The β-Blocker Carvedilol Prevented Ultraviolet-Mediated Damage of Murine Epidermal Cells and 3D Human Reconstructed Skin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
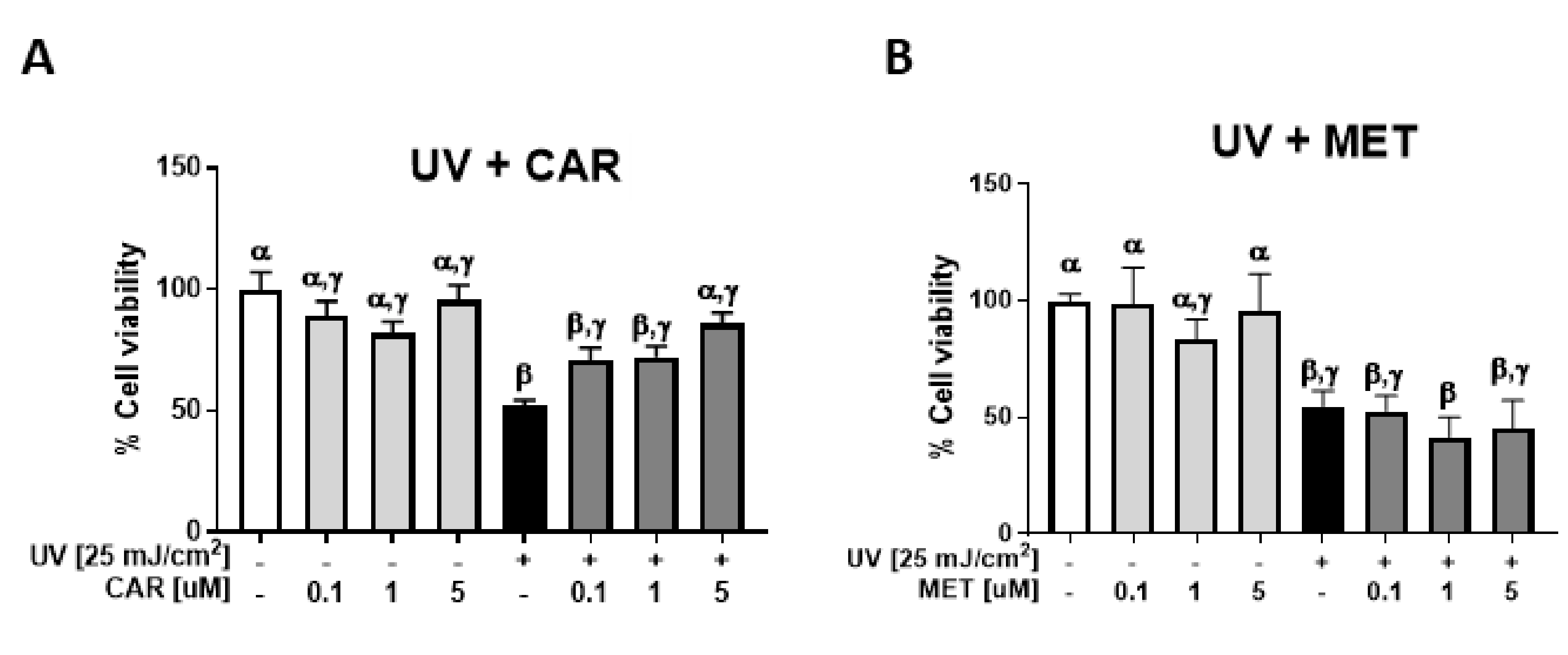
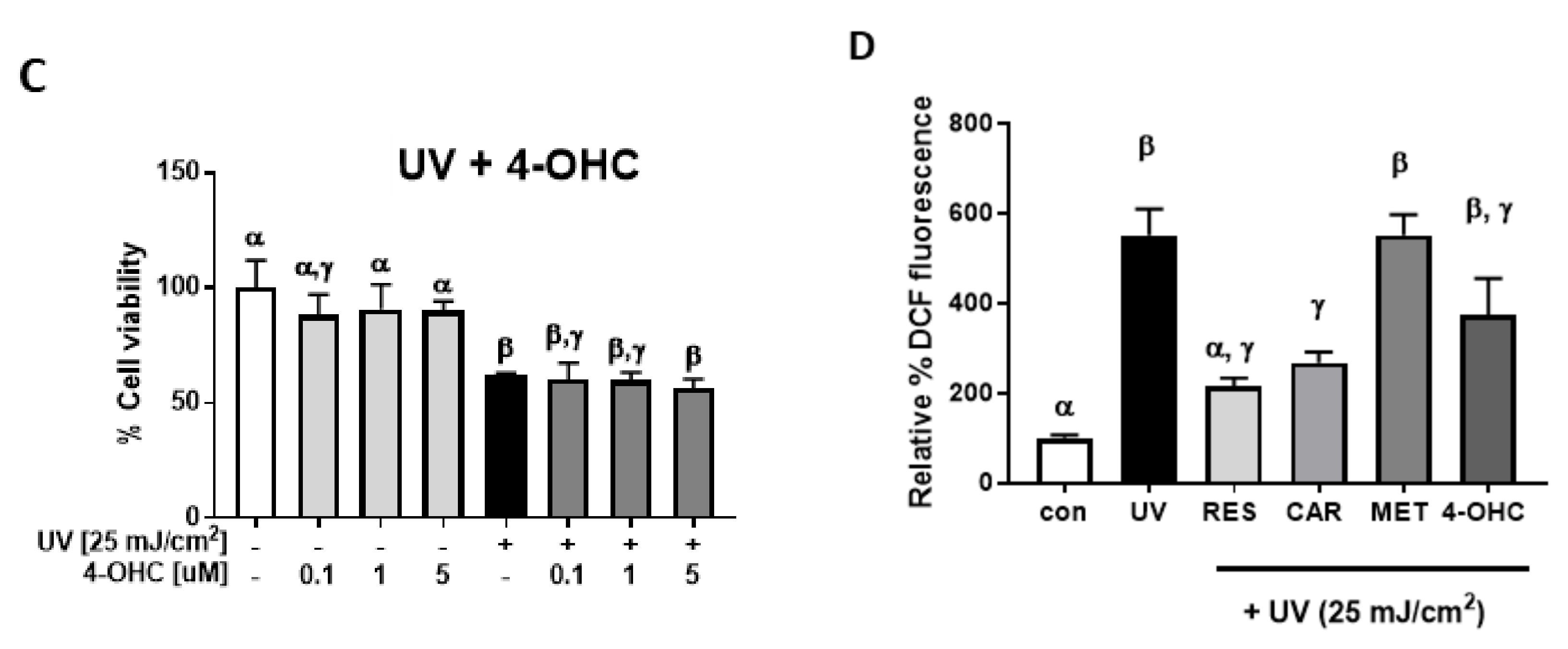
2.1. Protective Effects of Carvedilol on ultraviolet (UV)-Induced Epidermal Cell Death
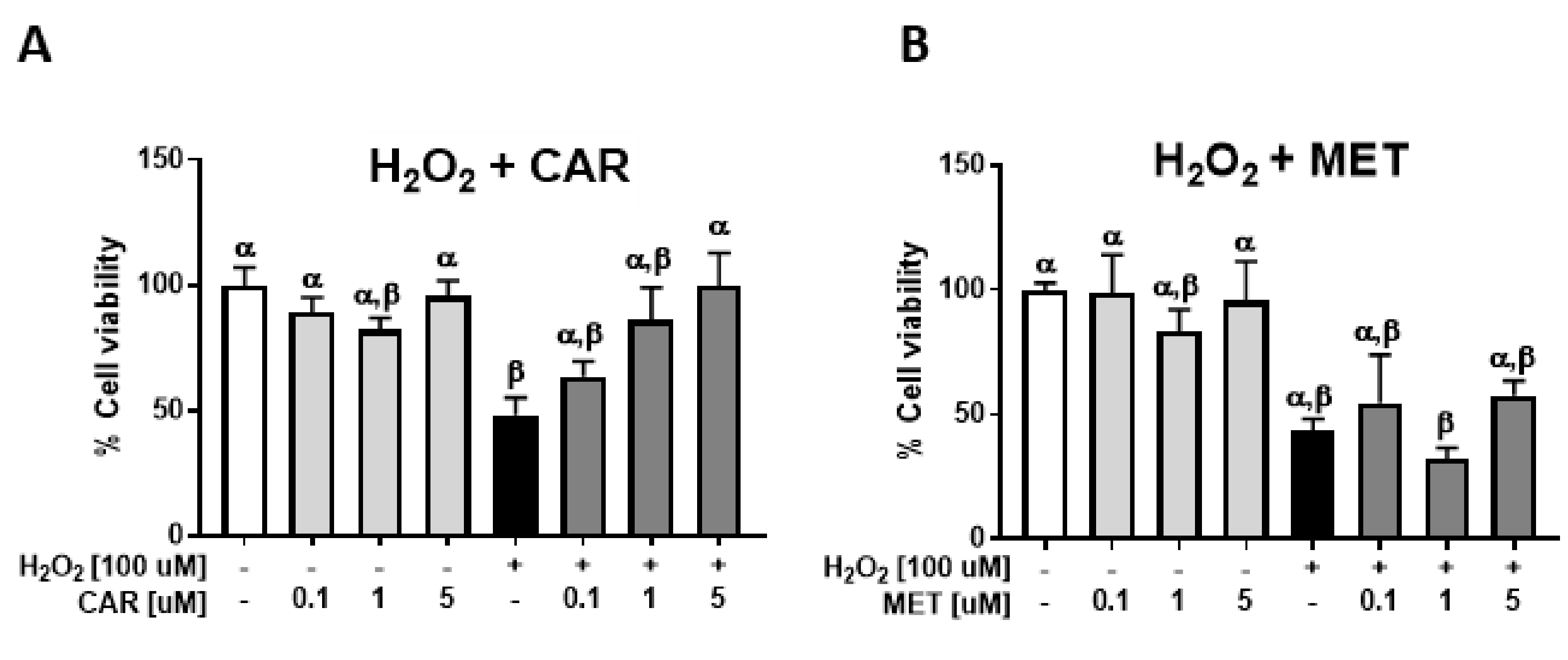
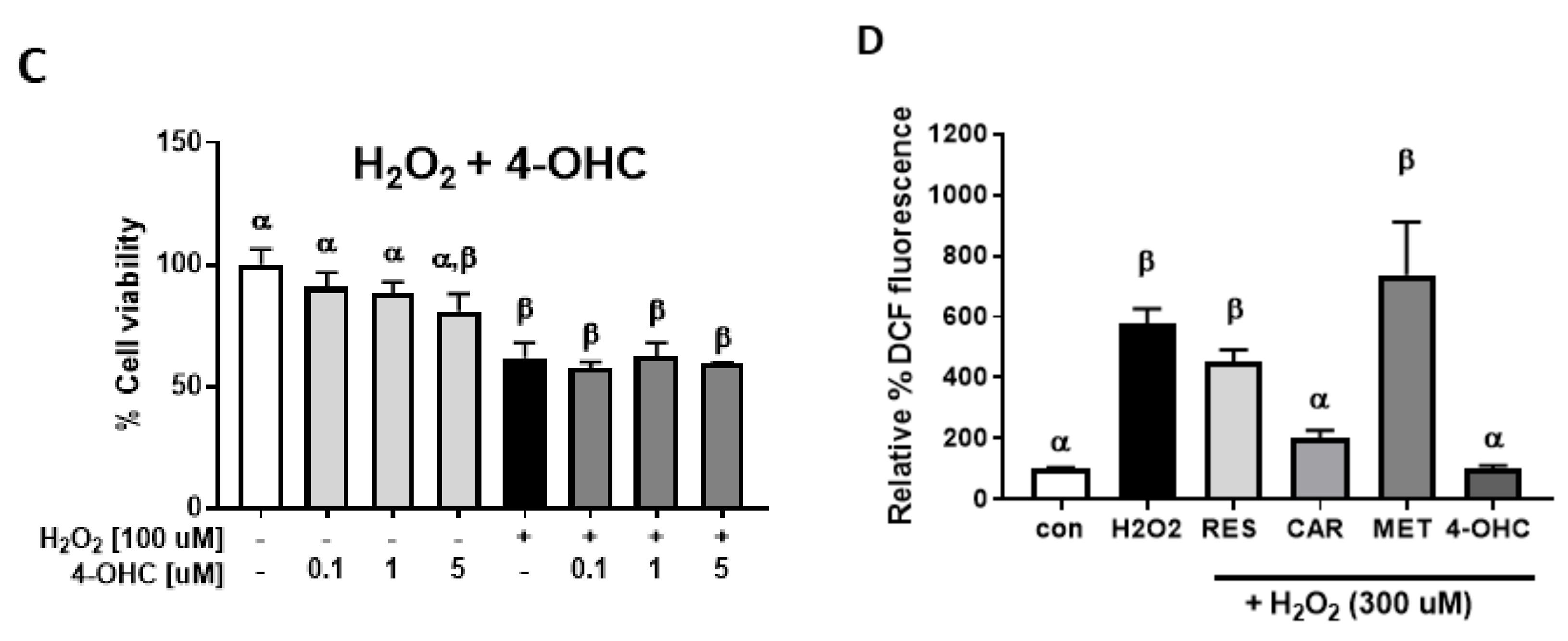
2.2. Protective Effects of Carvedilol on H2O2-Induced Epidermal Cell Death
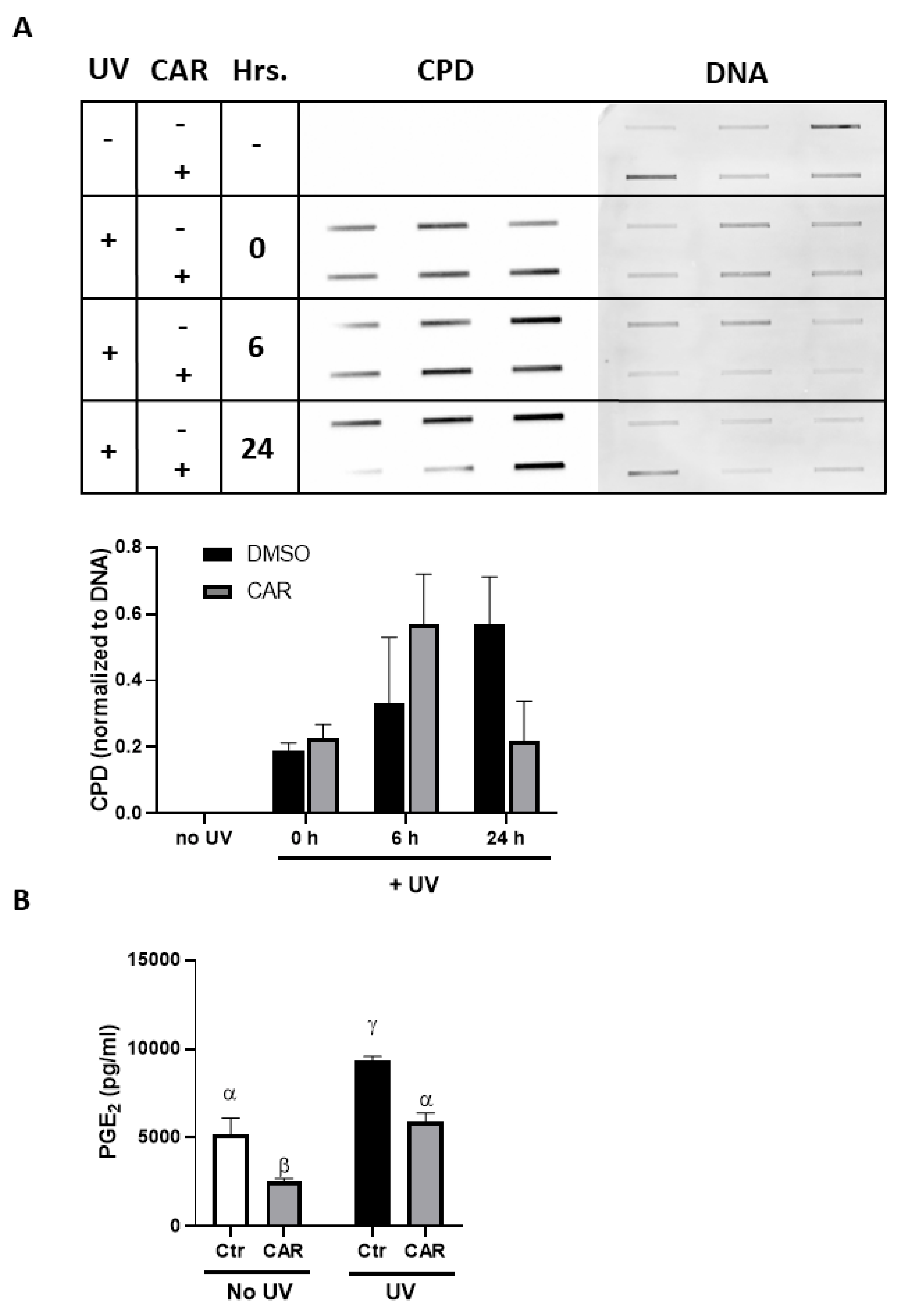
2.3. Effects of Carvedilol on UV-Induced Epidermal Cyclobutane Pyrimidine Dimer (CPD) Formation and PGE2 Secretion
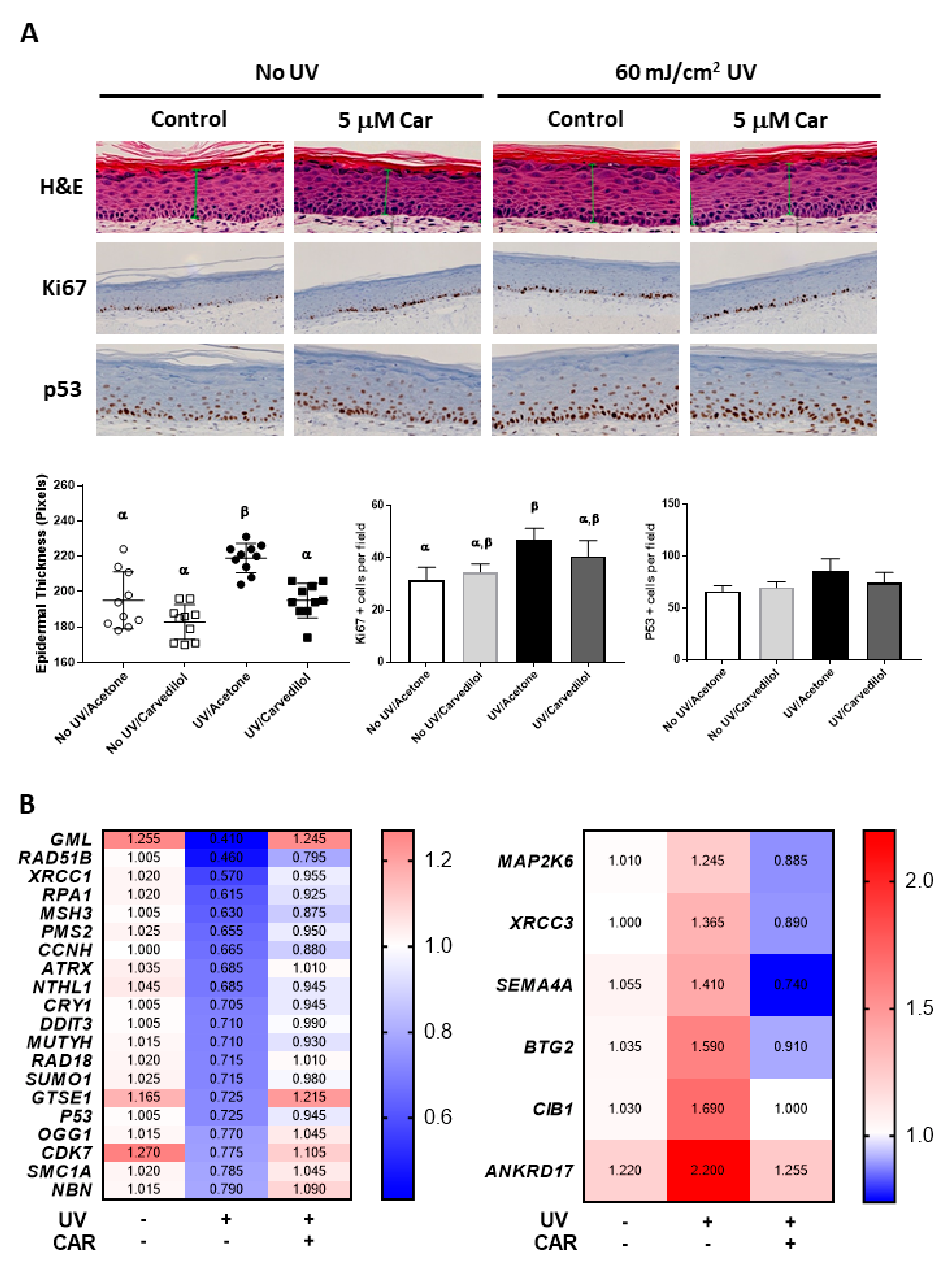
2.4. Effects of Carvedilol on UV-Induced Damage of 3D Reconstructed Human Skin
2.5. Effects of UV and Carvedilol on Expression of DNA Damage Signaling Genes by RT2 Profile Polymerase Chain Reaction (PCR) Array
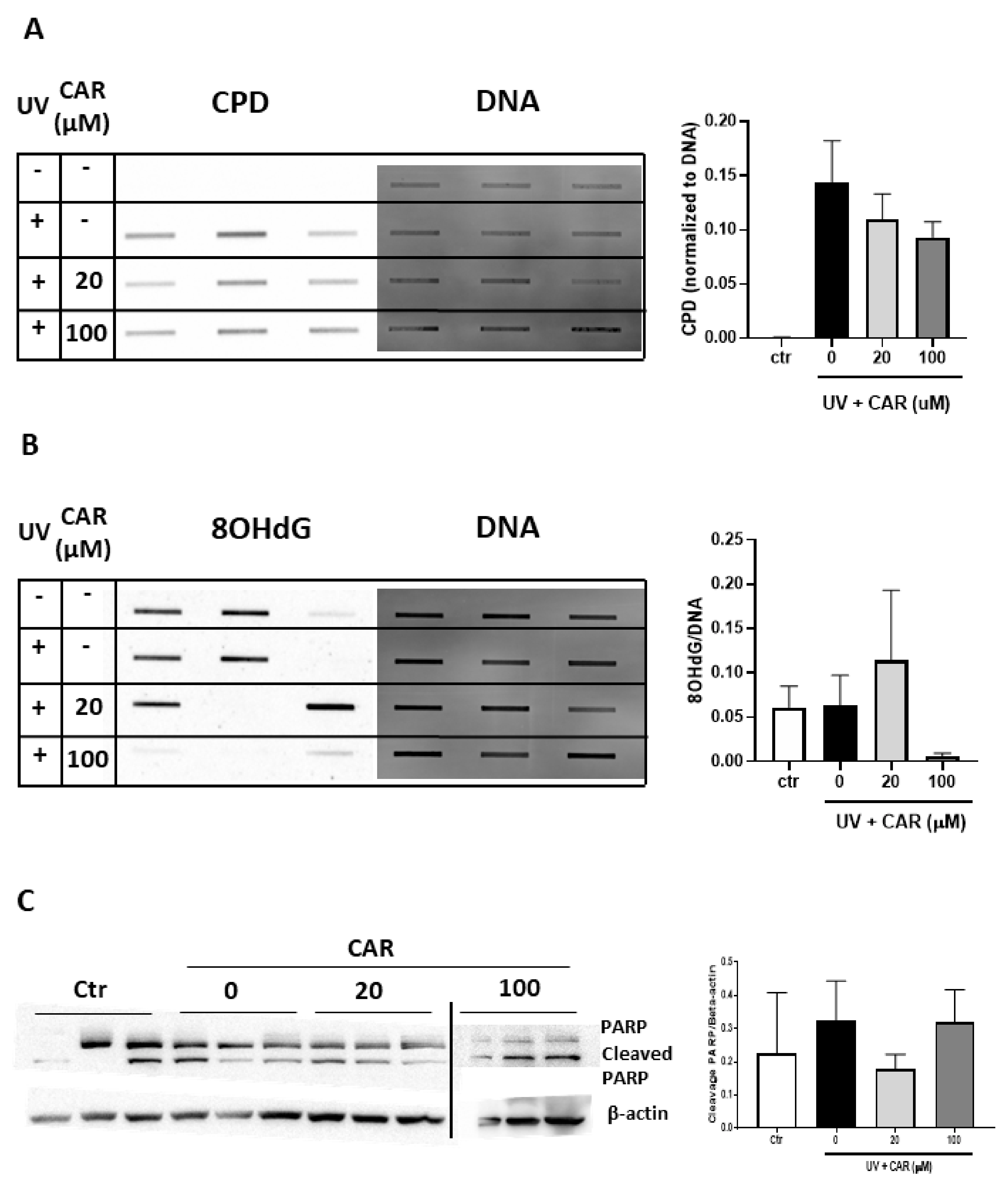
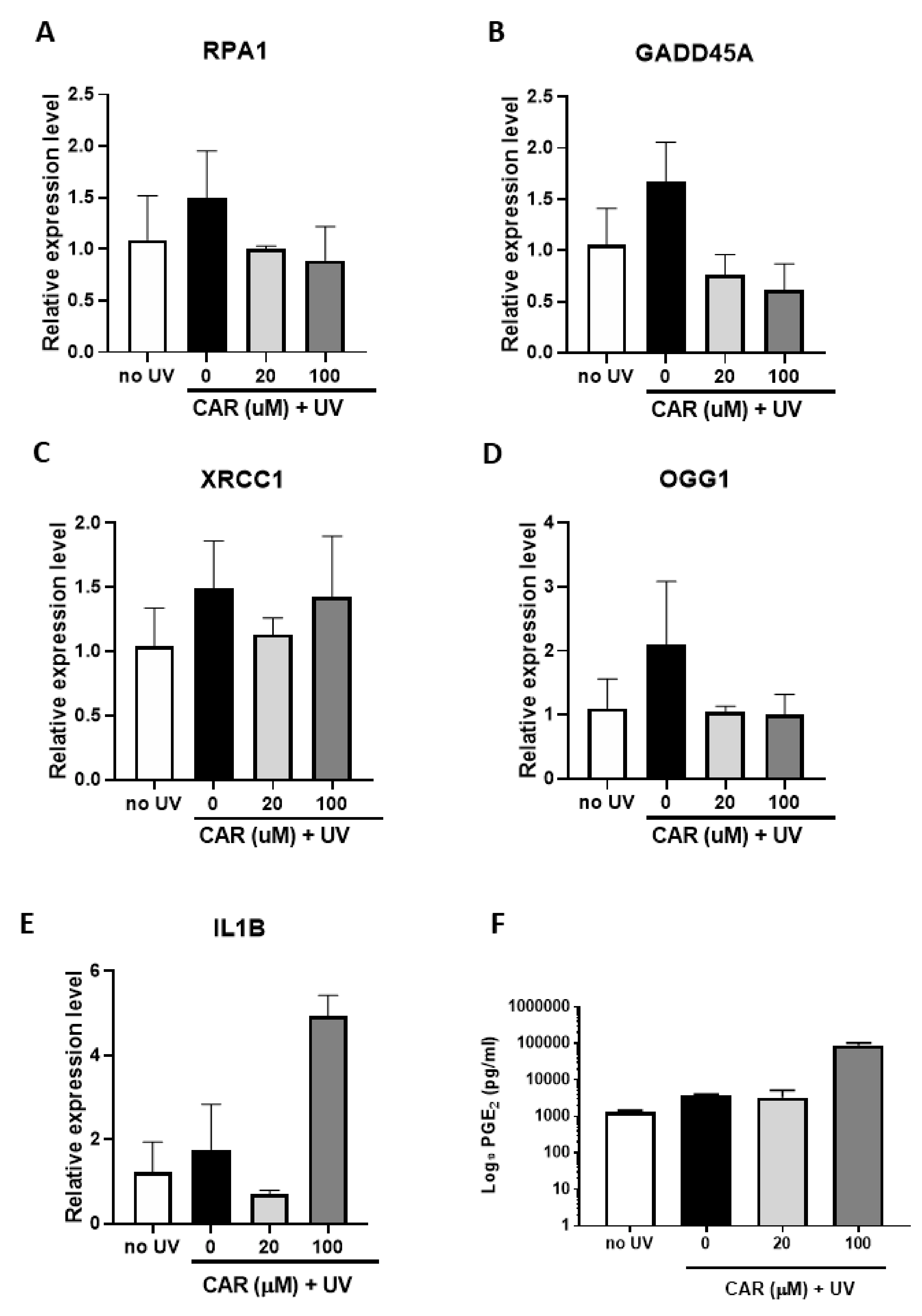
2.6. Effects of Higher Doses of Carvedilol on UV-Induced Damage in Reconstructed Human Skin
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Cell Culture
4.3. UV Light Source
4.4. Trypan Blue Exclusion Assay
4.5. Cellular Reactive Oxygen Species (ROS) Assay
4.6. Slot Blot Analysis for CPD and 8-Oxoguanine (8-OHdG)
4.7. Enzyme-Linked Immunosorbent Assay (ELISA) Assay for PGE2
4.8. 3D Human Reconstituted Skin Model
4.9. RNA Isolation and Quantitative PCR (qPCR)
4.10. Histological and Immunohistochemical (IHC) Analysis
4.11. Western Blot Analysis
4.12. Statistical Analysis
5. Conclusions
6. Patents
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| β-AR | β-Adrenergic receptor |
| 4-OHC | 4-Hydroxycarbazole |
| SD | Standard deviation |
| SE | Standard error |
| H2O2 | Hydrogen peroxide |
| UV | Ultraviolet |
| ROS | Reactive oxygen species |
| DMSO | Dimethylsulfoxide |
| CPD | Cyclobutane pyrimidine dimers |
| PGE2 | Prostaglandin E2 |
| EGF | Epidermal growth factor |
| FBS | Fetal bovine serum |
| 8-OHdG | 8-Hydroxy-2' -deoxyguanosine |
| IHC | Immunohistochemistry |
References
- Ong, H.T. Beta blockers in hypertension and cardiovascular disease. BMJ 2007, 334, 946–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, J.W.; Kokolus, K.M.; Reed, C.B.; Hylander, B.L.; Ma, W.W.; Repasky, E.A. A nervous tumor microenvironment: the impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response. Cancer Immunol. Immunother. 2014, 63, 1115–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, T.L.; Cheng, H.Y.; Lysko, P.G.; McKenna, P.J.; Feuerstein, R.; Gu, J.L.; Lysko, K.A.; Davis, L.L.; Feuerstein, G. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J. Pharmacol. Exp. Ther. 1992, 263, 92–98. [Google Scholar] [PubMed]
- Yue, T.L.; McKenna, P.J.; Ruffolo, R.R., Jr.; Feuerstein, G. Carvedilol, a new beta-adrenoceptor antagonist and vasodilator antihypertensive drug, inhibits superoxide release from human neutrophils. Eur. J. Pharmacol. 1992, 214, 277–280. [Google Scholar] [PubMed]
- Calo, L.A.; Semplicini, A.; Davis, P.A. Antioxidant and antiinflammatory effect of carvedilol in mononuclear cells of hypertensive patients. Am. J. Med. 2005, 118, 201–202. [Google Scholar] [CrossRef]
- Chang, A.; Yeung, S.; Thakkar, A.; Huang, K.M.; Liu, M.M.; Kanassatega, R.S.; Parsa, C.; Orlando, R.; Jackson, E.K.; Andresen, B.T. Prevention of skin carcinogenesis by the beta-blocker carvedilol. Cancer Prev. Res. 2015, 8, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.M.; Liang, S.; Yeung, S.; Oiyemhonlan, E.; Cleveland, K.H.; Parsa, C.; Orlando, R.; Meyskens, F.L., Jr.; Andresen, B.T.; Huang, Y. Topically Applied Carvedilol Attenuates Solar Ultraviolet Radiation Induced Skin Carcinogenesis. Cancer Prev. Res. 2017, 10, 598–606. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Liu, X.; Zhang, Q.; Yu, Z.; Gao, D. Carvedilol suppresses malignant proliferation of mammary epithelial cells through inhibition of the ROSmediated PI3K/AKT signaling pathway. Oncol. Rep. 2019, 41, 811–818. [Google Scholar]
- Cleveland, K.H.; Yeung, S.; Huang, K.M.; Liang, S.; Andresen, B.T.; Huang, Y. Phosphoproteome profiling provides insight into the mechanism of action for carvedilol-mediated cancer prevention. Mol. Carcinog. 2018, 57, 997–1007. [Google Scholar] [CrossRef]
- Cleveland, K.H.; Liang, S.; Chang, A.; Huang, K.M.; Chen, S.; Guo, L.; Huang, Y.; Andresen, B.T. Carvedilol inhibits EGF-mediated JB6 P+ colony formation through a mechanism independent of adrenoceptors. PLoS ONE 2019, 14, e0217038. [Google Scholar] [CrossRef]
- Dandona, P.; Ghanim, H.; Brooks, D.P. Antioxidant activity of carvedilol in cardiovascular disease. J. Hypertens. 2007, 25, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Lysko, P.G.; Webb, C.L.; Gu, J.L.; Ohlstein, E.H.; Ruffolo, R.R., Jr.; Yue, T.L. A comparison of carvedilol and metoprolol antioxidant activities in vitro. J. Cardiovasc. Pharmacol. 2000, 36, 277–281. [Google Scholar] [CrossRef]
- Bosch, R.; Philips, N.; Suarez-Perez, J.A.; Juarranz, A.; Devmurari, A.; Chalensouk-Khaosaat, J.; Gonzalez, S. Mechanisms of Photoaging and Cutaneous Photocarcinogenesis, and Photoprotective Strategies with Phytochemicals. Antioxidants 2015, 4, 248–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.M.; Huang, K.M.; Yeung, S.; Chang, A.; Zhang, S.; Mei, N.; Parsa, C.; Orlando, R.; Huang, Y. Inhibition of Neoplastic Transformation and Chemically-Induced Skin Hyperplasia in Mice by Traditional Chinese Medicinal Formula Si-Wu-Tang. Nutrients 2017, 9, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunaway, S.; Odin, R.; Zhou, L.; Ji, L.; Zhang, Y.; Kadekaro, A.L. Natural Antioxidants: Multiple Mechanisms to Protect Skin From Solar Radiation. Front. Pharmacol. 2018, 9, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, E.H.; Fukuda, N.; Matsumoto, T.; Katakawa, M.; Yamamoto, C.; Han, Y.; Ueno, T.; Kobayashi, N.; Matsumoto, K. Effects of the antioxidative beta-blocker celiprolol on endothelial progenitor cells in hypertensive rats. Am. J. Hypertens. 2008, 21, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Romeo, F.; Li, D.; Shi, M.; Mehta, J.L. Carvedilol prevents epinephrine-induced apoptosis in human coronary artery endothelial cells: Modulation of Fas/Fas ligand and caspase-3 pathway. Cardiovasc. Res. 2000, 45, 788–794. [Google Scholar] [CrossRef] [Green Version]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Aydinalp, A.; Okyay, K.; Tekin, A.; Bal, U.A.; Bayraktar, N.; Yildirir, A.; Muderrisoglu, H. Comparison of Carvedilol and Metoprolol for Preventing Contrast-Induced Nephropathy after Coronary Angiography. Cardiorenal. Med. 2015, 5, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Malig, T.C.; Ashkin, M.R.; Burman, A.L.; Barday, M.; Heyne, B.J.M.; Back, T.G. Comparison of free-radical inhibiting antioxidant properties of carvedilol and its phenolic metabolites. Med. Chem. Comm. 2017, 8, 606–615. [Google Scholar] [CrossRef]
- Park, K.H.; Park, D.R.; Kim, Y.W.; Nam, T.S.; Jang, K.Y.; Chung, H.T.; Kim, U.H. The Essential Role of Ca(2+) Signals in UVB-Induced IL-1beta Secretion in Keratinocytes. J. Invest. Dermatol. 2019, 139, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.L.; Le, M.N.; Miranda, C.L.; Reed, R.L.; Stevens, J.F.; Indra, A.K.; Ganguli-Indra, G. Photoprotective Properties of Isothiocyanate and Nitrile Glucosinolate Derivatives From Meadowfoam (Limnanthes alba) Against UVB Irradiation in Human Skin Equivalent. Front. Pharmacol. 2018, 9, 477. [Google Scholar] [CrossRef] [PubMed]
- Paz-Elizur, T.; Sevilya, Z.; Leitner-Dagan, Y.; Elinger, D.; Roisman, L.C.; Livneh, Z. DNA repair of oxidative DNA damage in human carcinogenesis: Potential application for cancer risk assessment and prevention. Cancer Lett. 2008, 266, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, N.; Daiber, A.; Forstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [Green Version]
- Reagan-Shaw, S.; Mukhtar, H.; Ahmad, N. Resveratrol imparts photoprotection of normal cells and enhances the efficacy of radiation therapy in cancer cells. Photochem. Photobiol. 2008, 84, 415–421. [Google Scholar] [CrossRef]
- Arab, H.H.; El-Sawalhi, M.M. Carvedilol alleviates adjuvant-induced arthritis and subcutaneous air pouch edema: Modulation of oxidative stress and inflammatory mediators. Toxicol. Appl. Pharmacol. 2013, 268, 241–248. [Google Scholar] [CrossRef]
- Yin, Y.; Li, W.; Son, Y.O.; Sun, L.; Lu, J.; Kim, D.; Wang, X.; Yao, H.; Wang, L.; Pratheeshkumar, P. Quercitrin protects skin from UVB-induced oxidative damage. Toxicol. Appl. Pharmacol. 2013, 269, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; Wang, Y.; Xue, Y.; He, L.; Li, Y.; Xiong, J. Low-dose UVB irradiation prevents MMP2-induced skin hyperplasia by inhibiting inflammation and ROS. Oncol. Rep. 2015, 34, 1478–1486. [Google Scholar] [CrossRef] [Green Version]
- Afaq, F.; Zaid, M.A.; Khan, N.; Dreher, M.; Mukhtar, H. Protective effect of pomegranate-derived products on UVB-mediated damage in human reconstituted skin. Exp. Dermatol. 2009, 18, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.O.; Wang, Y.; Stebbins, W.G.; Gao, D.; Zhou, X.; Phelps, R.; Lebwohl, M.; Wei, H. Photoprotective effect of isoflavone genistein on ultraviolet B-induced pyrimidine dimer formation and PCNA expression in human reconstituted skin and its implications in dermatology and prevention of cutaneous carcinogenesis. Carcinogenesis 2006, 27, 1627–1635. [Google Scholar] [CrossRef]
- Gruber, J.V.; Holtz, R.; In Yang, S. In vitro examination of an oleosome-based sun protection product on the influence of UVB-induced inflammation markers in human epidermal skin equivalent tissue model. J. Photochem. Photobiol. B 2018, 179, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.W.; Lee, H.W.; Park, S.H.; Hu, Y.; Wang, H.T.; Chen, L.C.; Rom, W.N.; Huang, W.C.; Lepor, H.; Wu, X.R. Aldehydes are the predominant forces inducing DNA damage and inhibiting DNA repair in tobacco smoke carcinogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E6152–E6161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.W.; Park, S.H.; Weng, M.W.; Wang, H.T.; Huang, W.C.; Lepor, H.; Wu, X.R.; Chen, L.C.; Tang, M.S. E-cigarette smoke damages DNA and reduces repair activity in mouse lung, heart, and bladder as well as in human lung and bladder cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1560–E1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Eberlin, S.; Clerici, S.P.; Abdalla, B.M. In vitro effects of infrared A radiation on the synthesis of MMP-1, catalase, superoxide dismutase and GADD45 alpha protein. Inflamm. Allergy. Drug. Targets. 2015, 14, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Jeong, S.A.; Choi, H.S.; Ro, S.; Lee, J.S.; Park, J.K. Protective effects of ginsenoside Rg2 and astaxanthin mixture against UVB-induced DNA damage. Anim. Cells Syst. 2018, 22, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, N.; Srivastava, S.K.; Arora, S.; Omar, Y.; Ijaz, Z.M.; Al-Ghadhban, A.; Deshmukh, S.K.; Carter, J.E.; Singh, A.P.; Singh, S. Comparative analysis of the relative potential of silver, Zinc-oxide and titanium-dioxide nanoparticles against UVB-induced DNA damage for the prevention of skin carcinogenesis. Cancer Lett. 2016, 383, 53–61. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Liang, S.; Shahid, A.; Andresen, B.T.; Huang, Y. The β-Blocker Carvedilol Prevented Ultraviolet-Mediated Damage of Murine Epidermal Cells and 3D Human Reconstructed Skin. Int. J. Mol. Sci. 2020, 21, 798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030798
Chen M, Liang S, Shahid A, Andresen BT, Huang Y. The β-Blocker Carvedilol Prevented Ultraviolet-Mediated Damage of Murine Epidermal Cells and 3D Human Reconstructed Skin. International Journal of Molecular Sciences. 2020; 21(3):798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030798
Chicago/Turabian StyleChen, Mengbing, Sherry Liang, Ayaz Shahid, Bradley T. Andresen, and Ying Huang. 2020. "The β-Blocker Carvedilol Prevented Ultraviolet-Mediated Damage of Murine Epidermal Cells and 3D Human Reconstructed Skin" International Journal of Molecular Sciences 21, no. 3: 798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030798