Practical Guidance in Genome-Wide RNA:DNA Triple Helix Prediction


1
Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119234 Moscow, Russia
2
Institute of Bioengineering, Research Center of Biotechnology, Russian Academy of Science, 117312 Moscow, Russia
3
Department of Biological and Medical Physics, Moscow Institute of Physics and Technology, 141701 Dolgoprudny, Russia
4
Department of Computational Biology, Vavilov Institute of General Genetics, Russian Academy of Science, 117971 Moscow, Russia
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(3), 830; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030830
Submission received: 17 December 2019
/
Revised: 21 January 2020
/
Accepted: 25 January 2020
/
Published: 28 January 2020
(This article belongs to the Special Issue Medical Genetics, Genomics and Bioinformatics)
Abstract
:Long noncoding RNAs (lncRNAs) play a key role in many cellular processes including chromatin regulation. To modify chromatin, lncRNAs often interact with DNA in a sequence-specific manner forming RNA:DNA triple helices. Computational tools for triple helix search do not always provide genome-wide predictions of sufficient quality. Here, we used four human lncRNAs (MEG3, DACOR1, TERC and HOTAIR) and their experimentally determined binding regions for evaluating triplex parameters that provide the highest prediction accuracy. Additionally, we combined triplex prediction with the lncRNA secondary structure and demonstrated that considering only single-stranded fragments of lncRNA can further improve DNA-RNA triplexes prediction.
1. Introduction
Long noncoding RNAs (lncRNAs) are usually defined as transcripts of more than 200 nt in length and demonstrating no protein-coding capacity. LncRNAs are often lowly expressed and highly tissue-specific as compared to protein-coding genes. These properties of lncRNAs lead to difficulties in the robust detection of their transcription as well as detailed reconstruction of the transcript structure [1,2]. Yet, a combination of several RNA-sequencing techniques improved the detection of lncRNAs [3,4]. Currently, the total number of lncRNAs annotated in the human and mouse genomes is close to the number of protein-coding genes [5,6]. The functional role of the majority of lncRNAs is still unclear. On the other hand, transcription of lncRNAs is regulated [3,7], supporting their functional importance. Indeed, it has been shown that lncRNAs function via surprisingly diverse molecular mechanisms on the transcriptional and posttranscriptional levels (reviewed in [8,9,10]), playing a key role in many cellular processes, including epigenetic regulation of transcription via interaction with chromatin [10]. Being located in the nucleus of mammalian cells [11] lncRNAs often mediate transcription by targeting chromatin-modifying complexes [12,13] or transcription factors (TFs) [14] to specific genomic loci. In addition to protein binding capacity, RNA has the capacity to form hydrogen bonds on both sides of the DNA strand. RNA forms both Watson–Crick and non-Watson–Crick pairs on the Watson–Crick face of DNA strand and Hoogsteen bonds on the other side of DNA strand [15]. As a result, lncRNAs use different molecular mechanisms to bind to the chromatin, including RNA binding to single-stranded DNA regions (known as R-loops) [16], co-transcriptional RNA:RNA interactions based on Watson-Crick pairing [17] and direct RNA:DNA hybridization via triple helices based on Hoogsteen interactions [18,19]. Hoogsteen or reverse Hoogsteen bonds are formed between a single-stranded nucleotide and a purine nucleotide in a double-stranded nucleic acid molecule. An RNA strand becomes parallel or antiparallel to the DNA strand [20]. UC motives in RNA strand tend to form parallel triplexes, AG motives tend to form antiparallel triplexes, while GU motives could form both [20]. Hoogsteen bonds are weaker than Watson-Crick bonds, resulting in Hoogsteen pairing rules being less strict [20].
There are several known cases of lncRNAs involved in chromatin regulation via formation of triple helices with DNA in specific regions. HOTAIR binds to DNA and recruits two chromatin-modifying complexes: PRC2 and LSD1-CoREST, leading to transcription repression in trans [21]. lncRNA ANRIL binds to DNA in cis and recruits PRC1 and PRC2, which in turn represses transcription [21]. LncRNA MEG3 regulates Wnt/β-catenin [22], VEGF [23] and TGF-β [13] pathways by formation of triple helices and in this way attracting PRC2 to target genes [13]. MEG3 ability to form triplexes was validated via different experimental methods [13]. Fendrr recruits PRC2 via RNA:DNA triplex formation and interacts with Trithorax group/Mixed lineage leukemia (TrxG/Mll) complex [12].
Yet, genome-wide prediction of RNA:DNA triplex-based interactions remains a challenging task with a great dependency on triplexes parameters and with a lot of false positive predictions [24]. Single-stranded ribonucleotide chains (including lncRNAs) tend to fold into thermodynamically stable structures [25]. In many cases, the secondary structure of lncRNAs dictates their function (reviewed in detail in [26]). According to the Hoogsteen rules an RNA region cannot form both duplexes and triplexes at the same time. This implies that triplexes can be formed by single-stranded RNA regions only. Indeed, some data indicate that the validated DNA binding domains of the MEG3 lncRNA might correspond to the mainly unpaired RNA regions in the experimentally identified secondary structure model [27]. In this work, we test if adding a predicted secondary structure of lncRNA in the triple helix search increases prediction specificity.
2. Results
2.1. Fitting Triplex Parameters
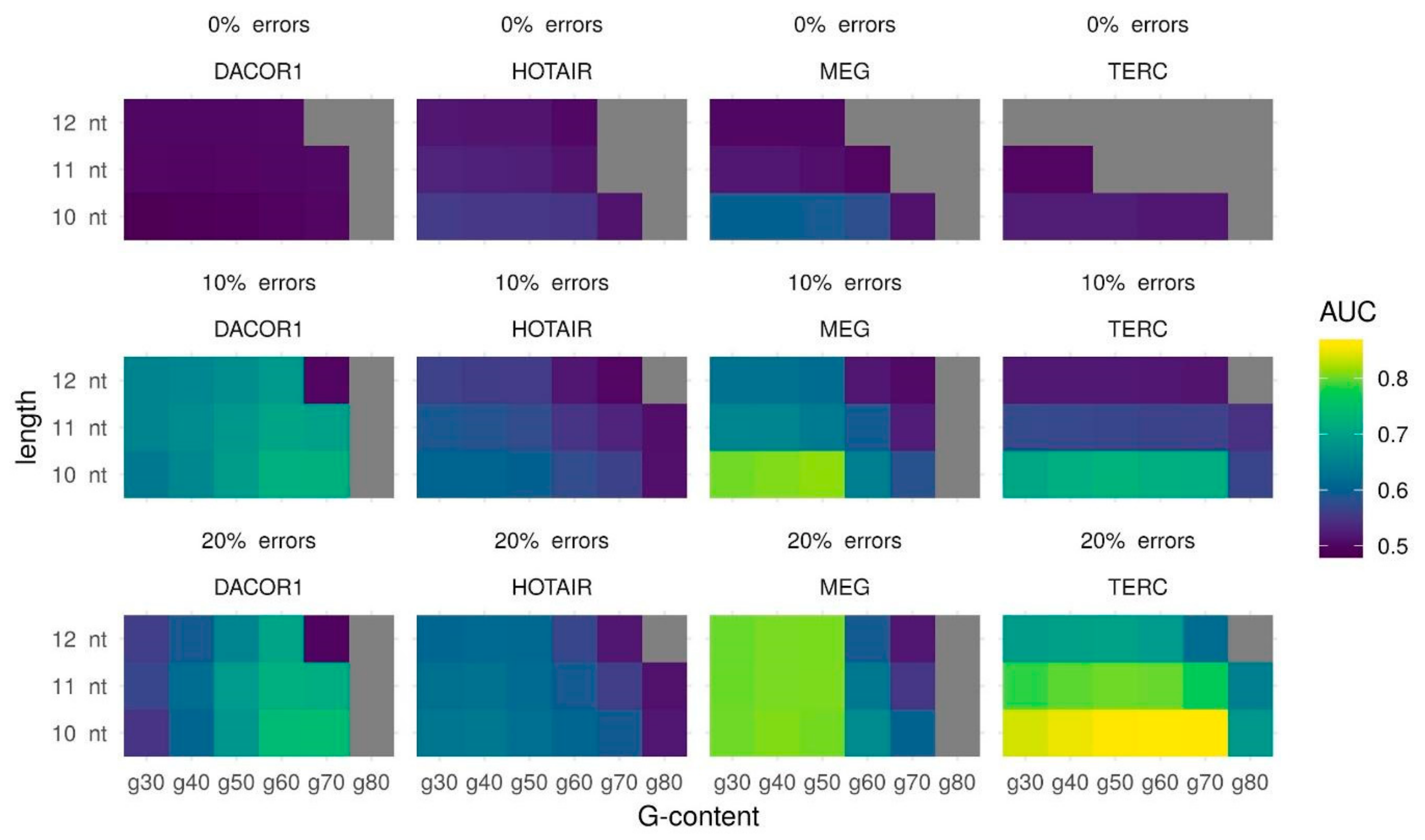
Known cases of lncRNA forming triplexes suggest that actual triple helices may vary in length and number of mismatches. Additionally, enrichment in GA-rich sequences is associated with the formation of stronger triple helices [13]. This makes it reasonable to tune triplexes parameters such as the minimum triplex length, the maximum error-rate, and the minimum G-content to see if there is a combination of the parameters that works best for the majority of lncRNAs with known binding regions. Areas Under the Curve (AUC) for several different parameter combinations are provided in Table S1 and Figure 1. These results clearly suggest that 10 nt as a minimum length of predicted triple helix gives the best AUC in all cases. No errors permitted leads to a very poor prediction quality: AUC close to 0.5 or lack of predicted triplexes. For three out of four lncRNAs (DACOR1, HOTAIR, and TERC) using 20% of the errors leads to the best results. For lncRNA MEG3 both 10% and 20% of the errors give similar results (AUC for 10% errors is 0.0044 higher than AUC for 20% errors). For the sake of uniformity, we suggest that 10 nt and 20% errors are the parameters that are likely to provide the best results. In terms of minimal G-content, lncRNA may be divided into two groups: those that form high (at least 70%) and low (at least 40%) G-content triplexes: TERC and DACOR1 vs. MEG3 and HOTAIR, respectively.
Summing up, we demonstrate that for lncRNAs TERC and DACOR1, the best AUC could be obtained with a minimum length of 10 nt, a maximum error rate of 20%, and a minimum G-content of 70% (AUC = 0.8601 and AUC = 0.7423, respectively), while for MEG3 and HOTAIR, the best AUC could be obtained with a minimum length of 10 nt, a maximum error rate of 20%, and a minimum G-content of 40% (AUC = 0.8078 and AUC = 0.6372, respectively) (Table 1). It should be noted that while a minimum length of 10 bp and a maximum error rate of 20% produce the best AUC for all lncRNAs, the minimum G-content can vary between lncRNAs splitting them into two groups (Table 1). As we see no reason to give any preference to one G-content threshold over another, we suggest to try both sets of parameters.
2.2. Secondary Structure Predictions
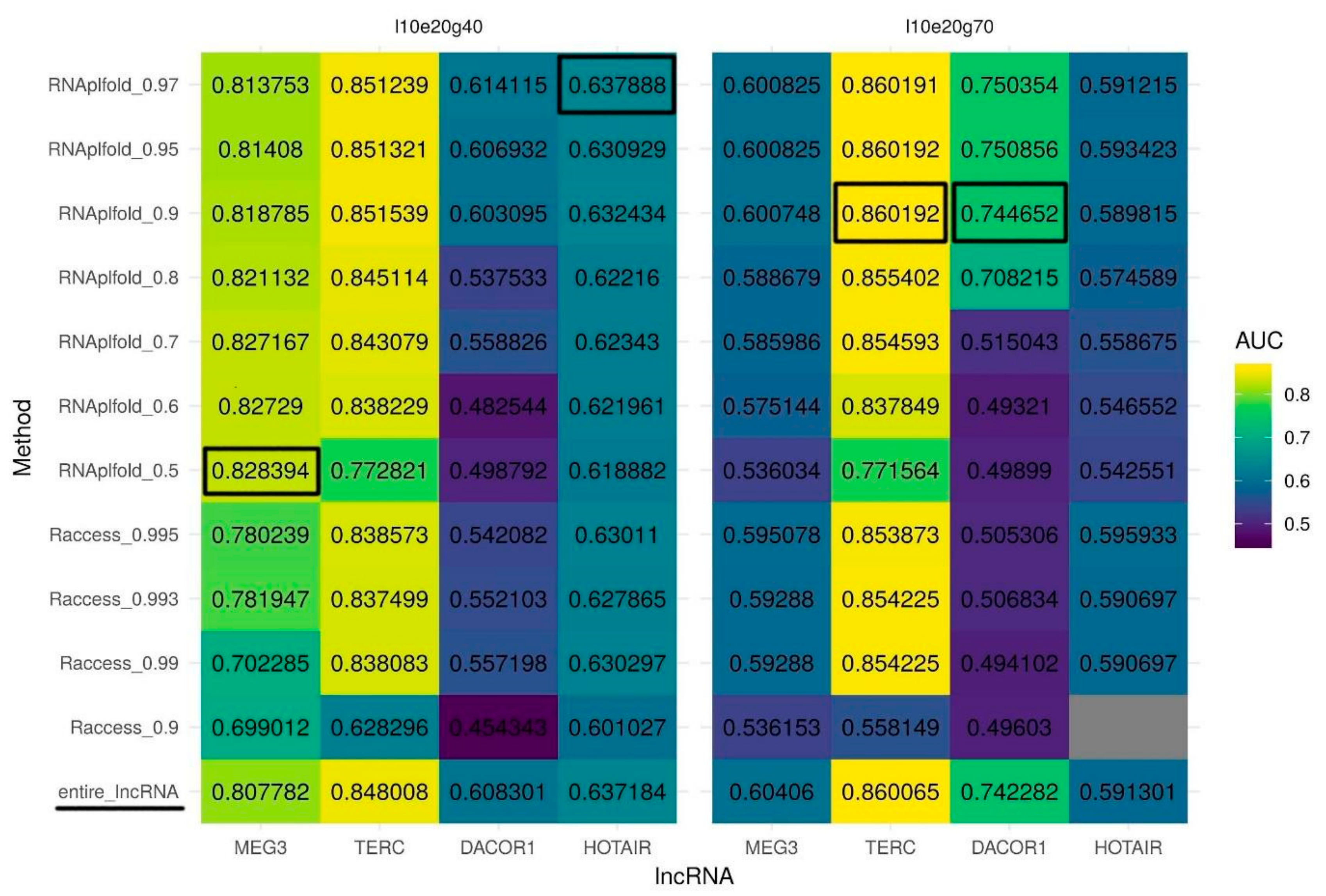
According to the Hoogsteen rules, only unpaired RNA nucleotides can participate in triplex formation. Thus, we speculate that information about the lncRNA secondary structure may reduce the number of false positive predictions and as a result may increase prediction quality (AUC). To identify unpaired regions within lncRNAs, we test two tools for secondary structure prediction: RNAplfold and Raccess. To run these tools, we used several different thresholds for nucleotide pairing probability and the best sets of Triplexator parameters determined above. If a nucleotide in a lncRNA was predicted to be paired it was masked by the N character and the modified sequence was used for triple helix prediction. Generally, structures predicted with RNAplfold outperform those predicted by Raccess in terms of AUC for triple helix predictions. Importantly, we observed that some parameters of RNA secondary structure prediction result in a huge decrease in the AUC values compared to the original unmasked sequence (Supplementary Figure S1). Yet, for MEG3, using only unpaired (single-stranded) regions of the lncRNA increases the AUC for triplex predictions for structures predicted by RNAplfold but not by Raccess (Figure 2, left panel). Surprisingly, to achieve the best quality of prediction for MEG3 we have to set the probability of pairing as low as 0.5 (RNAplfold). For other lncRNAs usage of solely single-stranded sequences predicted by RNAplfold but not by Racceess also improves the quality of triplexes prediction in most of the cases. For DACOR1 and TERC (Figure 2, right panel), the highest improvement of triplex prediction is achieved when a threshold for a probability of pairing is set to 0.95, while for HOTAIR (Figure 2, left panel), the best results are achieved with a pairing probability threshold of 0.97.
RNAplfold also outperforms Raccess based on partial AUC at 10% FPR in almost all the cases (Supplementary Figure S2) (for the reference, partial AUC at 10% FPR sequences for a random classifier is 0.5%). Partial AUC for MEG3 (the best parameters determined above: l10e20g40, RNAplfold pairing probability 0.5) is increased dramatically (from 4.76% to 6.05%) as compared to the increase in full AUC suggesting an improvement in the prediction of triplexes with the highest normalized frequencies (tpot). Partial AUC for DACOR1 (the best parameters determined above: l10e20g70, RNAplfold pairing probability 0.95) is also increased (from 2.7% to 3.55%), supporting the biggest improvement for the sequences with the highest normalized frequencies of predicted triplexes. Partial AUC for TERC increases only slightly (from 3.696% to 3.704%). Partial AUC for HOTAIR is not increased with the previously determined parameters (l10e20g40, RNAplfold pairing probability 0.97), while it shows a slight increase (from 1.01% to 1.07%) when the RNAplfold pairing probability threshold is set to 0.8.
Summing up, we demonstrate that RNAplfold outperforms Raccess in terms of AUC and partial AUC for 10% FPR triplexes predicted. Three thresholds for probability of pairing lead to best results: 0.5 for MEG3, 0.95 for DACOR1 and TERC, and 0.97 for HOTAIR.
2.3. Triplex Prediction Using Secondary Structure
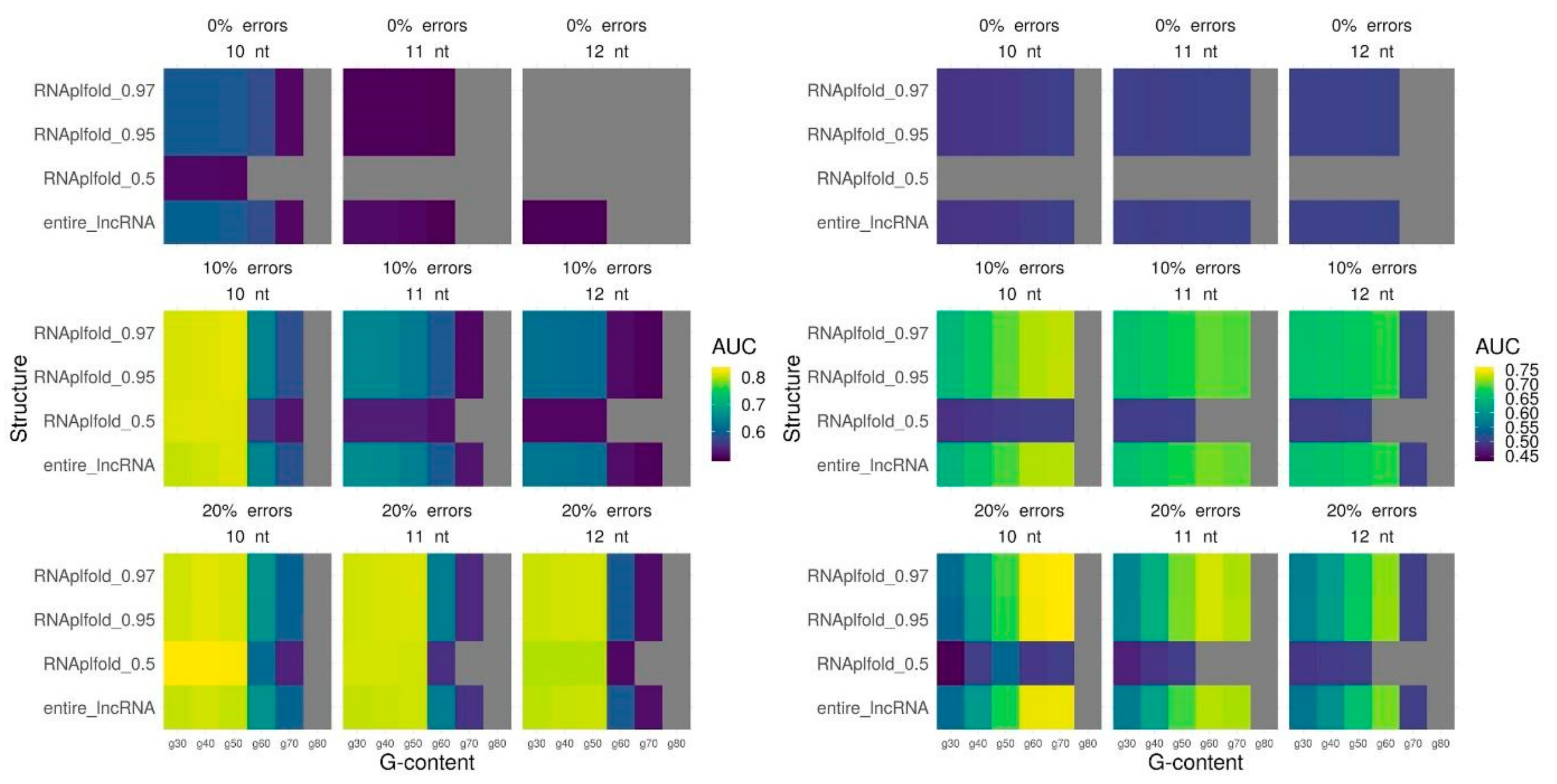
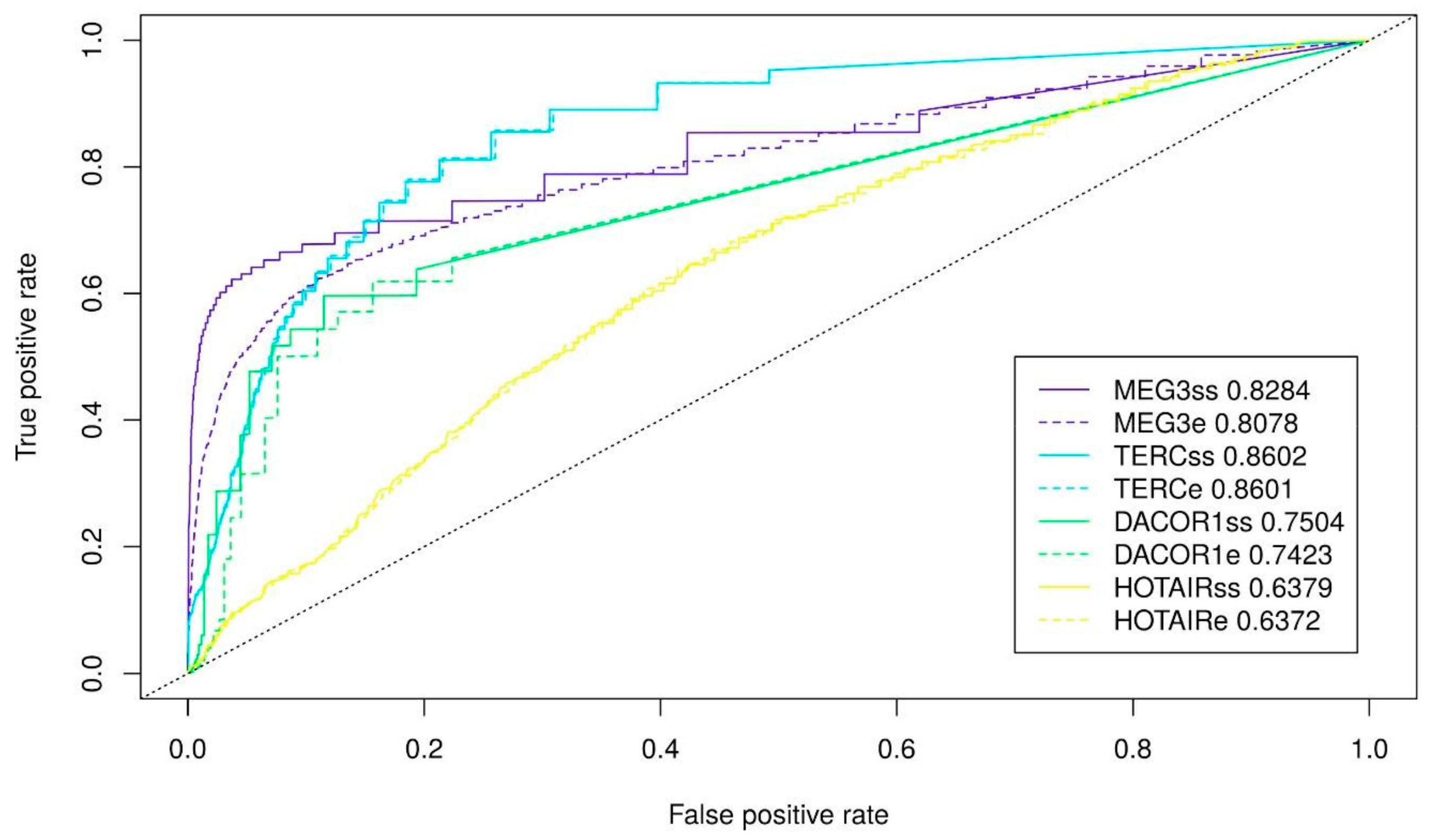
Finally, we fit the parameters for triplex prediction in combination with the probability of nucleotide pairing based on the secondary structure for all lncRNAs. We vary a minimum length, maximum error-rate, and minimum G-content for predicted triplexes and used three different RNAplfold thresholds (0.5, 0.95, 0.97) that performed best for the whole lncRNAs. In the majority of the cases, parameters performed the best for the entire lncRNA appears to be the best parameters also when only single-stranded lncRNA regions are used for triplex prediction: a minimum length of 10 nt and a minimum error-rate of 20% (Figure 3, Supplementary Figure S3). A tendency of a lncRNA to form a low G-content (40%) or high G-content (70%) triplexes also retains (Figure 3, Supplementary Figure S3). To conclude, we achieve the best quality of triplexes prediction using the following set of parameters: for MEG3 (l10e20g40 and RNAplfold pairing probability of 0.5), for TERC (l10e20g70 and RNAplfold pairing probability of 0.95), for DACOR1 (l10e20g40 and RNAplfold pairing probability of 0.95), and for HOTAIR (l10e20g70 and RNAplfold pairing probability of 0.97). It should be noted that the most significant improvement is achieved for MEG3. The ROC (receiver operating characteristic)-curves with the best prediction quality are provided on Figure 4.
2.4. DNA Binding Domains (DBDs) and Single-Stranded Fragments
We further investigate why the usage of the secondary structure is the most beneficial in the case of MEG3. To do so we detect DNA binding domains (DBDs) - fragments of lncRNAs that form the majority of triplexes with the target DNA regions - using Triplex Domain Finder (TDF) [28] both for entire lncRNAs and for single-stranded fragments. In the case of the entire MEG3 (Figure 5, bottom) TDF found a lot of long DBDs that form a significant number of triplexes with the target DNA regions. Yet, many of these DBDs contain nucleotides with a relatively high probability of pairing, thus, considering only single-stranded fragments (Figure 5, top) leads to removal or shrinkage of such DBDs. Remaining DBDs form triplexes with a lot of experimentally validated RNA:DNA interacting regions. Two of the previously reported DBDs: 20–38 nt and 971–999 nt [13,28] increase their z-score. In the case of MEG3, usage of the secondary structure reduces false positive predictions leading to an improvement of the overall prediction quality. In the case of DACOR1 (Figure 6), only two DBDs are detected for the entire lncRNA. For a single-stranded variant of lncRNA, the DBD with the low number of predicted triplexes is removed, presumably reducing the number of false positives. For the entire TERC and HOTAIR (Supplementary Figures S4 and S5) several DBDs are detected and the majority of them are kept intact or slightly shortened if only a single-stranded RNA is considered. As a result, only a very moderate increase in AUC is observed.
3. Discussion
Prediction of triple helix structures genome-wide is a challenging task [24]. Although Triplexator [29] outperforms an alternative approach provided by LongTarget [30], the prediction quality is still to be improved [24]. Recently, a high accuracy model for triplex prediction based on a neuronal network has been proposed [31]. However, it requires data on experimental lncRNA binding for training which is available only for a small number of lncRNAs.
Computational genome-wide prediction of triplexes formed by a particular lncRNA is complicated by the presence of universal triplex target sites (TTS) - regions that are capable of triplex formation with almost any expressed lncRNA [32]. Similarly, we found universal DNA binding domains (DBDs). The most significant one is located at the 3′ end of HOTAIR and forms even more triplexes with random DNA fragments than with the HOTAIR target regions obtained in the ChOP-seq experiment (Table S2). Additionally, it has been shown that HOTAIR can form not only triplexes but also duplexes with single-stranded DNA [17]. We speculate that having two different mechanisms of binding may contribute to the low quality of triplex prediction in the case of HOTAIR.
Interestingly, to increase the quality of MEG3 triplex prediction, we had to use a relatively low threshold for base pairing in RNAplfold. It should be noticed that the entire MEG3 has lots of DBDs (and lots of triplex forming oligos (TFOs) - fragments that potentially form triplexes with any possible DNA – that can be obtained from Triplexator (Table S3). The majority of these DBDs cover only a small fraction of MEG3 target sequences. It seems unlikely that all these DBDs or TFOs represent real triplexes suggesting the need to use stricter RNAplfold base pairing threshold (0.5). While DACOR1 and TERC have only a few TFOs (or DBDs) (Table S3) when the best triplex parameters are used (Table 1) and these DBDs are present in many target regions, suggesting that those DBDs are more likely to be functional. Since HOTAIR has two mechanisms of binding, we do not recommend to base any conclusions on the results of this lncRNA.
It should be noted that our approach has several limitations. First, experimental techniques such as ChOP-seq and ChIRP-seq detect only the regions of DNA that interact with a particular lncRNA but do not uncover the interaction mechanisms. For several particular lncRNA:DNA triplex formation has been validated experimentally in vitro [13], but it is difficult to scale this approach to genome- and transcriptome-wide levels. Since ChOP-seq and ChIRP-seq do not uncover the interaction mechanisms of lncRNA and DNA regions, we cannot be absolutely sure that all experimentally detected RNA-DNA interacting regions indeed form triplexes. To overcome this limitation, a new experimental approach that uncovers only interactions via triple helices has been proposed [33]. Yet, this approach is capable only of detecting a pool of DNA-interacting RNAs and vice versa, a pool of RNA-interacting DNA fragments but not the DNA-RNA interacting pairs. To the best of our knowledge, an experimental methods of detecting DNA-RNA triple helices genome- and transcriptome-wide are yet to be developed.
The second limitation of our approach is that Triplexator takes into account only Hoogsteen pairing rules summing up the number of nucleotides that are involved into triplex formation. Yet, other factors could significantly affect triplex fprmation (for example, the G-C nucleotides form stronger triplexes as compared to A-T nucleotides [34]), and therefore, should also be considered in the model.
Third, in this work, we used predicted, rather than experimentally validated RNA secondary structures. Unfortunately, for the majority of lncRNAs for which RNA-DNA binding data is available, experimentally validated structures have not been reported. Moreover, it is known that lncRNA structures are dynamic and adopt multiple conformations; for example, several secondary structures that have been experimentally detected are known for the A-repeat section of lncRNA Xist [35]. Multiple bioinformatic tools for RNA structure prediction have been developed, yet their results differ dramatically and it is not always clear how to choose a predicted structure for a particular RNA. RNAsubopt tool [36] calculates suboptimal secondary RNA structures that might help to find regions that are functional and therefore unpaired in all suboptimal structures. Alternatively, only one suboptimal structure may form a triplex. At this stage, further research is needed to determine a strategy for incorporation of suboptimal structures into the model. Considering dynamic folding and unfolding of different RNA structures (using, for example, thermodynamic constraints on structure folding) might also improve the quality of triplex predictions. Yet, to the best of our knowledge, tools for prediction of RNA structure dynamics have not been developed. Increasing the accuracy of RNA structure prediction, consideration of multiple conformations and RNA structure dynamic could potentially benefit the triplex prediction model.
Fourth, experimental data on RNA-DNA binding is currently available for only a few lncRNAs. Additional information on genome-wide lncRNA binding will provide the possibility to re-evaluate our model and to improve RNA-DNA triplex prediction accuracy.
Although the methods for experimental detection of genome and transcriptome-wide RNA-DNA interactions are being developed (GRID-seq [37], MARGI [38], RADICL-seq [39]), they have limited capacity of detection interactions for low expressed RNAs. Due to the finite sequencing depth mostly contacts between DNA and nascent RNA as well as contacts for RNAs with high expression are detected [24]. High quality computational predictions of RNA-DNA interactions may improve the detection of interactions of low-expressed RNAs. Genome- and transcriptome-wide prediction of high confidence triple helices could help selecting RNA-DNA pairs for small-scale experimental validation with electrophoretic mobility shift assay (EMSA), immunostaining with anti-triplex antibody and other methods. Being experimentally validated particular RNA-DNA interactions may contribute to understanding the mechanisms of human diseases, since, for example, MEG3 and HOTAIR are associated with various cancers (gastric cancer [40], hepatocellular carcinoma [41], cervical cancer [42], etc.).
In this work we focused on the triplexes formed by lncRNA and DNA but they are not the only molecules that can form triplexes. There are several reported cases of triplex formation between mRNA and miRNA (for example, E2F1 mRNA with miR-205–5p and miR-342–3p [43]). miRNAs are also capable to form triplexes with DNA [44], as well as DNA can form triplexes with itself [45]. As Triplexator predicts forming Hoogsteen or reverse Hoogsteen nucleotides interactions we do not see any reason for using it only for lncRNA and DNA. The only constraint is that nucleotide sequence must be at least 10 nt in length.
Summing up, we believe that relaxed parameters for triplex prediction as well as usage of only single-stranded RNA regions and exclusion of regions with an extremely high probability of pairing may improve prediction quality.
4. Materials and Methods
4.1. lncRNA Selection and Experimental Data on lncRNA Binding
For this analysis, we used four lncRNAs—MEG3 (ENST00000451743.6), TERC (ENST00000363312.1), DACOR1 (TCONS_00023265) and HOTAIR (ENST00000424518.5) - for which triplex formation was experimentally validated as well as data of genome-wide binding was available. Triplexes formation was validated for MEG3 in vitro with electrophoretic mobility shift assay (EMSA), circular dichroism (CD) spectroscopy, cell transfection with biotin-labelled MEG3 triplex-forming oligos (TFO) and in vivo with immunostaining with anti-triplex antibody [13]. HOTAIR triplexes formation was validated via electrophoretic mobility shift assay (EMSA) [46]. TERC ability to form triplexes in vitro was observed via EMSA and melting temperature [47]. HOTAIR, TERC and DACOR1 lncRNAs were also used for evaluating deep learning mode for triplexes prediction [31].
For all lncRNAs, the binding regions (ChIRP-seq and ChOP-seq peaks) were obtained from a corresponding paper (Table 2) and converted to hg38 using liftOver if needed. We used detected peaks for each lncRNA as true positives and random sequences of the same median length from hg38 obtained by bedtools as true negatives.
4.2. LncRNA Secondary Structure Prediction
For each lncRNA, we predicted a secondary structure reflecting nucleotides state as being paired or unpaired using Raccess [50] and RNAplfold [51]. Raccess provides energy of pairing (kcal/mol) for a particular nucleotide. The probability of pairing can be calculated using the formula , where . RNAplfold provides a probability of pairing for each nucleotide pair. A nucleotide is considered as paired if it is paired with at least one other nucleotide with a probability above a given threshold. For further predictions paired nucleotides were masked as “N” in the sequence of the particular lncRNA to simulate their inability to bind DNA.
4.3. Triple Helix Prediction
We used Triplexator [29] and TDF [28] command-line tools for RNA:DNA triplex prediction. LncRNA and DNA sequences in FASTA format were provided as input. We fitted several triplex parameters such as: minimum triplex length, maximum error-rate, G-content. We do not disregard repeat and low-complex regions as suggested by default settings of Triplexator. For Triplexator predictions, we used a tpot as a measure of triplex prediction quality. It reflects a number of predicted triplexes for each RNA:DNA pair normalized by DNA and lncRNA sequence length. For each lncRNA, TDF provides DNA binding Domains (DBDs), exact regions that form triplexes, their p-value, and z-score, calculated based on 10,000 random samplings from the human genome.
4.4. ROC-Curves and AUC
To estimate the quality of predictions we used ROC-curves and AUC measures. We ranked ChIRP-seq and ChOP-seq peaks as well as random background sequences based on tpot considering peaks with no triplexes predicted, constructed ROC-curves and calculated AUC using ROCR package [52]. We also investigated the sequences with the highest normalized frequencies of predicted triplexes (highest tpot) using partial AUC for 10% of FPR.
5. Conclusions
To conclude, triplexes prediction quality dramatically depends on triplexes parameters. To achieve the highest accuracy, we suggest to use two sets of Triplexator parameters: minimum length of potential triplexes of 10 nt, maximum error-rate of 20% and minimum G-content of 40% or 70%. Additionally, we recommend to mask RNA regions with the highest probability of pairing (0.95–0.97) in a lncRNA that has a few potential TFOs. Yet, if the number of TFO is high, as low as 0.5 probability of pairing may be beneficial.
Supplementary Materials
Supplementary materials can be found at https://0-www-mdpi-com.brum.beds.ac.uk/1422-0067/21/3/830/s1.
Author Contributions
E.M., Y.A.M., and I.A. formulated and evaluated the ideas. E.M. conducted the research and wrote the original draft. I.A. reviewed and edited the draft. Y.A.M. supervised the research, reviewed and edited the original draft. All authors have read and agreed to the published version of the manuscript.
Funding
I.A. and Y.A.M. were funded by RSF grant 18-14-00240.
Conflicts of Interest
Authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genome Res. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, R.; Andersen, P.R.; Valen, E.; Core, L.J.; Bornholdt, J.; Boyd, M.; Jensen, T.H.; Sandelin, A. Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat. Commun. 2014, 5, 5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hon, C.-C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.L.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagarde, J.; Uszczynska-Ratajczak, B.; Santoyo-Lopez, J.; Gonzalez, J.M.; Tapanari, E.; Mudge, J.M.; Steward, C.A.; Wilming, L.; Tanzer, A.; Howald, C.; et al. Extension of human lncRNA transcripts by RACE coupled with long-read high-throughput sequencing (RACE-Seq). Nat. Commun. 2016, 7, 12339. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.M.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Forrest, A.R.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; De Hoon, M.J.L.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar]
- Alam, T.; Medvedeva, Y.A.; Jia, H.; Brown, J.B.; Lipovich, L.; Bajic, V.B. Promoter Analysis Reveals Globally Differential Regulation of Human Long Non-Coding RNA and Protein-Coding Genes. PLoS ONE 2014, 9, e109443. [Google Scholar] [CrossRef] [Green Version]
- Böhmdorfer, G.; Wierzbicki, A.T. Control of Chromatin Structure by Long Noncoding RNA. Trends Cell Boil. 2015, 25, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Jandura, A.; Krause, H.M. The New RNA World: Growing Evidence for Long Noncoding RNA Functionality. Trends Genet. 2017, 33, 665–676. [Google Scholar] [CrossRef]
- Qian, X.; Zhao, J.; Yeung, P.Y.; Zhang, Q.C.; Kwok, C.K. Revealing lncRNA Structures and Interactions by Sequencing-Based Approaches. Trends Biochem. Sci. 2019, 44, 33–52. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Morales, D.R.; Thomas, K.; Presser, A.; Bernstein, B.E.; Van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [Green Version]
- Grote, P.; Herrmann, B.G. The long non-coding RNA Fendrr links epigenetic control mechanisms to gene regulatory networks in mammalian embryogenesis. RNA Boil. 2013, 10, 1579–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA–DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.-Y.; Bogu, G.K.; Soh, B.S.; Stanton, L.W. The Long Noncoding RNA RMST Interacts with SOX2 to Regulate Neurogenesis. Mol. Cell 2013, 51, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, J.A.; Westhof, E. The Dynamic Landscapes of RNA Architecture. Cell 2009, 136, 604–609. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chédin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Meredith, E.K.; Balas, M.M.; Sindy, K.; Haislop, K.; Johnson, A.M. An RNA matchmaker protein regulates the activity of the long noncoding RNA HOTAIR. RNA 2016, 22, 995–1010. [Google Scholar] [CrossRef] [Green Version]
- Postepska-Igielska, A.; Giwojna, A.; Gasri-Plotnitsky, L.; Schmitt, N.; Dold, A.; Ginsberg, D.; Grummt, I. LncRNA Khps1 Regulates Expression of the Proto-oncogene SPHK1 via Triplex-Mediated Changes in Chromatin Structure. Mol. Cell 2015, 60, 626–636. [Google Scholar] [CrossRef]
- O’Leary, V.B.; Ovsepian, S.V.; Carrascosa, L.G.; Buske, F.A.; Radulović, V.; Niyazi, M.; Moertl, S.; Trau, M.; Atkinson, M.J.; Anastasov, N. PARTICLE, a Triplex-Forming Long ncRNA, Regulates Locus-Specific Methylation in Response to Low-Dose Irradiation. Cell Rep. 2015, 11, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Syed, J.; Sugiyama, H. RNA-DNA Triplex Formation by Long Noncoding RNAs. Cell Chem. Boil. 2016, 23, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Angrand, P.-O.; Vennin, C.; Le Bourhis, X.; Adriaenssens, E. The role of long non-coding RNAs in genome formatting and expression. Front. Genet. 2015, 6, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Lu, X. Decreased expression of MEG3 contributes to retinoblastoma progression and affects retinoblastoma cell growth by regulating the activity of Wnt/β-catenin pathway. Tumor Boil. 2015, 37, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Gordon, F.E.; Nutt, C.L.; Cheunsuchon, P.; Nakayama, Y.; Provencher, K.A.; Rice, K.A.; Zhou, Y.; Zhang, X.; Klibanski, A. Increased expression of angiogenic genes in the brains of mouse meg3-null embryos. Endocrinology 2010, 151, 2443–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonov, I.V.; Mazurov, E.; Borodovsky, M.; A Medvedeva, Y. Prediction of lncRNAs and their interactions with nucleic acids: Benchmarking bioinformatics tools. Briefings Bioinform. 2018, 20, 551–564. [Google Scholar] [CrossRef]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar] [CrossRef]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Boil. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Sherpa, C.; Rausch, J.W.; Le Grice, S.F.J. Structural characterization of maternally expressed gene 3 RNA reveals conserved motifs and potential sites of interaction with polycomb repressive complex 2. Nucleic Acids Res. 2018, 46, 10432–10447. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.-C.; Hänzelmann, S.; Cetin, N.S.; Frank, S.; Zajzon, B.; Derks, J.-P.; Akhade, V.S.; Ahuja, G.; Kanduri, C.; Grummt, I.; et al. Detection of RNA–DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 2019, 47, e32. [Google Scholar] [CrossRef] [Green Version]
- Buske, F.A.; Bauer, D.C.; Mattick, J.S.; Bailey, T.L. Triplexator: Detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012, 22, 1372–1381. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Zhang, H.; Liu, H.; Zhu, H. LongTarget: A tool to predict lncRNA DNA-binding motifs and binding sites via Hoogsteen base-pairing analysis. Bioinformatics 2015, 31, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Chainani, P.; White, T.; Yang, J.; Liu, Y.; Soibam, B. Deep learning identifies genome-wide DNA binding sites of long noncoding RNAs. RNA Boil. 2018, 15, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Antonov, I.; Medvedeva, Y.A. Purine-rich low complexity regions are potential RNA binding hubs in the human genome. F1000Research 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Sentürk, C.N.; Cetin, N.S.; Kuo, C.C.; Ribarska, T.; Li, R.; Costa, I.G. Isolation and genome-wide characterization of cellular DNA:RNA triplex structures. Nucleic Acids Res. 2019, 47, 2306–2321. [Google Scholar] [CrossRef]
- Kunkler, C.N.; Hulewicz, J.P.; Hickman, S.C.; Wang, M.C.; McCown, P.J.; A Brown, J. Stability of an RNA•DNA-DNA triple helix depends on base triplet composition and length of the RNA third strand. Nucleic Acids Res. 2019, 47, 7213–7222. [Google Scholar] [CrossRef] [PubMed]
- Maenner, S.; Blaud, M.; Fouillen, L.; Savoye, A.; Marchand, V.; Dubois, A.; Sanglier-Cianférani, S.; Van Dorsselaer, A.; Clerc, P.; Avner, P.; et al. 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Boil. 2010, 8, e1000276. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Boil. 2011, 6, 26. [Google Scholar] [CrossRef]
- Li, X.; Zhou, B.; Chen, L.; Gou, L.-T.; Li, H.; Fu, X.-D. GRID-seq reveals the global RNA–chromatin interactome. Nat. Biotechnol. 2017, 35, 940–950. [Google Scholar] [CrossRef]
- Sridhar, B.; Rivas-Astroza, M.; Nguyen, T.C.; Chen, W.; Yan, Z.; Cao, X. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Curr. Biol. 2017, 27, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, A.; Agostini, F.; Suzuki, A.M.; Hashimoto, K.; Pascarella, G.; Gimenez, J.; Roos, L.; Nash, A.J.; Ghilotti, M.; Cameron, C.J.; et al. RADICL-seq identifies general and cell type-specific principles of genome-wide RNA-chromatin interactions. BioRxiv 2019, 681924. [Google Scholar]
- Li, T.; Mo, X.; Fu, L.; Xiao, B.; Guo, J. Molecular mechanisms of long noncoding RNAs on gastric cancer. Oncotarget 2016, 7, 8601–8612. [Google Scholar] [CrossRef] [Green Version]
- Abbastabar, M.; Sarfi, M.; Golestani, A.; Khalili, E. lncRNA involvement in hepatocellular carcinoma metastasis and prognosis. EXCLI J. 2018, 17, 900–913. [Google Scholar] [PubMed]
- Peng, L.; Yuan, X.; Jiang, B.; Tang, Z.; Li, G.-C. LncRNAs: Key players and novel insights into cervical cancer. Tumor Boil. 2015, 37, 2779–2788. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Gupta, S.K.; Schmitz, U.; Marquardt, S.; Knoll, S.; Spitschak, A.; Wolkenhauer, O.; Pützer, B.M.; Vera, J. MiR-205-5p and miR-342-3p cooperate in the repression of the E2F1 transcription factor in the context of anticancer chemotherapy resistance. Theranostics 2018, 8, 1106–1120. [Google Scholar] [CrossRef] [PubMed]
- Paugh, S.W.; Coss, D.R.; Bao, J.; Laudermilk, L.T.; Grace, C.R.; Ferreira, A.M. MicroRNAs Form Triplexes with Double Stranded DNA at Sequence-Specific Binding Sites; a Eukaryotic Mechanism via which microRNAs Could Directly Alter Gene Expression. PLoS Comput. Biol. 2016, 12, e1004744. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wang, G.; Vasquez, K.M. New Perspectives on DNA and RNA Triplexes As Effectors of Biological Activity. PLoS Genet. 2015, 11, e1005696. [Google Scholar] [CrossRef] [Green Version]
- Kalwa, M.; Hänzelmann, S.; Otto, S.; Kuo, C.-C.; Franzen, J.; Joussen, S.; Fernandez-Rebollo, E.; Rath, B.; Koch, C.; Hofmann, A.; et al. The lncRNA HOTAIR impacts on mesenchymal stem cells via triple helix formation. Nucleic Acids Res. 2016, 44, 10631–10643. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, Y.; Ge, Y.; Liu, J.; Zhao, Y. TERC promotes cellular inflammatory response independent of telomerase. Nucleic Acids Res. 2019, 47, 8084–8095. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell. 2011, 44, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Merry, C.R.; Forrest, M.E.; Sabers, J.N.; Beard, L.; Gao, X.-H.; Hatzoglou, M.; Jackson, M.W.; Wang, Z.; Markowitz, S.D.; Khalil, A.M. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 2015, 24, 6240–6253. [Google Scholar] [CrossRef]
- Kiryu, H.; Terai, G.; Imamura, O.; Yoneyama, H.; Suzuki, K.; Asai, K. A detailed investigation of accessibilities around target sites of siRNAs and miRNAs. Bioinformatics 2011, 27, 1788–1797. [Google Scholar] [CrossRef]
- Bernhart, S.H.; Hofacker, I.L.; Stadler, P.F. Local RNA base pairing probabilities in large sequences. Bioinformatics 2006, 22, 614–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 2005, 21, 3940–3941. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Triplexes predictions for four lncRNAs (TERC, MEG3, DACOR1, HOTAIR) and their experimentally validated target regions with different Triplexator parameters: minimum triplex length, maximum error rate, and minimum G-content. Prediction quality is measured using the AUC (color scheme). Gray color represents the absence of predicted triplexes.
Figure 1.
Triplexes predictions for four lncRNAs (TERC, MEG3, DACOR1, HOTAIR) and their experimentally validated target regions with different Triplexator parameters: minimum triplex length, maximum error rate, and minimum G-content. Prediction quality is measured using the AUC (color scheme). Gray color represents the absence of predicted triplexes.

Figure 2.
Quality of triplexes predictions with the two sets of parameters. (left panel) The best set of parameters for lncRNAs MEG3 and HOTAIR (l10e20g40: minimum length of 10 bp, maximum error rate of 20%, minimum G-content of 40%, Table 1); (right panel) The best set of parameters for lncRNAs TERC and DACOR1 (l10e20g70: minimum length of 10 bp, maximum error rate of 20%, minimum G-content of 70%, Table 1). Two secondary structure prediction tools were evaluated: RNAplfold and Raccess with a wide range of thresholds. The last row (entire_lncRNA) corresponds to a quality of triplexes prediction with no secondary structure being used. Prediction quality is measured by an AUC (colored scheme). Gray color represents the lack of predicted triplexes.
Figure 2.
Quality of triplexes predictions with the two sets of parameters. (left panel) The best set of parameters for lncRNAs MEG3 and HOTAIR (l10e20g40: minimum length of 10 bp, maximum error rate of 20%, minimum G-content of 40%, Table 1); (right panel) The best set of parameters for lncRNAs TERC and DACOR1 (l10e20g70: minimum length of 10 bp, maximum error rate of 20%, minimum G-content of 70%, Table 1). Two secondary structure prediction tools were evaluated: RNAplfold and Raccess with a wide range of thresholds. The last row (entire_lncRNA) corresponds to a quality of triplexes prediction with no secondary structure being used. Prediction quality is measured by an AUC (colored scheme). Gray color represents the lack of predicted triplexes.

Figure 3.
Triplexes prediction quality with different triplexes parameters: minimum length (nt), maximum error-rate, minimum G-content for the entire lncRNA and three RNAplfold thresholds for selection of single-stranded fragments. Prediction quality is measured by AUC (colored scheme), gray color means no triplexes predicted. MEG3 lncRNA (left panel) DACOR1 lncRNA (right panel).
Figure 3.
Triplexes prediction quality with different triplexes parameters: minimum length (nt), maximum error-rate, minimum G-content for the entire lncRNA and three RNAplfold thresholds for selection of single-stranded fragments. Prediction quality is measured by AUC (colored scheme), gray color means no triplexes predicted. MEG3 lncRNA (left panel) DACOR1 lncRNA (right panel).

Figure 4.
ROC-curves for the best cases of triplexes prediction and corresponding AUC. MEG3ss, and MEG3e stand for single-stranded fragments and the entire MEG3, respectively (l10e20g40, RNAplfold pairing probability 0.5). TERCss and TERCe stand for single-stranded fragments and the entire TERC, respectively (l10e20g70, RNAplfold pairing probability 0.95). DACOR1ss and DACOR1e stand for single-stranded fragments and the entire of DACOR1, respectively (l10e20g70, RNAplfold pairing probability 0.95). HOTAIRss and HOTAIRe stand for single-stranded fragments and the entire HOTAIR, respectively (l10e20g40, RNAplfold pairing probability 0.97).
Figure 4.
ROC-curves for the best cases of triplexes prediction and corresponding AUC. MEG3ss, and MEG3e stand for single-stranded fragments and the entire MEG3, respectively (l10e20g40, RNAplfold pairing probability 0.5). TERCss and TERCe stand for single-stranded fragments and the entire TERC, respectively (l10e20g70, RNAplfold pairing probability 0.95). DACOR1ss and DACOR1e stand for single-stranded fragments and the entire of DACOR1, respectively (l10e20g70, RNAplfold pairing probability 0.95). HOTAIRss and HOTAIRe stand for single-stranded fragments and the entire HOTAIR, respectively (l10e20g40, RNAplfold pairing probability 0.97).

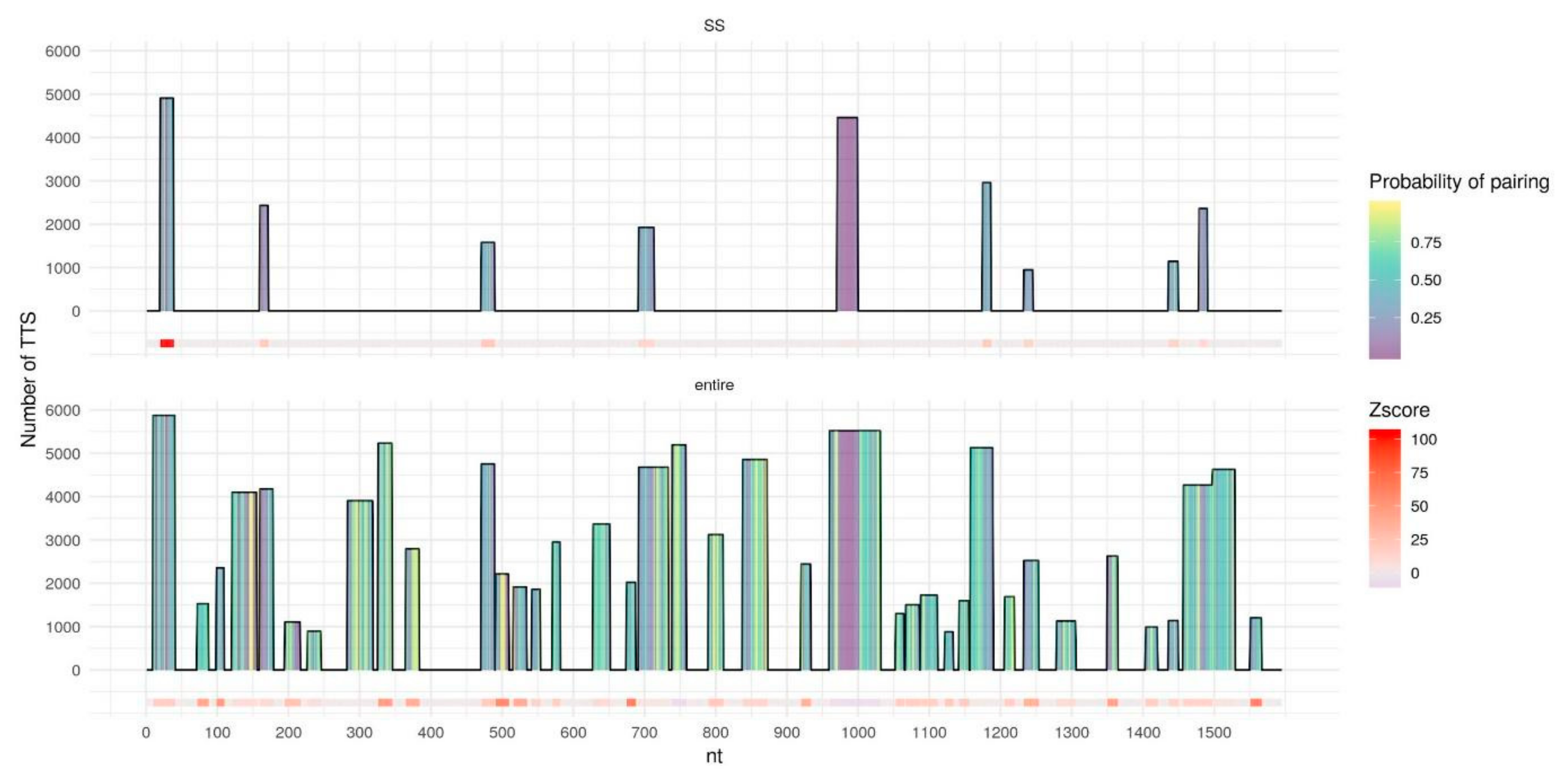
Figure 5.
DNA binding domains (DBDs) of the entire MEG3 (bottom) and of single-stranded fragments of MEG3 (top), predicted by TDF with MEG3 best parameters (l10e20g40). The horizontal axis represents MEG3 in length, columns represent DBDs with height corresponding to the number of DNA peaks (Triplex target sites, TTS) with predicted triplexes for the particular DBD. DBDs columns are filled with color by the probability of pairing for each nucleotide predicted by RNAplfold. DBDs in the MEG3 sequence are colored by z-score for a particular DBD calculated by TDF based on random samplings from the human genome; z-score = 0 corresponds to no DBDs found.
Figure 5.
DNA binding domains (DBDs) of the entire MEG3 (bottom) and of single-stranded fragments of MEG3 (top), predicted by TDF with MEG3 best parameters (l10e20g40). The horizontal axis represents MEG3 in length, columns represent DBDs with height corresponding to the number of DNA peaks (Triplex target sites, TTS) with predicted triplexes for the particular DBD. DBDs columns are filled with color by the probability of pairing for each nucleotide predicted by RNAplfold. DBDs in the MEG3 sequence are colored by z-score for a particular DBD calculated by TDF based on random samplings from the human genome; z-score = 0 corresponds to no DBDs found.

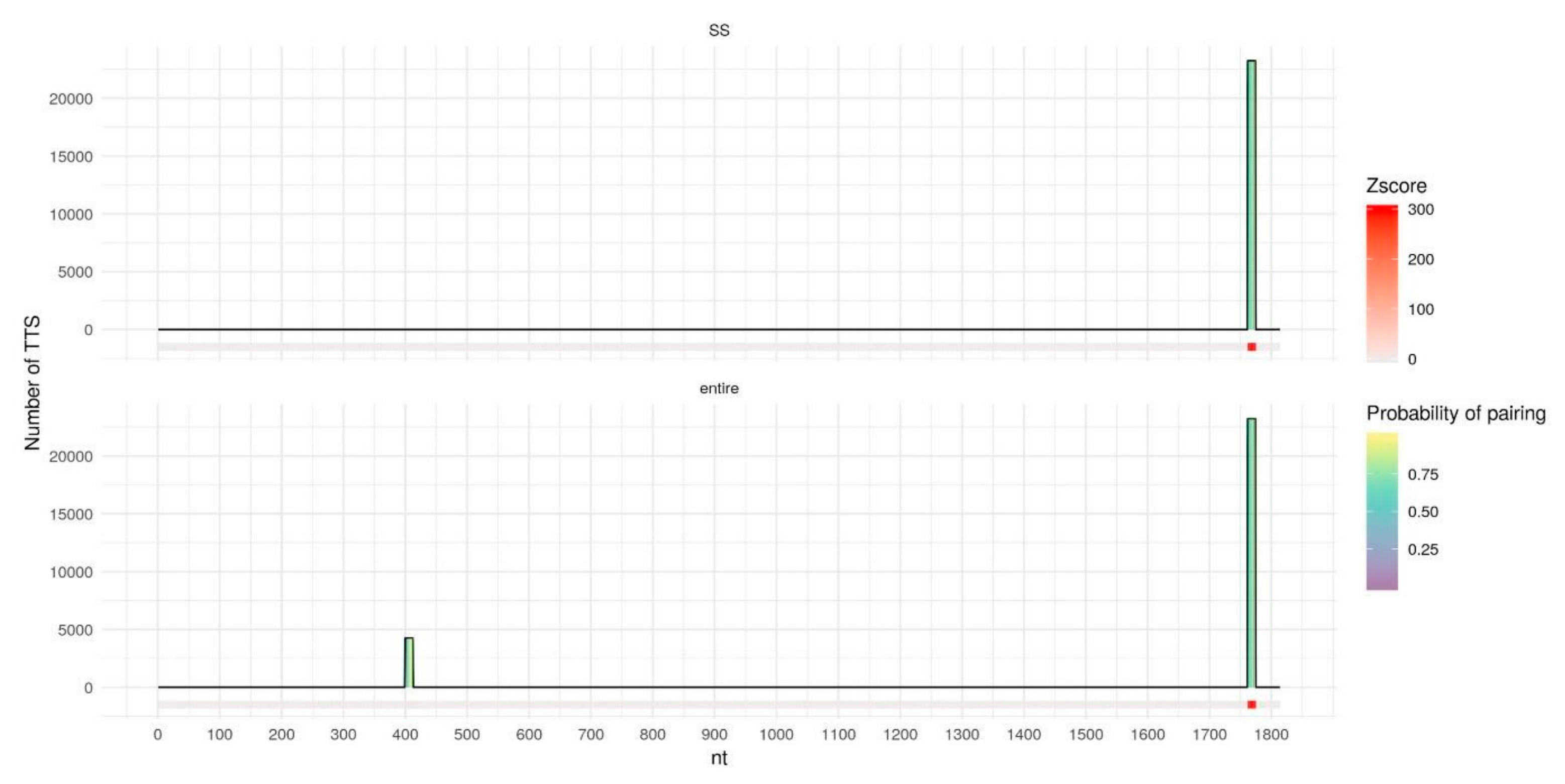
Figure 6.
DNA binding domains (DBDs) of the entire DACOR1 (bottom) and of single-stranded fragments of DACOR1 (top), predicted by TDF with DACOR1 best parameters (l10e20g70). The horizontal axis represents DACOR1 in length, columns represent DBDs with height corresponding to the number of DNA peaks (Triplex target sites, TTS) with predicted triplexes for the particular DBD. DBDs columns are filled with color by the probability of pairing for each nucleotide predicted by RNAplfold. DBDs in the DACOR1 sequence are colored by z-score for a particular DBD calculated by TDF based on 10,000 random samplings from the human genome; z-score = 0 corresponds to no DBDs found.
Figure 6.
DNA binding domains (DBDs) of the entire DACOR1 (bottom) and of single-stranded fragments of DACOR1 (top), predicted by TDF with DACOR1 best parameters (l10e20g70). The horizontal axis represents DACOR1 in length, columns represent DBDs with height corresponding to the number of DNA peaks (Triplex target sites, TTS) with predicted triplexes for the particular DBD. DBDs columns are filled with color by the probability of pairing for each nucleotide predicted by RNAplfold. DBDs in the DACOR1 sequence are colored by z-score for a particular DBD calculated by TDF based on 10,000 random samplings from the human genome; z-score = 0 corresponds to no DBDs found.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Triplexator parameters that generated the best results for each of the four analyzed lncRNAs. Overall, we recommend using a minimum length of 10 bp, a maximum error-rate of 20%, and test a minimum G-content of 40% and 70%.
Table 1.
Triplexator parameters that generated the best results for each of the four analyzed lncRNAs. Overall, we recommend using a minimum length of 10 bp, a maximum error-rate of 20%, and test a minimum G-content of 40% and 70%.
| lncRNA | Min Length (nt) | Max Error-Rate (%) | Min G-Content (%) | AUC | Symbol |
|---|---|---|---|---|---|
| MEG3 | 10 | 20 | 40 | 0.8078 | l10 e20g40 |
| HOTAIR | 10 | 20 | 40 | 0.6372 | l10e20g40 |
| TERC | 10 | 20 | 70 | 0.8601 | l10e20g70 |
| DACOR1 | 10 | 20 | 70 | 0.7423 | l10e20g70 |
Table 2.
Summary statistics and data sources for lncRNAs used in this study.
| lncRNA | lncRNA ID | lncRNA Length | Number of DNA Peaks | Median Peaks Length | Method | Ref |
|---|---|---|---|---|---|---|
| MEG3 | ENST00000451743.6 | 1595 nt | 6798 | 400 nt | ChOP-seq | [13] |
| TERC | ENST00000363312.1 | 451 nt | 2198 | 756 nt | ChIRP-seq | [48] |
| DACOR1 | TCONS_00023265 | 1814 nt | 40213 | 279 nt | ChIRP-seq | [49] |
| HOTAIR | ENST00000424518.5 | 2421 nt | 832 | 678 nt | ChIRP-seq | [48] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Matveishina, E.; Antonov, I.; Medvedeva, Y.A. Practical Guidance in Genome-Wide RNA:DNA Triple Helix Prediction. Int. J. Mol. Sci. 2020, 21, 830. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030830
AMA Style
Matveishina E, Antonov I, Medvedeva YA. Practical Guidance in Genome-Wide RNA:DNA Triple Helix Prediction. International Journal of Molecular Sciences. 2020; 21(3):830. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030830
Chicago/Turabian StyleMatveishina, Elena, Ivan Antonov, and Yulia A. Medvedeva. 2020. "Practical Guidance in Genome-Wide RNA:DNA Triple Helix Prediction" International Journal of Molecular Sciences 21, no. 3: 830. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030830
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.