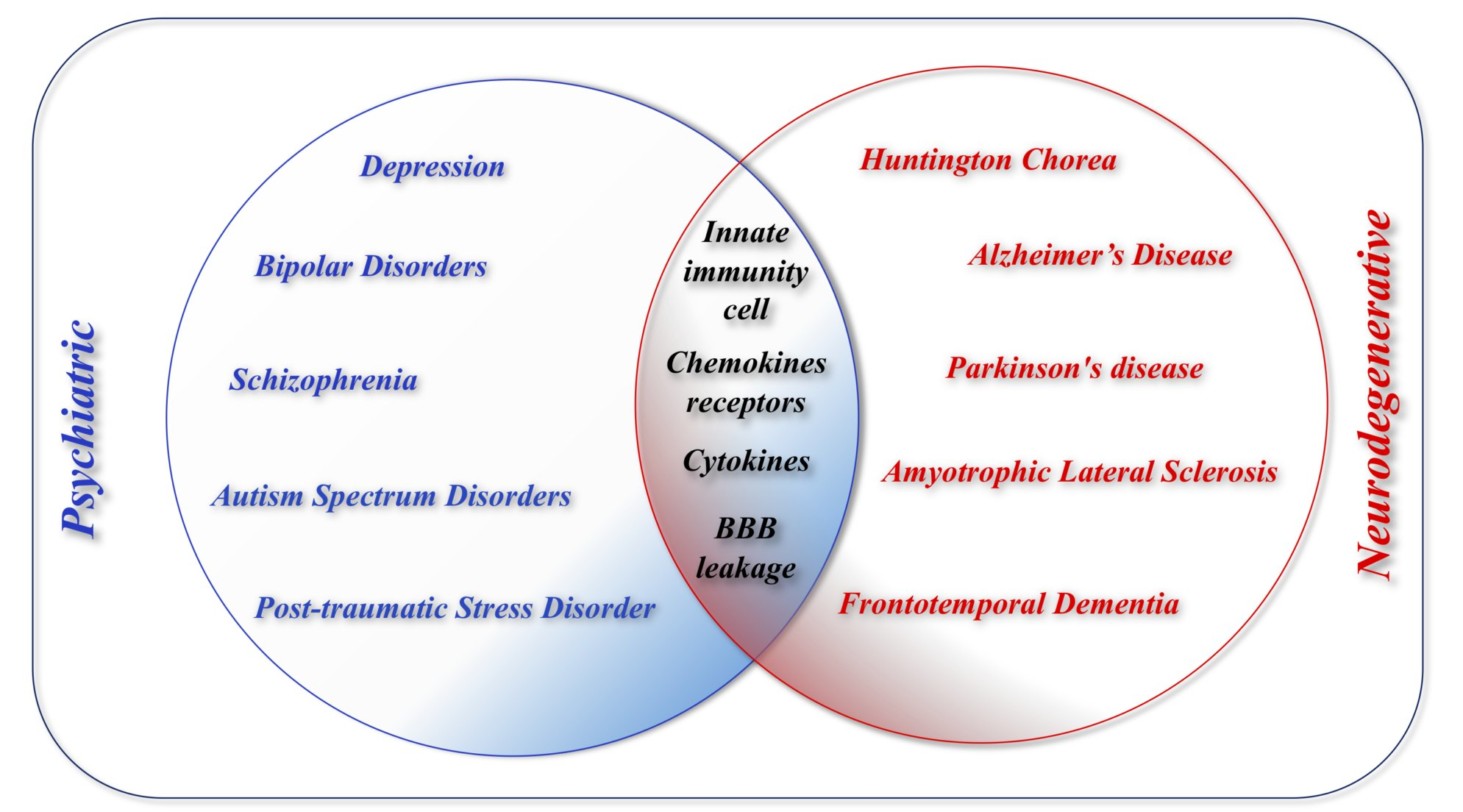{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
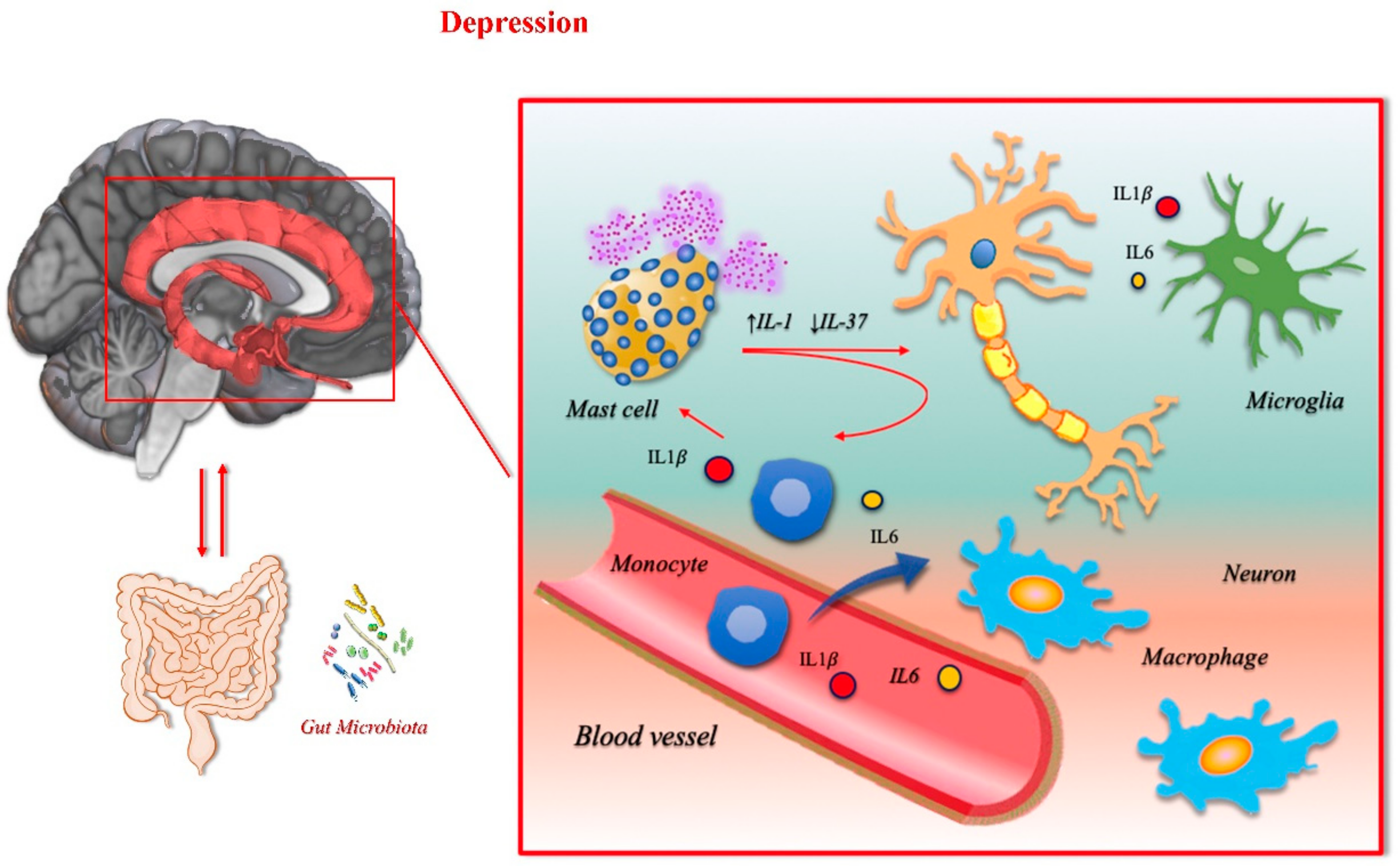{kind=link}
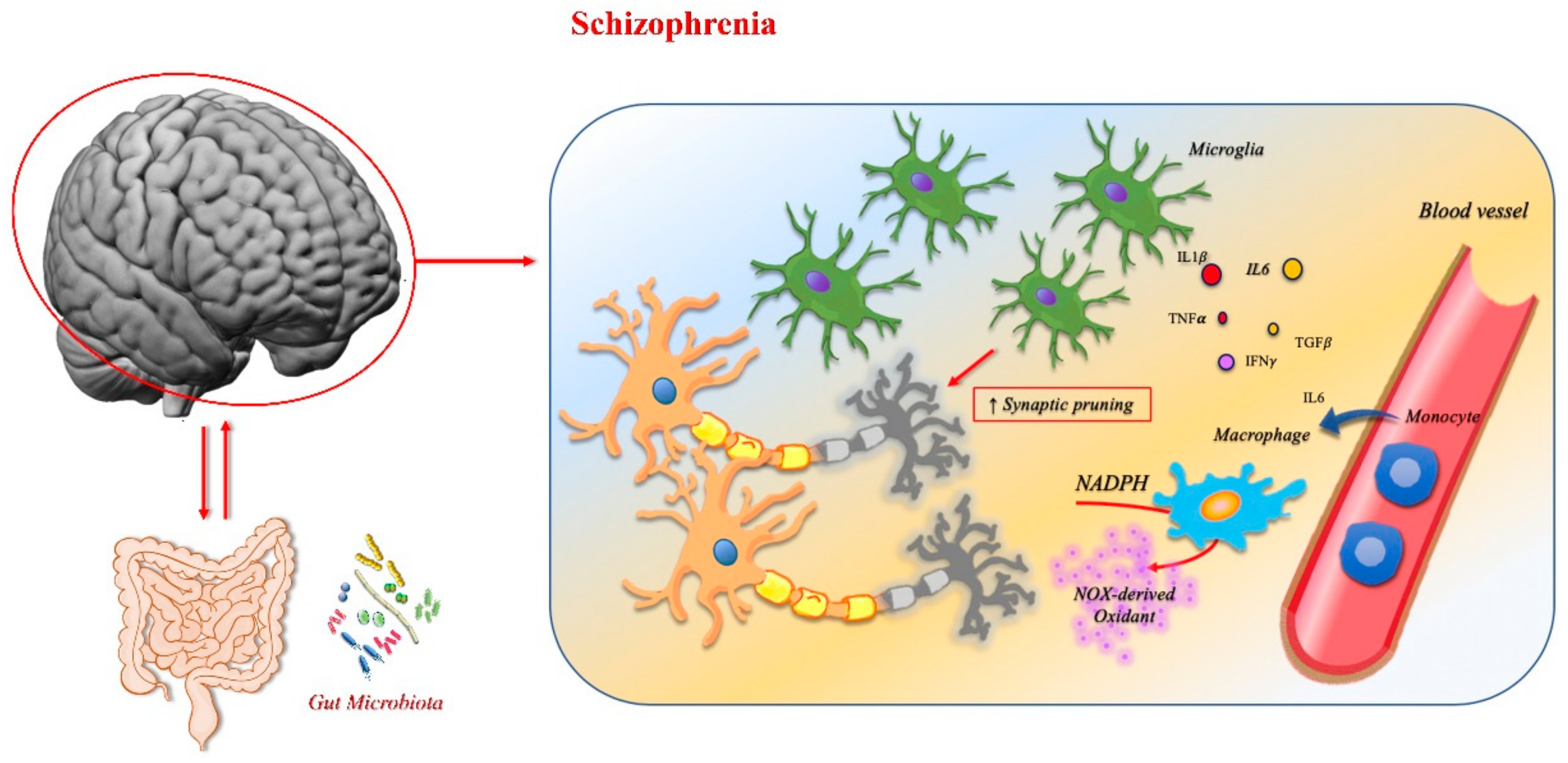{kind=link}
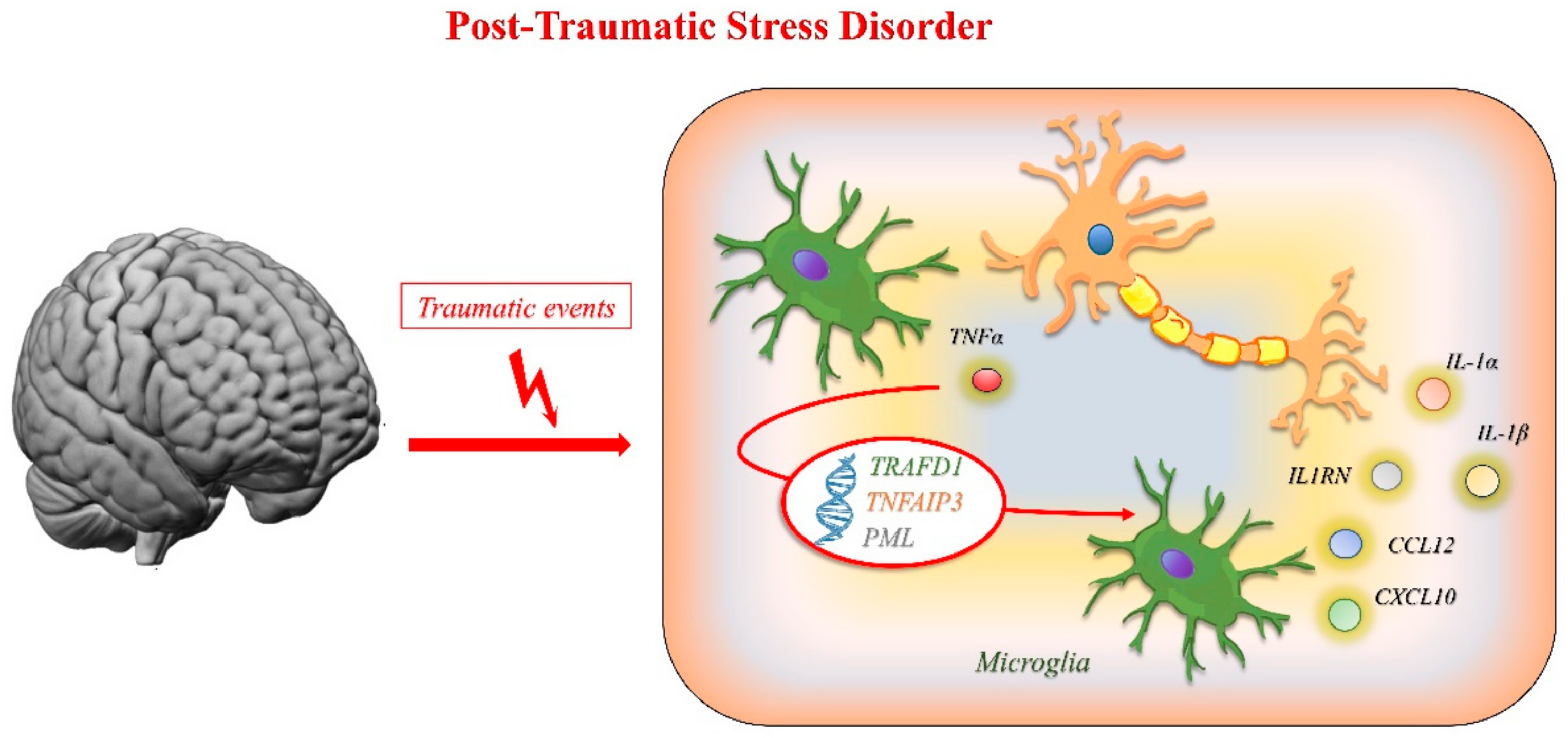{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Cytokine or Chemokine | Main Functions of Cytokine | Cytokine Source | Pathology [References] |
|---|---|---|---|---|
| IFNα | Interferon alpha | ↑ NK cells and CTL functions Influence isotype switching | Activate macrophages, monocytes | AD [10] Depression [203] |
| IFNγ | Interferon gamma | ↑ NK cells and CTL functions ↑ APC production of IL-12 ↓ IL-4 production Influence isotype switching | Activate macrophages | PD [55] Schizophrenia [249] |
| IL-1 | Interleukin-1 | Pro-inflammatory ↑ Acute phase response | Macrophages, neutrophils, epithelial and endothelial cells | AD [10] PD [90] Depression [200] Schizophrenia [249] |
| IL-2 | Interleukin-2 | Th1 cytokine ↑ T and B cell activation ↑ NK cell proliferation and production of TNF, IFNγ Required for Treg cell differentiation | Activated T-cells | Depression [199] |
| Il-6 | Interleukin-6 | Pro-inflammatory ↑ Acute phase response | Activated phagocytes, Endothelial cells, Some activated T cells | AD [10] PD [244] Depression [201] Schizophrenia [249] PTSD [267] |
| IL-8 | Interleukin-8 | CXC chemokine ↑ Neutrophil chemotaxis and degranulation | All cell types encountering TNF, IL-1 or bacterial endotoxin | Schizophrenia [261] |
| IL-12 | Interleukin-12 | Required for Th1 cell differentiation ↑ production of IFN γ by macrophages, activated Th1 cells, NK cells ↑ DC and macrophage cytokines secretion ↑ CTL and NK cytotoxicity Influences isotype switching | Activated macrophages, Dentritic cells, neutrophils, monocytes, B cells | Schizophrenia [249] |
| TNF | Tumor Necrosis Factor | Potent inflammatory, immunoregulatory, cytotoxic, antiviral, pro-coagulatory, and growth stimulatory effects | Many types of activated hematopoietic and non-hematopoietic cells | AD [10] PD [264] Depression [203,204] Schizophrenia [249] PTSD [270] ASD [283] |
| TGFβ | Transforming growth factor-β | Anti-inflammatory, immunosuppressive | Most activated hematopoietic cells; some non-hematopoietic cells | AD [10] Schizophrenia [249] |
| CXCL10 (IP-10) | C-X-C motif chemokine 10 (Interferon gamma-induced protein 10) | Chemoattractant for monocytes, macrophages, T cells, NK cells, and DC | Several cell types in response to IFN-γ: Monocytes, endothelial cells and fibroblasts | PTSD [270] |
| CXCL12 (SDF-1) | C-X-C motif chemokine 12 (stromal cell-derived factor 1) | Chemoattractant for lymphocytes, macrophages, hematopoietic cells. In neuroinflammation attraction of leukocytes across the blood brain barrier. | Leucocytes, endothelial cells, glial cells and neurons. | PD [55] |
| CCL12 | Chemokine (C-C motif) ligand 12 | Chemoattractant for eosinophils, monocytes and lymphocytes. | Leucocytes, endothelial cells and fibroblasts | PTSD [270] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novellino, F.; Saccà, V.; Donato, A.; Zaffino, P.; Spadea, M.F.; Vismara, M.; Arcidiacono, B.; Malara, N.; Presta, I.; Donato, G. Innate Immunity: A Common Denominator between Neurodegenerative and Neuropsychiatric Diseases. Int. J. Mol. Sci. 2020, 21, 1115. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031115
Novellino F, Saccà V, Donato A, Zaffino P, Spadea MF, Vismara M, Arcidiacono B, Malara N, Presta I, Donato G. Innate Immunity: A Common Denominator between Neurodegenerative and Neuropsychiatric Diseases. International Journal of Molecular Sciences. 2020; 21(3):1115. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031115
Chicago/Turabian StyleNovellino, Fabiana, Valeria Saccà, Annalidia Donato, Paolo Zaffino, Maria Francesca Spadea, Marco Vismara, Biagio Arcidiacono, Natalia Malara, Ivan Presta, and Giuseppe Donato. 2020. "Innate Immunity: A Common Denominator between Neurodegenerative and Neuropsychiatric Diseases" International Journal of Molecular Sciences 21, no. 3: 1115. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031115