Disruption of the Unique ABCG-Family NBD:NBD Interface Impacts Both Drug Transport and ATP Hydrolysis


Abstract
:1. Introduction
2. Results
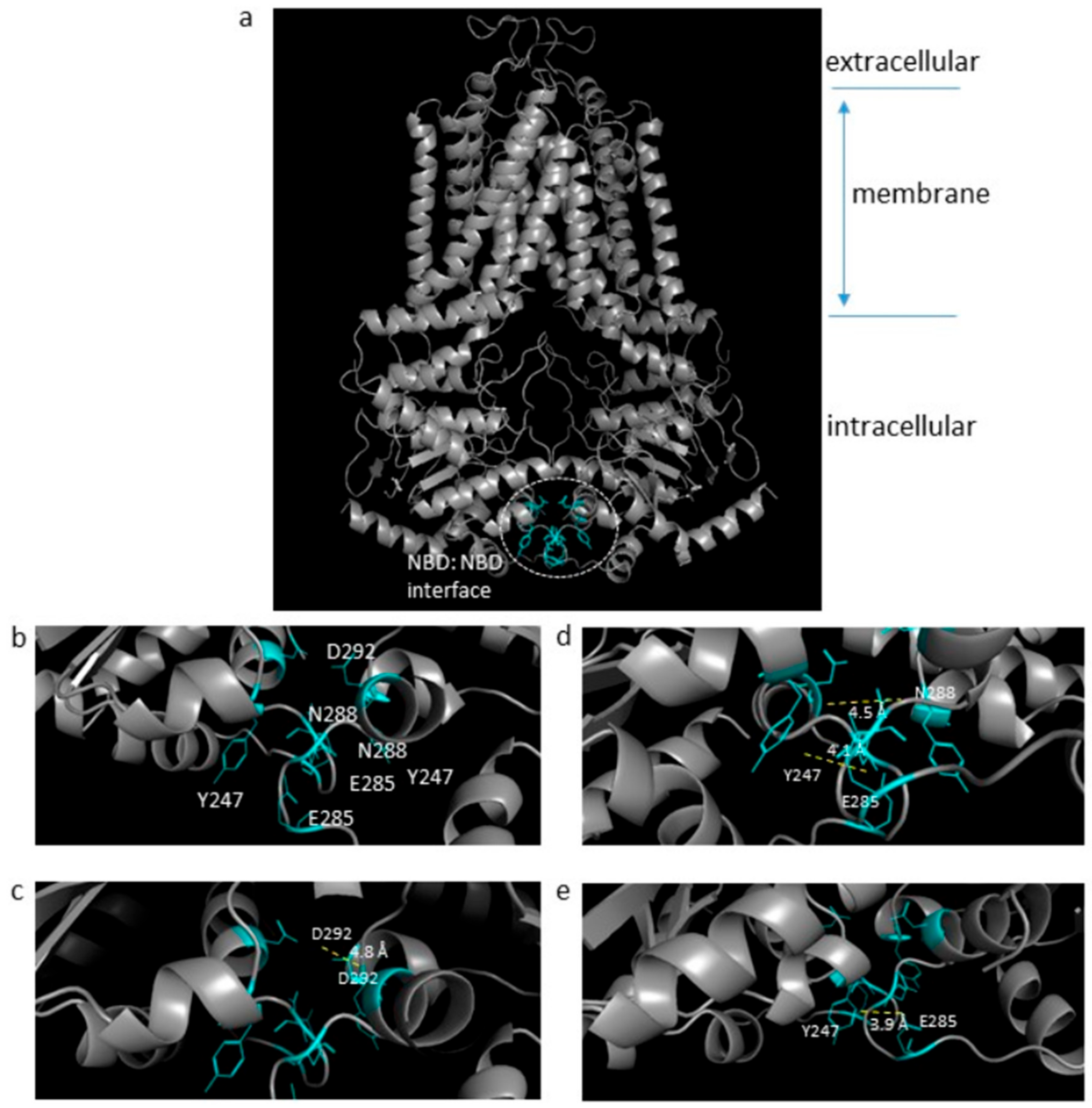
2.1. The ABCG Family Has A Novel NBD:NBD Interface
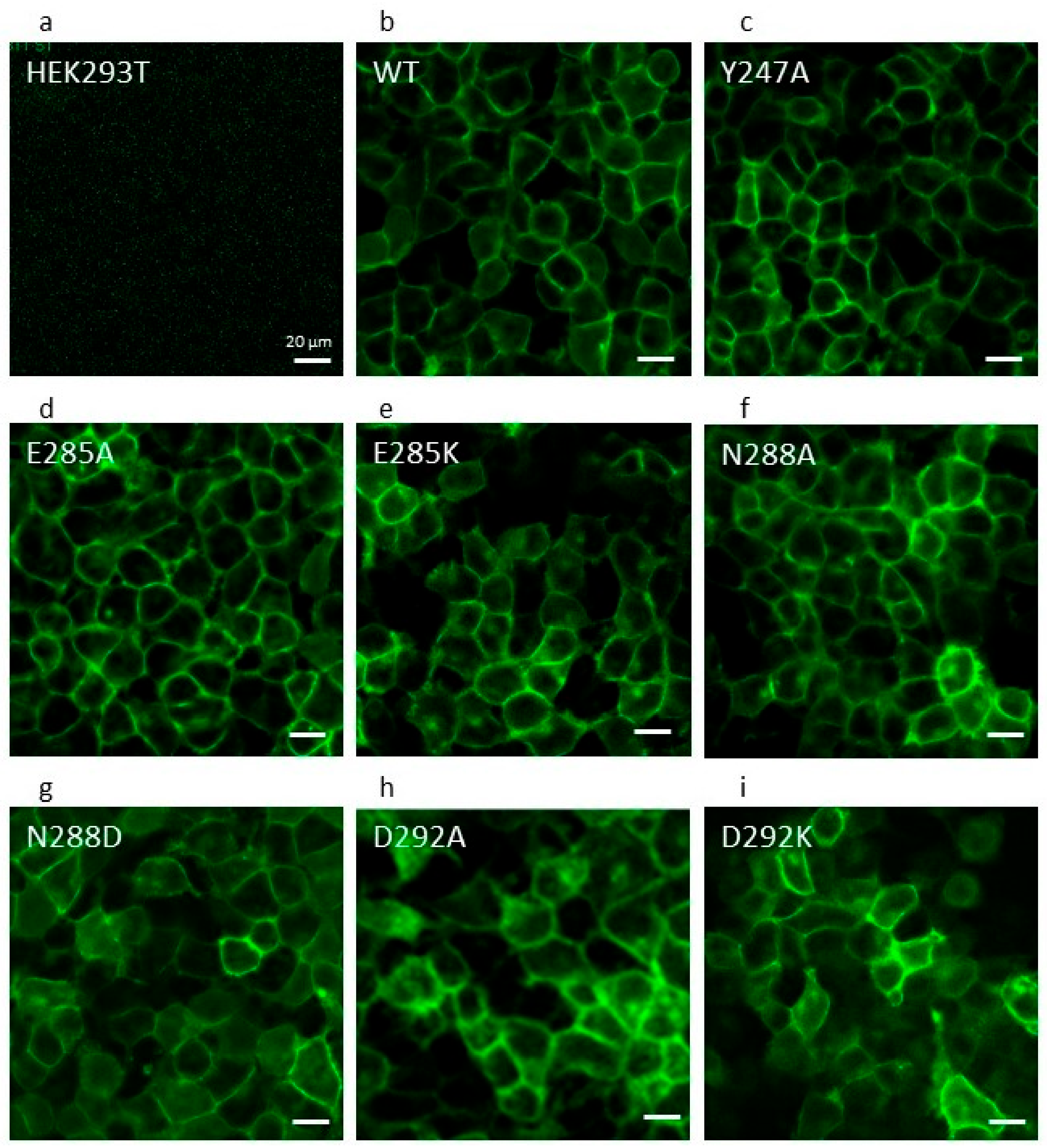
2.2. The ABCG2 Mutation N288D Fails to Traffic to the Plasma Membrane
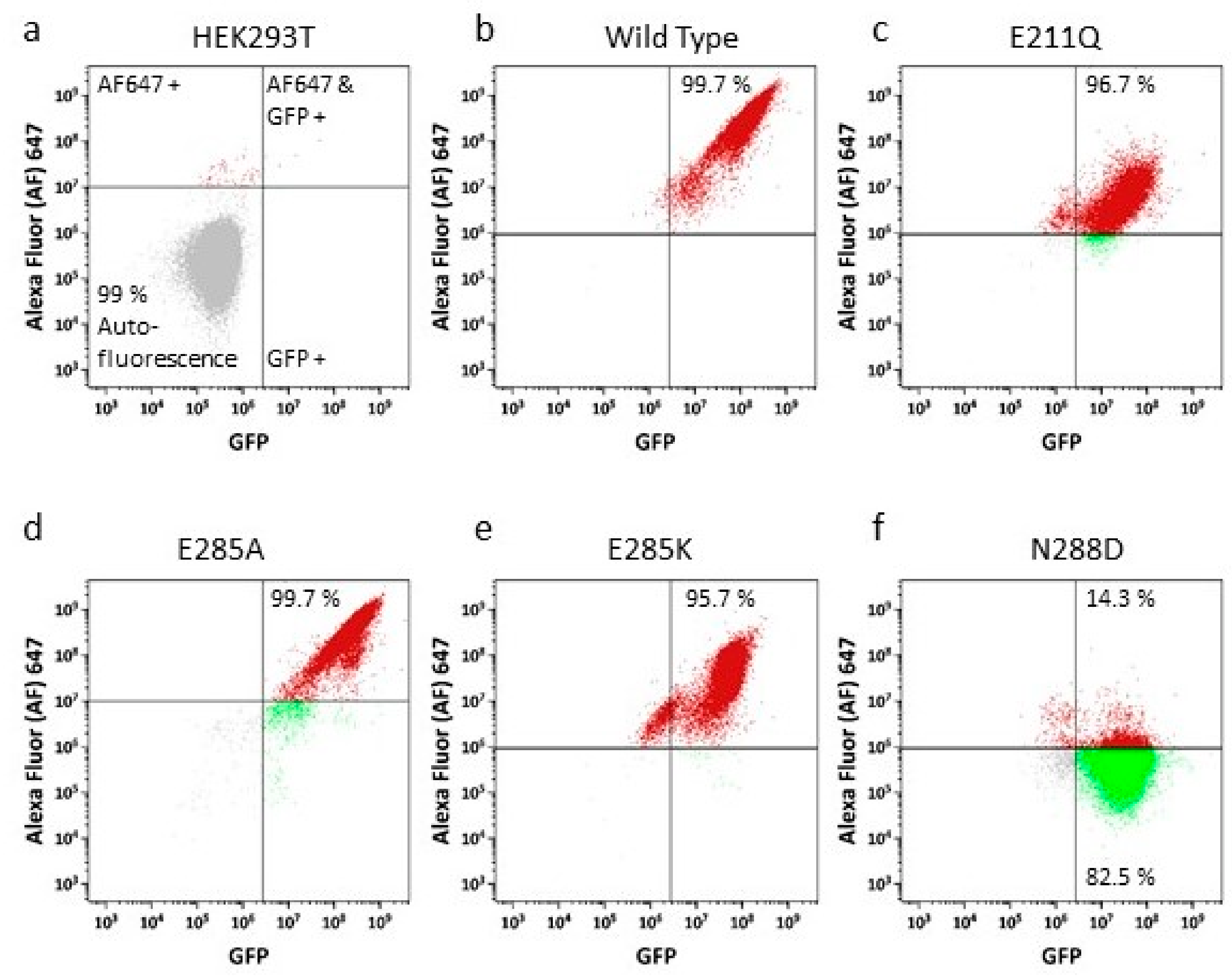
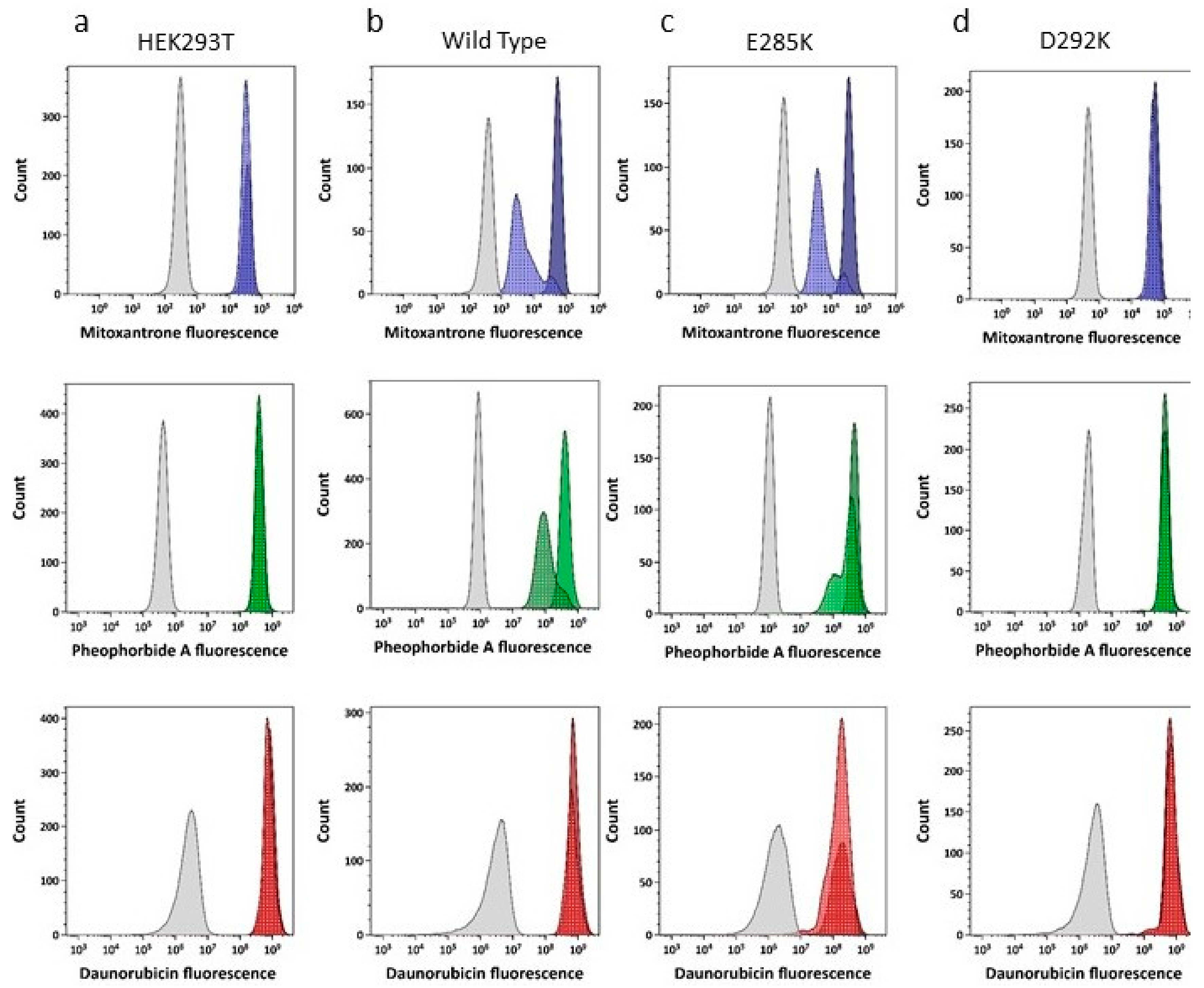
2.3. Drug Transport Is Altered in NBD Interface Mutants
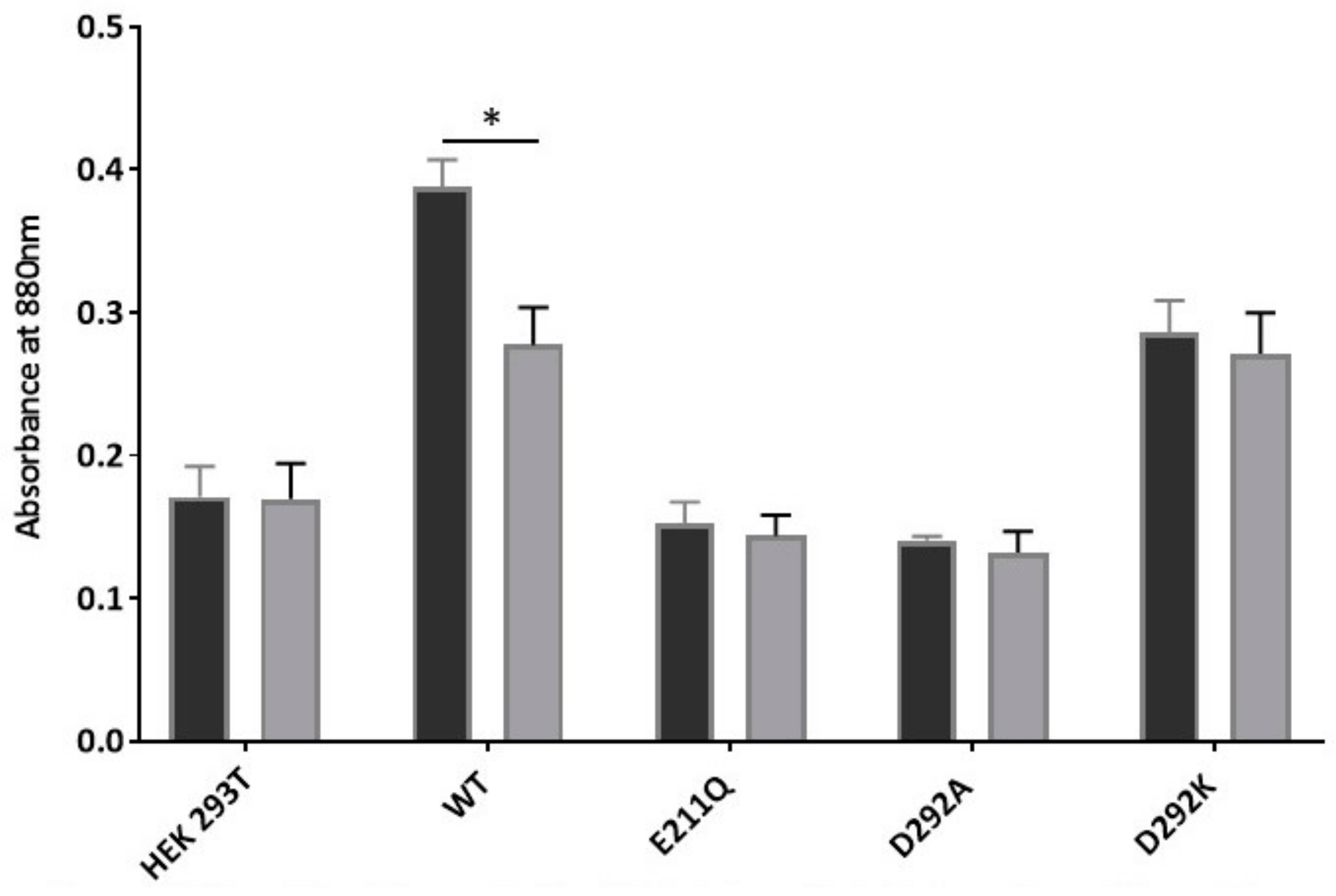
2.4. Abrogation of Drug Transport in D292 Mutants Is Due to Inhibition of ATPase Activity
3. Discussion
4. Materials and Methods
4.1. Site Directed Mutagenesis
4.2. Cell Culture
4.3. Transfection and Selection of Stable Cell Lines
4.4. Cell Imaging
4.5. Drug Transport Analysis
4.6. Cell Surface Expression Analysis
4.7. Membrane Preparation and ATPase Assay
4.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP binding cassette |
| NBD | Nucleotide binding domain |
| TMD | Transmembrane domain |
| MX | Mitoxantrone |
| PhA | Pheophorbide A |
| DNR | Daunorubicin |
| MDR | Multidrug resistance |
| HEK | Human embryonic kidney |
| DMEM | Dulbecco’s modified Eagle medium |
| FCS | Fetal calf serum |
| PEI | Polyethyleneimine |
| DMSO | Dimethylsulfoxide |
| ANOVA | Analysis of variance |
References
- Damiani, D.; Tiribelli, M.; Geromin, A.; Michelutti, A.; Cavallin, M.; Sperotto, A.; Fanin, R. ABCG2 overexpression in patients with acute myeloid leukemia: Impact on stem cell transplantation outcome. Am. J. Hematol. 2015, 90, 784–789. [Google Scholar] [CrossRef]
- Benderra, Z.; Faussat, A.M.; Sayada, L.; Perrot, J.Y.; Tang, R.; Chaoui, I.; Morjani, H.; Marzac, C.; Marie, J.P.; Legrand, O. MRP3, BCRP, and P-Glycoprotein Activities are Prognostic Factors in Adult Acute Myeloid Leukemia. Clin. Cancer Res. 2005, 11, 7764–7772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Heuvel-Eibrink, M.M.; Wiemer, E.A.C.; Prins, A.; Meijerink, J.P.P.; Vossebeld, P.J.M.; Van Der Holt, B.; Pieters, R.; Sonneveld, P. Increased expression of the breast cancer resistance protein (BCRP) in relapsed or refractory acute myeloid leukemia (AML). Leukemia 2002, 16, 833–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannangara, D.R.; Phipps-Green, A.J.; Dalbeth, N.; Stamp, L.K.; Williams, K.M.; Graham, G.G.; Day, R.O.; Merriman, T.R. Hyperuricaemia: Contributions of urate transporter ABCG2 and the fractional renal clearance of urate. Ann. Rheum. Dis. 2016, 75, 1363–1366. [Google Scholar] [CrossRef] [Green Version]
- Heyes, N.; Kapoor, P.; Kerr, I.D. Polymorphisms of the Multidrug Pump ABCG2: A Systematic Review of Their Effect on Protein Expression, Function, and Drug Pharmacokinetics. Drug Metab. Dispos. 2018, 46, 1886–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, O.M.; Tukaye, D.N.; Cui, J.; Greenwell, P.; Constantoulakis, L.M.; Parker, B.S.; Rao, A.; Köttgen, M.; Maloney, P.C.; Guggino, W.B. Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, 5223–5228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, O.M.; Köttgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Köttgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [Green Version]
- Ding, R.; Jin, S.; Pabon, K.; Scotto, K.W. A role for ABCG2 beyond drug transport: Regulation of autophagy. Autophagy 2016, 12, 737–751. [Google Scholar] [CrossRef] [Green Version]
- Deuring, J.J.; de Haar, C.; Koelewijn, C.L.; Kuipers, E.J.; Peppelenbosch, M.P.; van der Woude, C.J. Absence of ABCG2-mediated mucosal detoxification in patients with active inflammatory bowel disease is due to impeded protein folding. Biochem. J. 2012, 441, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Ross, D.D.; Nakanishi, T.; Bailey-Dell, K.; Zhou, S.; Mercer, K.E.; Sarkadi, B.; Sorrentino, B.P.; Schuetz, J.D. The Stem Cell Marker Bcrp/ABCG2 Enhances Hypoxic Cell Survival through Interactions with Heme. J. Biol. Chem. 2004, 279, 24218–24225. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D.; Haider, A.J.; Gelissen, I.C. The ABCG family of membrane-associated transporters: You don’t have to be big to be mighty. Br. J. Pharmacol. 2011, 164, 1767–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopfner, K.P. Rad50/SMC proteins and ABC transporters: Unifying concepts from high-resolution structures. Curr. Opin. Struct. Biol. 2003, 13, 249–255. [Google Scholar] [CrossRef]
- Wong, K.; Briddon, S.J.; Holliday, N.D.; Kerr, I.D. Plasma membrane dynamics and tetrameric organisation of ABCG2 transporters in mammalian cells revealed by single particle imaging techniques. Biochim. Biophys. Acta 2016, 1863, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Haider, A.J.; Briggs, D.; Self, T.J.; Chilvers, H.L.; Holliday, N.D.; Kerr, I.D. Dimerization of ABCG2 Analysed by Bimolecular Fluorescence Complementation. PLoS ONE 2011, 6, e25818. [Google Scholar] [CrossRef] [Green Version]
- Leimanis, M.L.; Georges, E. ABCG2 membrane transporter in mature human erythrocytes is exclusively homodimer. Biochem. Biophys. Res. Commun. 2007, 354, 345–350. [Google Scholar] [CrossRef]
- Bhatia, A.; Schäfer, H.J.; Hrycyna, C.A. Oligomerization of the Human ABC Transporter ABCG2: Evaluation of the Native Protein and Chimeric Dimers. Biochemistry 2005, 44, 10893–10904. [Google Scholar] [CrossRef]
- Horsey, A.J.; Cox, M.H.; Sarwat, S.; Kerr, I.D. The multidrug transporter ABCG2: Still more questions than answers. Biochem. Soc. Trans. 2016, 44, 824–830. [Google Scholar] [CrossRef] [Green Version]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kinch, L.N.; Borek, D.M.; Wang, J.; Wang, J.; Urbatsch, I.L.; Xie, X.S.; Grishin, N.V.; Cohen, J.C.; Otwinowski, Z.; et al. Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature 2016, 533, 561–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, P.; Horsey, A.J.; Cox, M.H.; Kerr, I.D. ABCG2: Does resolving its structure elucidate the mechanism? Biochem. Soc. Trans. 2018, 46, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.H.; Kapoor, P.; Briggs, D.A.; Kerr, I.D. Residues contributing to drug transport by ABCG2 are localised to multiple drug-binding pockets. Biochem. J. 2018, 475, 1553–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khunweeraphong, N.; Stockner, T.; Kuchler, K. The structure of the human ABC transporter ABCG2 reveals a novel mechanism for drug extrusion. Sci. Rep. 2017, 7, 13767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laszlo, L.; Sarkadi, B.; Hegedus, T. Jump into a New Fold-A Homology Based Model for the ABCG2/BCRP Multidrug Transporter. PLoS ONE 2016, 11, e0164426. [Google Scholar] [CrossRef] [Green Version]
- McDevitt, C.A.; Crowley, E.; Hobbs, G.; Starr, K.J.; Kerr, I.D.; Callaghan, R. Is ATP binding responsible for initiating drug translocation by the multidrug transporter ABCG2? FEBS J. 2008, 275, 4354–4362. [Google Scholar] [CrossRef]
- Smith, P.C.; Karpowich, N.; Millen, L.; Moody, J.E.; Rosen, J.; Thomas, P.J.; Hunt, J.F. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 2002, 10, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Kerr, I.D. Structure and association of ATP-binding cassette transporter nucleotide-binding domains. Biochim. Biophys. Acta 2002, 1561, 47–64. [Google Scholar] [CrossRef] [Green Version]
- Zaitseva, J.; Jenewein, S.; Jumpertz, T.; Holland, I.B.; Schmitt, L. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 2005, 24, 1901–1910. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Li, G.; Gu, H.M.; Zhang, D.W. Characterization of the Role of a Highly Conserved Sequence in ATP Binding Cassette Transporter G (ABCG) Family in ABCG1 Stability, Oligomerization, and Trafficking. Biochemistry 2013, 52, 9497–9509. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Van Loevezijn, A.; Lakhai, J.M.; Van Der Valk, M.; Van Tellingen, O.; Reid, G.; Schellens, J.H.M.; Koomen, G.J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar] [PubMed]
- Robey, R.W.; Honjo, Y.; Morisaki, K.; Nadjem, T.A.; Runge, S.; Risbood, M.; Poruchynsky, M.S.; Bates, S.E. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br. J. Cancer 2003, 89, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Buitelaar, M.; Wagenaar, E.; Van Der Valk, M.A.; Scheffer, G.L.; Scheper, R.J.; Plösch, T.; Kuipers, F.; Elferink, R.P.; Rosing, H.; et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc. Natl. Acad. Sci. USA 2002, 99, 15649–15654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.X.; Li, C.Z.; Palaniyandi, K.; Magtibay, P.M.; Homolya, L.; Sarkadi, B.; Chang, X.B. Effects of Putative Catalytic Base Mutation E211Q on ABCG2-Mediated Methotrexate Transport. Biochemistry 2009, 48, 9122–9131. [Google Scholar] [CrossRef] [Green Version]
- Haider, A.J.; Cox, M.H.; Jones, N.; Goode, A.J.; Bridge, K.S.; Wong, K.; Briggs, D.; Kerr, I.D. Identification of residues in ABCG2 affecting protein trafficking and drug transport, using co-evolutionary analysis of ABCG sequences. Biosci. Rep. 2015, 35, e00241. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Wakabayashi-Nakao, K.; Tamura, A.; Toyoda, Y.; Koshiba, S.; Ishikawa, T. Disruption of N-linked glycosylation enhances ubiquitin-mediated proteasomal degradation of the human ATP-binding cassette transporter ABCG2. FEBS J. 2009, 276, 7237–7252. [Google Scholar] [CrossRef]
- Ejendal, K.F.; Diop, N.K.; Schweiger, L.C.; Hrycyna, C.A. The nature of amino acid 482 of human ABCG2 affects substrate transport and ATP hydrolysis but not substrate binding. Protein Sci. 2006, 15, 1597–1607. [Google Scholar] [CrossRef] [Green Version]
- Macalou, S.; Robey, R.W.; Jabor Gozzi, G.; Shukla, S.; Grosjean, I.; Hegedus, T.; Ambudkar, S.V.; Bates, S.E.; Di Pietro, A. The linker region of breast cancer resistance protein ABCG2 is critical for coupling of ATP-dependent drug transport. Cell. Mol. Life Sci. 2016, 73, 1927–1937. [Google Scholar] [CrossRef] [Green Version]
- Boussif, O.; Lezoualc’H, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chifflet, S.; Torriglia, A.; Chiesa, R.; Tolosa, S. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: Application to lens ATPases. Anal. Biochem. 1988, 168, 1–4. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Mitoxantrone | Pheophorbide A | Daunorubicin |
|---|---|---|---|
| HEK293T | 0.21 ± 0.07 | −0.07 ± 0.05 | 0.03 ± 0.01 |
| WT-ABCG2 | 11.90 ± 1.88 | 2.45 ± 0.48 | 0.03 ± 0.03 |
| E211Q | 1.22 ** ± 0.20 | −0.05 ** ± 0.07 | −0.02 ± 0.08 |
| Y247A | 12.19 ± 1.18 | 5.63 ** ± 0.05 | 0.20 ± 0.05 |
| E285A | 8.51 ± 1.53 | 3.05 ± 0.88 | 0.08 ± 0.03 |
| E285K | 20.26 ** ± 2.85 | 7.20 ** ± 1.06 | 0.26 ** ± 0.06 |
| N288A | 11.53 ± 2.40 | 4.37 ± 0.17 | 0.19 ± 0.04 |
| D292A | 1.46 ** ± 0.90 | 0.13 * ± 0.10 | 0.12 ± 0.04 |
| D292K | 0.81 ** ± 0.07 | 0.10 * ± 0.17 | 0.05 ± 04 |
| R482A | 0.53 ** ± 0.05 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapoor, P.; Briggs, D.A.; Cox, M.H.; Kerr, I.D. Disruption of the Unique ABCG-Family NBD:NBD Interface Impacts Both Drug Transport and ATP Hydrolysis. Int. J. Mol. Sci. 2020, 21, 759. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030759
Kapoor P, Briggs DA, Cox MH, Kerr ID. Disruption of the Unique ABCG-Family NBD:NBD Interface Impacts Both Drug Transport and ATP Hydrolysis. International Journal of Molecular Sciences. 2020; 21(3):759. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030759
Chicago/Turabian StyleKapoor, Parth, Deborah A. Briggs, Megan H. Cox, and Ian D. Kerr. 2020. "Disruption of the Unique ABCG-Family NBD:NBD Interface Impacts Both Drug Transport and ATP Hydrolysis" International Journal of Molecular Sciences 21, no. 3: 759. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030759