Comparative Transcriptome Analysis of Different Dendrobium Species Reveals Active Ingredients-Related Genes and Pathways

Abstract
:1. Introduction
2. Results
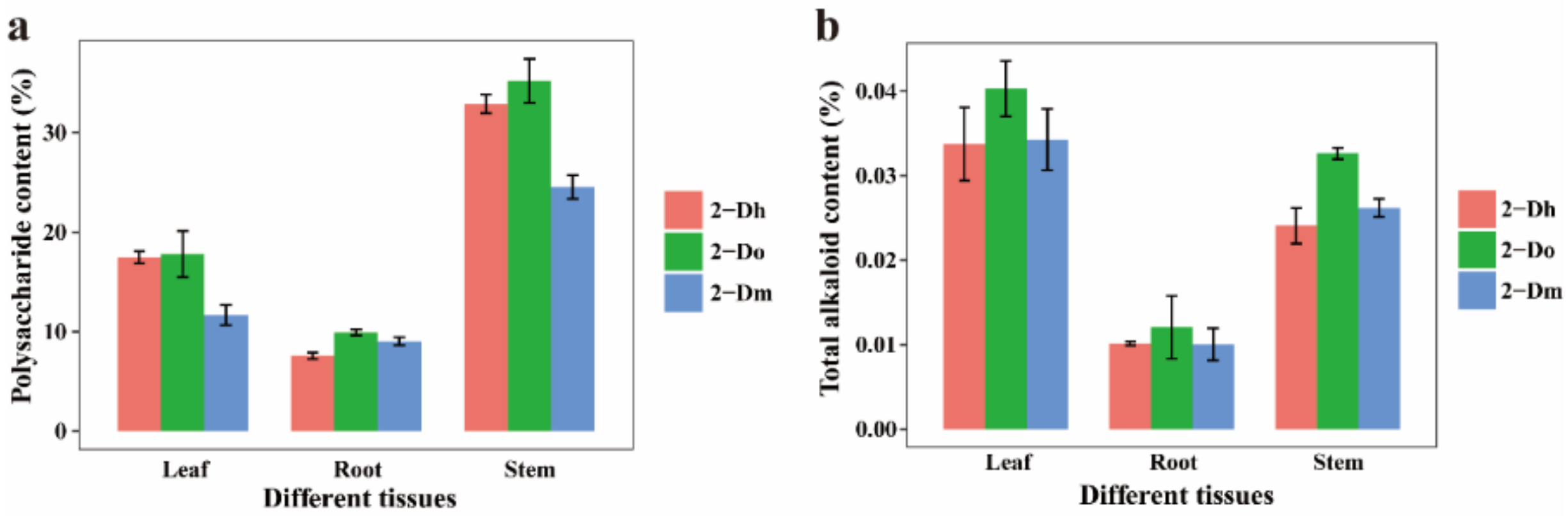
2.1. Determination of Total Alkaloid and Polysaccharide Contents in Different Species and Different Tissues
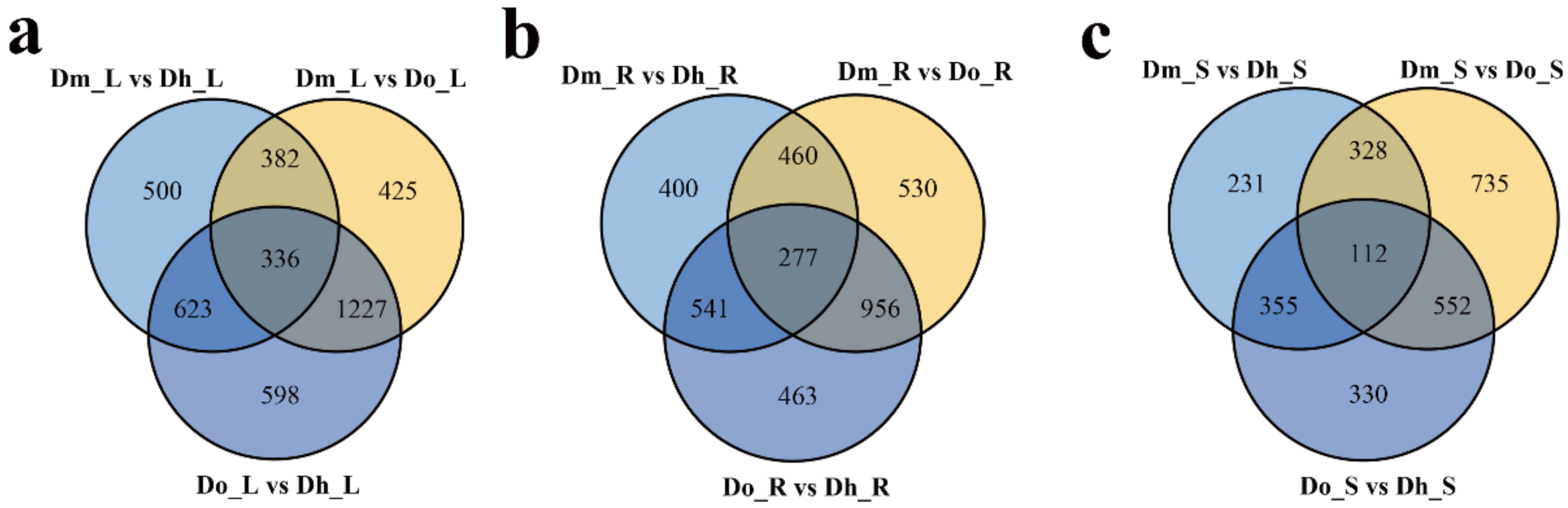
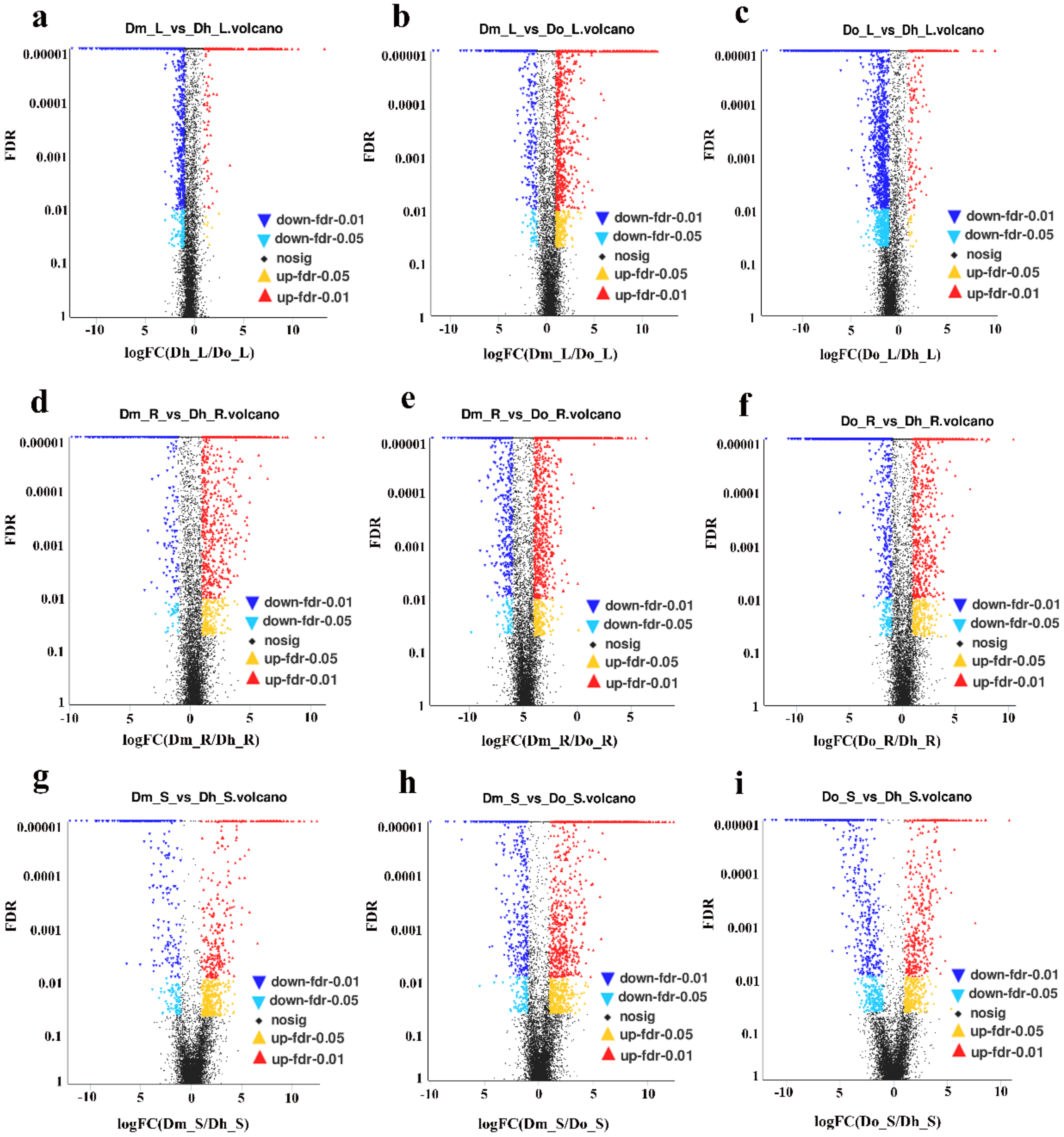
2.2. Differential Expression Analysis
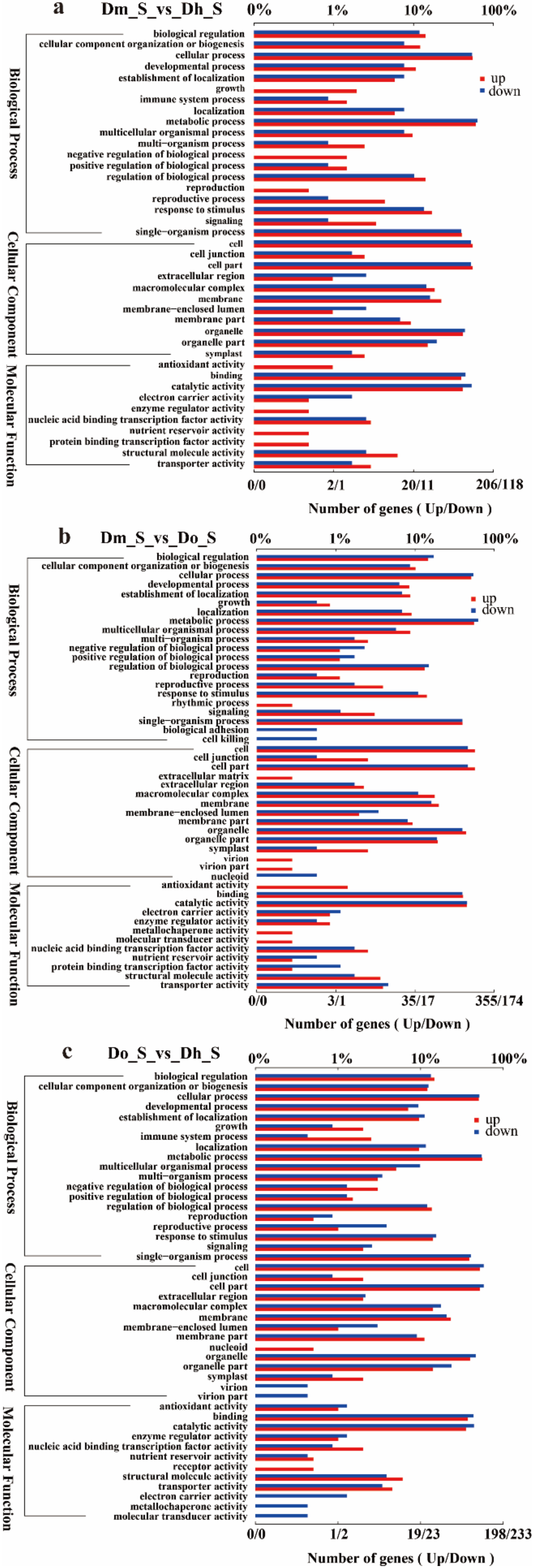
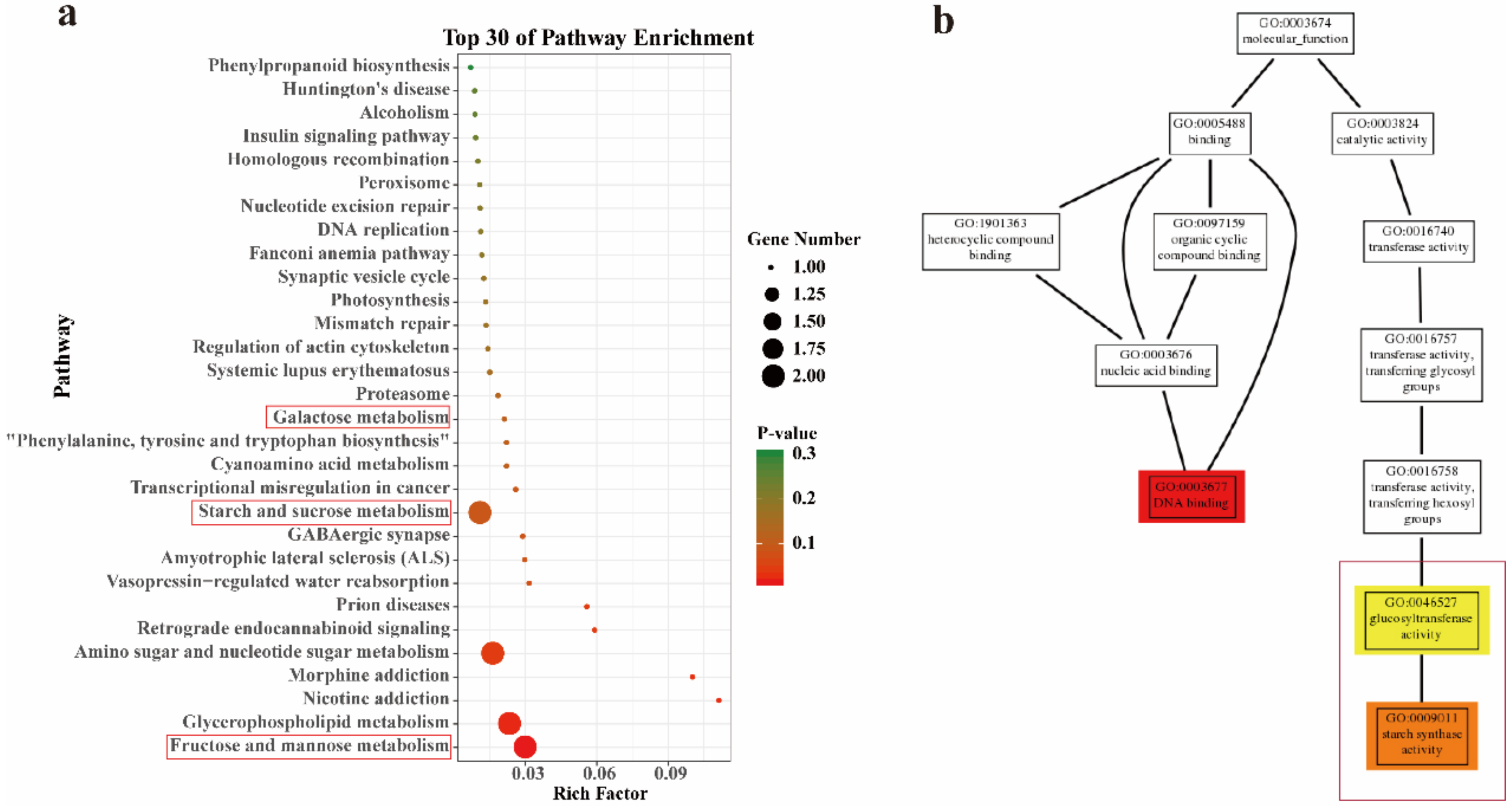
2.3. GO and KEGG of DEGs Annotation and Enrichment Analysis
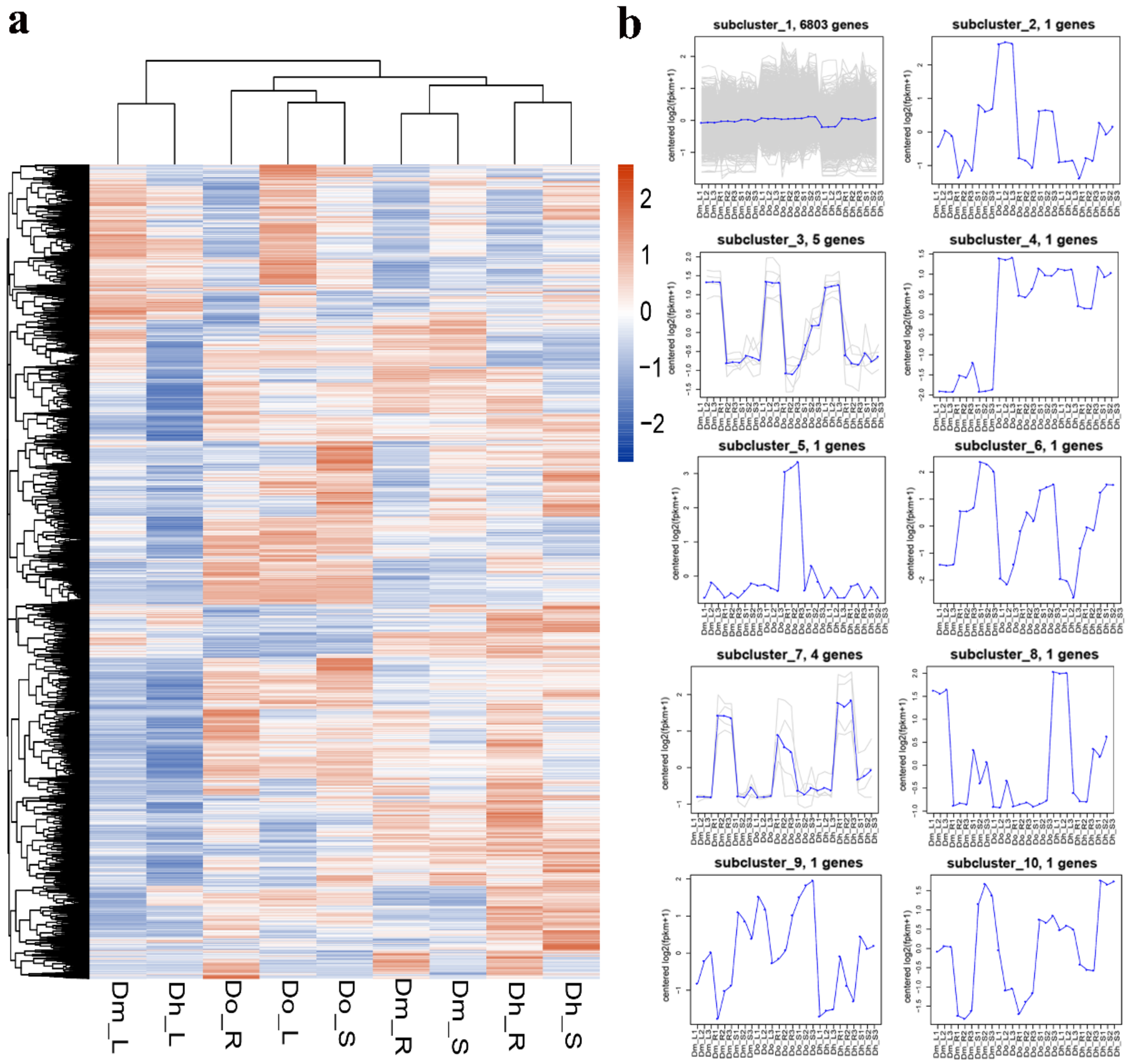
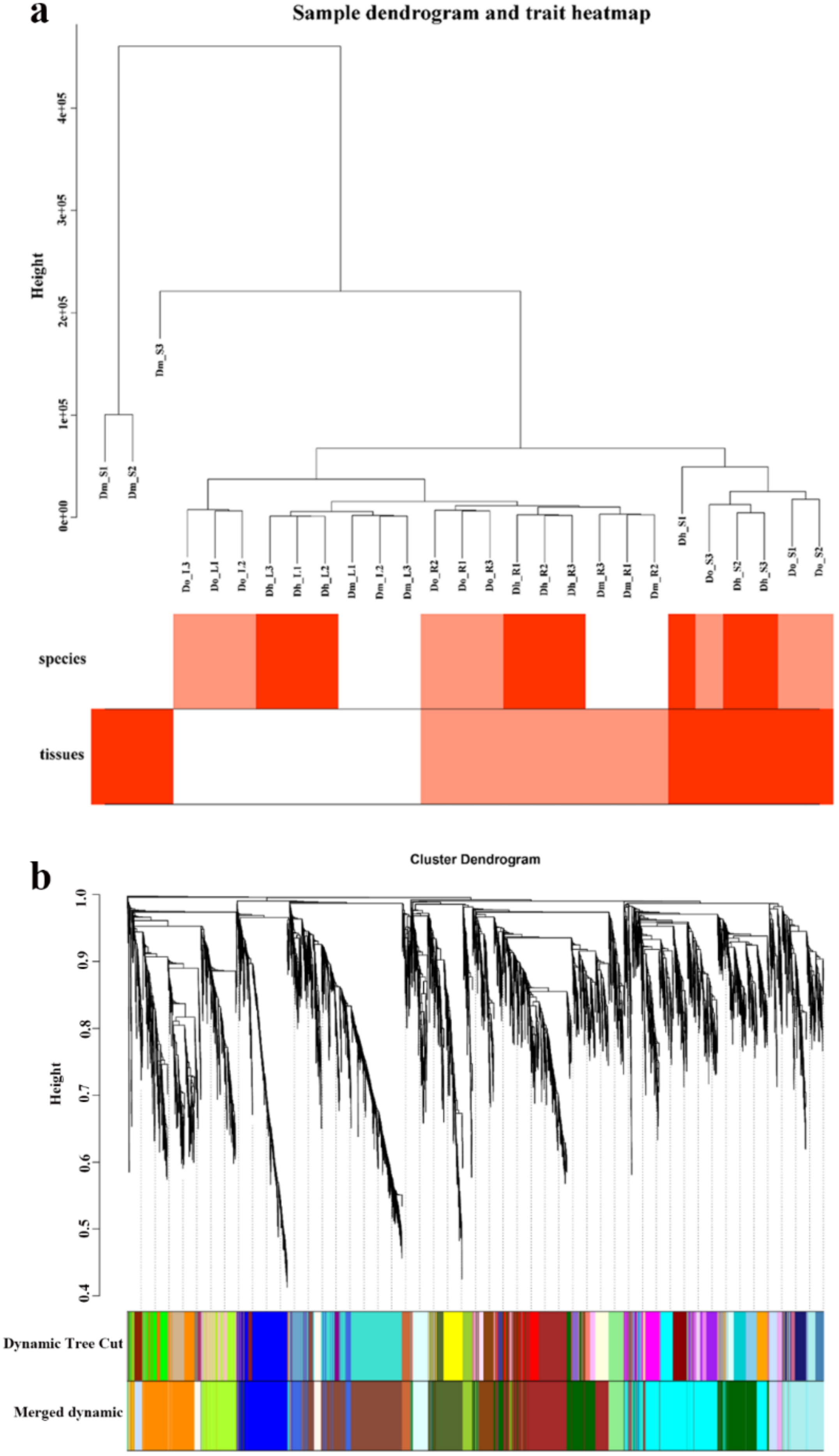
2.4. WGCNA Analysis
2.4.1. Construction of Gene Co-Expression Network of Dendrobium
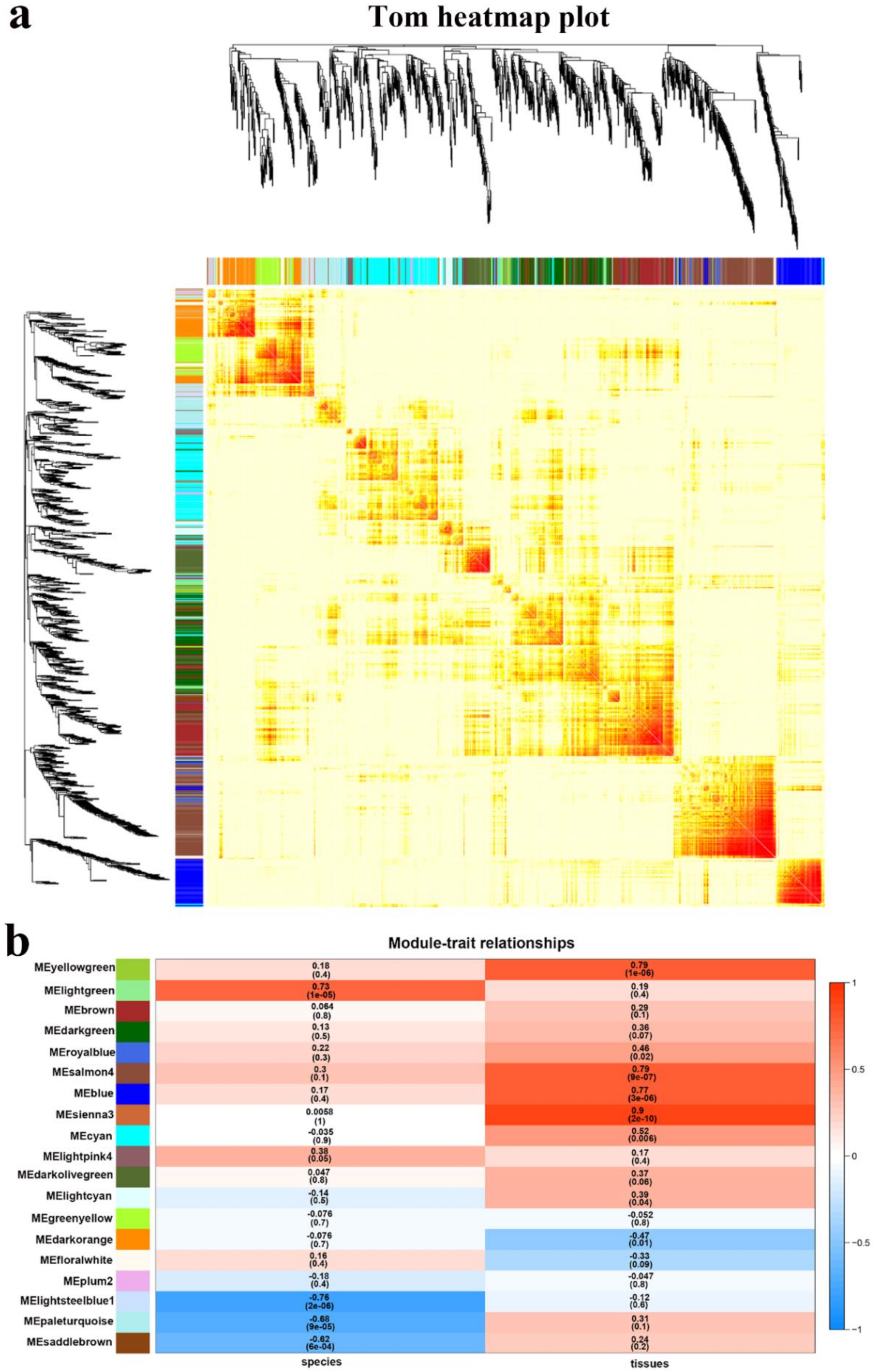
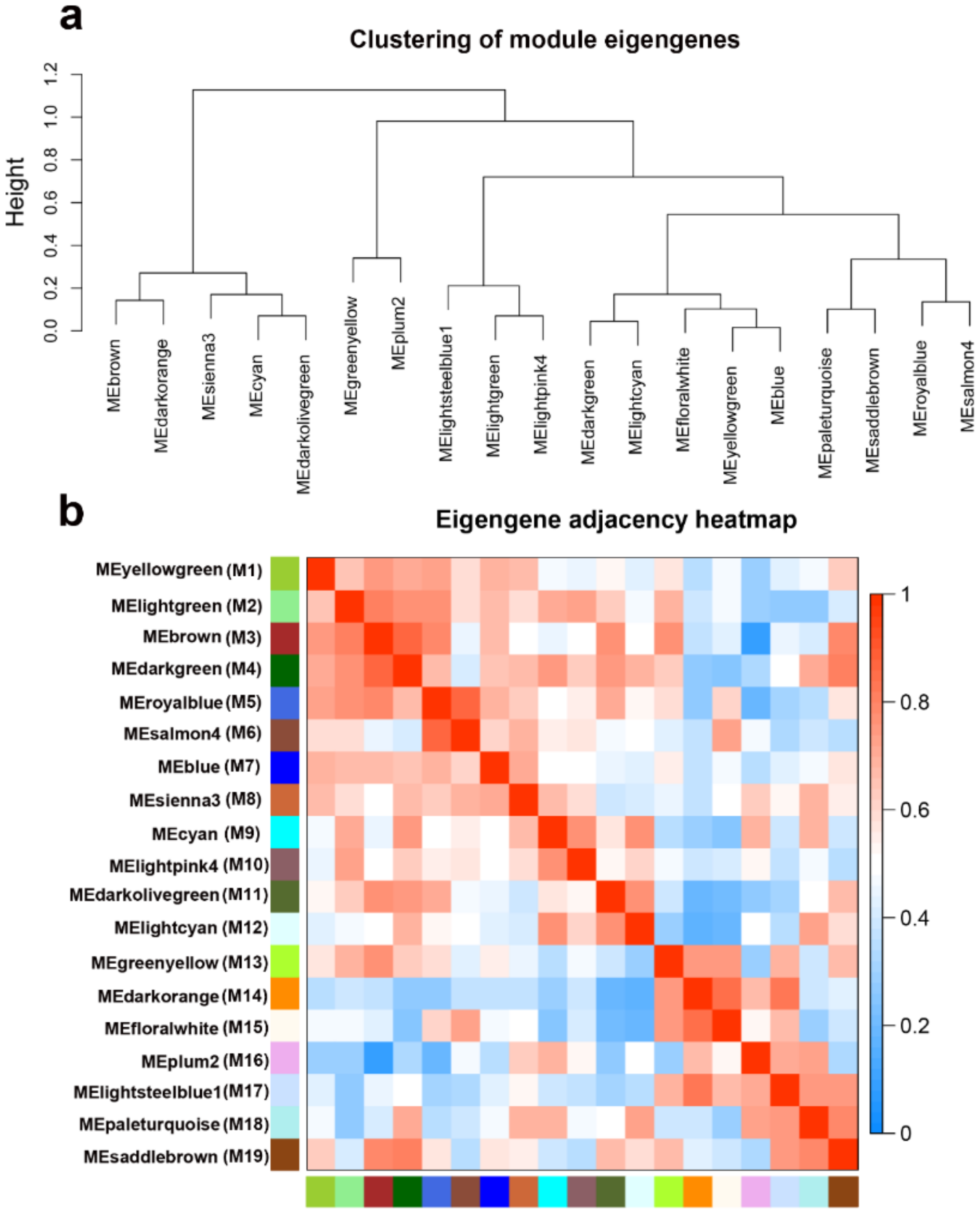
2.4.2. Interaction Analysis of Co-Expression Module
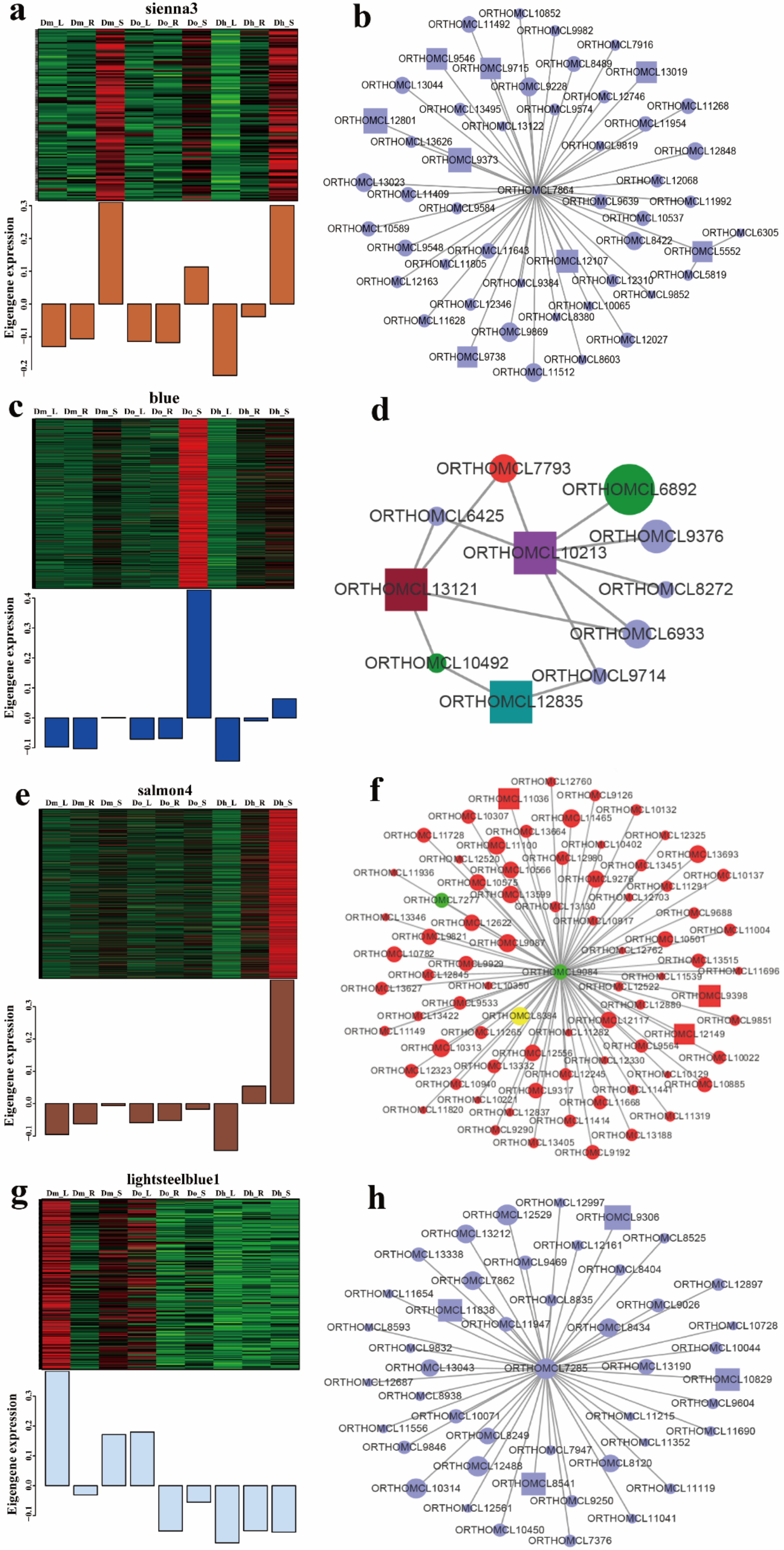
2.4.3. Functional Enrichment Analysis of Genes in Interested Modules
2.4.4. Module Visualize and Hub Genes
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Polysaccharide and Total Alkaloid Contents Determination
4.3. Identification of Orthologous Genes
4.4. Differential Gene Expression Analysis
4.5. WGCNA Analysis
4.5.1. Construction of Weighted Gene Co-Expression Networks and Identification of Modules
4.5.2. Interactions Analysis of Co-Expression Modules
4.5.3. Functional Enrichment Analysis of the Key Module
4.5.4. Identification of Hub Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wood, H.P. The Dendrobiums; ARG Gantner Verlag Timber Press: Portland, OR, USA, 2006. [Google Scholar]
- Zotz, G. The systematic distribution of vascular epiphytes—A critical update. Bot. J. Linn. Soc. 2013, 171, 453–481. [Google Scholar] [CrossRef] [Green Version]
- Lian, W.; Xu, J. The chromosome number in Dendrobium I. Ten species. J. Wuhan Bot. Res. 1989, 7, 112–114. [Google Scholar]
- Zhitao, N.; Shuying, Z.; Jiajia, P.; Ludan, L.; Jing, S.; Xiaoyu, D. Comparative analysis of Dendrobium plastomes and utility of plastomic mutational hotspots. Sci. Rep. 2017, 7, 2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Zhao, T.; Sheng, Y.; Zheng, T.; Fu, L.; Zhang, Y. Dendrobium officinale kimura et migo: A review on its ethnopharmacology, phytochemistry, pharmacology, and industrialization. Evid. Based Complement. Altern. Med. 2017, 2017, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-C.; Ku, A.F.; Tseng, Y.-Y.; Yang, W.-B.; Fang, J.-M.; Wong, C.-H. 6, 8-di-c-glycosyl flavonoids from Dendrobium huoshanense. J. Nat. Prod. 2010, 73, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.B.; Liu, J.; Wong, J.H.; Ye, X.; Sze, S.C.W.; Tong, Y.; Zhang, K.Y. Review of research on Dendrobium, a prized folk medicine. Appl. Microbiol. Biotechnol. 2012, 93, 1795–1803. [Google Scholar] [CrossRef]
- Jin, Q.; Jiao, C.; Sun, S.; Song, C.; Cai, Y.; Lin, Y.; Fan, H.; Zhu, Y. Metabolic analysis of medicinal Dendrobium officinale and Dendrobium huoshanense during different growth years. PLoS ONE 2016, 11, e0146607. [Google Scholar] [CrossRef]
- Hsieh, Y.S.; Chien, C.; Liao, S.K.; Liao, S.F.; Hung, W.T.; Yang, W.B.; Lin, C.C.; Cheng, T.J.; Chang, C.C.; Fang, J.M.; et al. Structure and bioactivity of the polysaccharides in medicinal plant Dendrobium huoshanense. Bioorg. Med. Chem. 2008, 16, 6054–6068. [Google Scholar] [CrossRef]
- Luo, J.P.; Deng, Y.Y.; Zha, X.Q. Mechanism of polysaccharides from Dendrobium huoshanense. On streptozotocin-induced diabetic cataract. Pharm. Biol. 2008, 46, 243–249. [Google Scholar] [CrossRef]
- Kaufmann, K.; Wellmer, F.; Muino, J.M.; Ferrier, T.; Wuest, S.E.; Kumar, V.; Serrano-Mislata, A.; Madueno, F.; Krajewski, P.; Meyerowitz, E.M.; et al. Orchestration of floral initiation by apetala1. Science 2010, 328, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Lou, Q.; Liu, Y.; Qi, Y.; Jiao, S.; Tian, F.; Jiang, L.; Wang, Y. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower colour in grape hyacinth. J. Exp. Bot. 2014, 65, 3157–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, J.C.; Haring, M.A.; Tunen, A.J.V.; Schuurink, R.C. Odorant1 regulates fragrance biosynthesis in petunia flowers. Plant. Cell 2005, 17, 1612–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Yu, M.; Jia, Z.; Song, X.; Liang, Y.; Zhang, J. Analysis of Dendrobium huoshanense transcriptome unveils putative genes associated with active ingredients synthesis. BMC Genom. 2018, 19, 978. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, J.; Kallman, J.; Liu, X.; Meng, M.; Lin, J. Polysaccharide biosynthetic pathway profiling and putative gene mining of Dendrobium moniliforme using RNA-seq in different tissues. BMC Plant. Biol 2019, 19, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Li, Y.; Li, C.; Luo, H.; Wang, L.; Qian, J.; Luo, X.; Xiang, L.; Song, J.; Sun, C.; et al. Analysis of the Dendrobium officinale transcriptome reveals putative alkaloid biosynthetic genes and genetic markers. Gene 2013, 527, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Khaitovich, P.; Weiss, G.; Lachmann, M.; Hellmann, I.; Enard, W.; Muetzel, B.; Wirkner, U.; Ansorge, W.; Pääbo, S. A neutral model of transcriptome evolution. PloS Biol. 2004, 2, e132. [Google Scholar] [CrossRef]
- Barabási, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Carter, S.L.; Brechbuhler, C.M.; Griffin, M.; Bond, A.T. Gene co-expression network topology provides a framework for molecular characterization of cellular state. Bioinformatics 2004, 20, 2242–2250. [Google Scholar] [CrossRef] [Green Version]
- Yook, S.H.; Oltvai, Z.N.; Barabási, A.L. Functional and topological characterization of protein interaction networks. Proteomics 2004, 4, 928–942. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 2007, 1, 54. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Li, C.H.; Huang, J. Systematic identification of cell-wall related genes in populus based on analysis of functional modules in co-expression network. PLoS ONE 2014, 9, e95176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dam, S.; Cordeiro, R.; Craig, T.; van Dam, J.; Wood, S.H.; de Magalhães, J.P. Genefriends: An online co-expression analysis tool to identify novel gene targets for aging and complex diseases. BMC Genom. 2012, 13, 535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tully, J.P.; Hill, A.E.; Ahmed, H.M.; Whitley, R.; Skjellum, A.; Mukhtar, M.S. Expression-based network biology identifies immune-related functional modules involved in plant defense. BMC Genom. 2014, 15, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Gong, Q.; Bohnert, H.J. An Arabidopsis gene network based on the graphical gaussian model. Genome Res. 2007, 17, 1614–1625. [Google Scholar] [CrossRef] [Green Version]
- Oldham, M.C.; Horvath, S.S.; Geschwind, D.H. Conservation and evolution of gene co-expression networks in human and chimpanzee brains. Proc. Natl. Acad. Sci. USA 2006, 103, 17973–17978. [Google Scholar] [CrossRef] [Green Version]
- Takeshi, O.; Shinpei, H.; Masayuki, S.; Motoshi, S.; Hiroyuki, O.; Kengo, K. Coxpresdb: A database of coexpressed gene networks in mammals. Nucleic Acids Res. 2007, 36, D77–D82. [Google Scholar]
- Lee, J.M.; Sonnhammer, E.L. Genomic gene clustering analysis of pathways in eukaryotes. Genome Res. 2003, 13, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Presson, A.P.; Sobel, E.M.; Papp, J.C.; Suarez, C.J.; Whistler, T.; Rajeevan, M.S.; Vernon, S.D.; Horvath, S. Integrated weighted gene co-expression network analysis with an application to chronic fatigue syndrome. BMC Syst. Biol. 2008, 2, 95. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. Wgcna: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Amrine, K.C.; Blanco-Ulate, B.; Cantu, D. Discovery of core biotic stress responsive genes in Arabidopsis by weighted gene co-expression network analysis. PLoS ONE 2015, 10, e0118731. [Google Scholar] [CrossRef] [Green Version]
- Borges, R.M.; Bessiere, J.M.; Ranganathan, Y. Diel variation in fig volatiles across syconium development: Making sense of scents. J. Chem. Ecol. 2013, 39, 630–642. [Google Scholar] [CrossRef]
- El-Sharkawy, I.; Liang, D.; Xu, K. Transcriptome analysis of an apple (Malus × Domestica) yellow fruit somatic mutation identifies a gene network module highly associated with anthocyanin and epigenetic regulation. J. Exp. Bot. 2015, 66, 7359–7376. [Google Scholar] [CrossRef] [Green Version]
- Smita, S.; Katiyar, A.; Pandey, D.M.; Chinnusamy, V.; Bansal, K.C. Identification of conserved drought stress responsive gene-network across tissues and developmental stages in rice. Bioinformation 2013, 9, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Langfelder, P.; Fuller, T.; Dong, J.; Li, A.; Hovarth, S. Weighted gene coexpression network analysis: State of the art. J. Biopharm. Stat. 2010, 20, 281–300. [Google Scholar] [CrossRef] [PubMed]
- Greenham, K.; Guadagno, C.R.; Gehan, M.A.; Mockler, T.C.; Weinig, C.; Ewers, B.E.; McClung, C.R. Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in Brassica rapa. Elife 2017, 6, e29655. [Google Scholar] [CrossRef] [PubMed]
- Hollender, C.A.; Kang, C.; Darwish, O.; Geretz, A.; Matthews, B.F.; Slovin, J.; Alkharouf, N.; Liu, Z. Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant. Physiol. 2014, 165, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Chen, G.; Zhang, H.; Qian, Q.; Ding, Y. Comparative transcript profiling of alloplasmic male-sterile lines revealed altered gene expression related to pollen development in rice (Oryza sativa l.). BMC Plant. Biol. 2016, 16, 175. [Google Scholar] [CrossRef] [Green Version]
- Farcuh, M.; Li, B.; Rivero, R.M.; Shlizerman, L.; Sadka, A.; Blumwald, E. Sugar metabolism reprogramming in a non-climacteric bud mutant of a climacteric plum fruit during development on the tree. J. Exp. Bot. 2017, 68, 5813–5828. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Cheng, S.; Zhou, X.; Chen, Z.; Kim, S.U.; Tan, J.; Zheng, J.; Xu, F.; Zhang, W.; Liao, Y.; et al. A global survey of full-length transcriptome of Ginkgo biloba reveals transcript variants involved in flavonoid biosynthesis. Ind. Crop. Prod. 2019, 139, 11547. [Google Scholar] [CrossRef]
- Oldham, M.C.; Konopka, G.; Iwamoto, K.; Langfelder, P.; Kato, T.; Horvath, S.; Geschwind, D.H. Functional organization of the transcriptome in human brain. Nat. Neurosci. 2008, 11, 1271. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends Plant. Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Saito, M.; Yamada, E.; Fujita, K.; Kakizaki, Y.; Nishihara, M. Isolation and characterization of gtmybp3 and gtmybp4, orthologues of R2R3-MYB transcription factors that regulate early flavonoid biosynthesis, in gentian flowers. J. Exp. Bot. 2012, 63, 6505–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Ning, Y.; Zhang, W.; Liao, Y.; Li, L.; Cheng, H.; Cheng, S. An R2R3-MYB transcription factor as a negative regulator of the flavonoid biosynthesis pathway in ginkgo biloba. Funct. Integr. Genom. 2014, 14, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ma, P.; Yang, D.; Li, W.; Liang, Z.; Liu, Y.; Liu, F. Cloning and characterization of a putative R2R3 MYB transcriptional repressor of the rosmarinic acid biosynthetic pathway from salvia miltiorrhiza. PLoS ONE 2013, 8, e73259. [Google Scholar] [CrossRef] [Green Version]
- Qi, T.; Huang, H.; Wu, D.; Yan, J.; Qi, Y.; Song, S.; Xie, D. Arabidopsis della and jaz proteins bind the WD-repeat/bHLH/MYB complex to modulate gibberellin and jasmonate signaling synergy. Plant. Cell 2014, 26, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- Nakata, M.; Mitsuda, N.; Herde, M.; Koo, A.J.; Moreno, J.E.; Suzuki, K.; Howe, G.A.; Ohme-Takagi, M. A bhlh-type transcription factor, ABA-inducible bhlh-type transcription factor/ja-associated myc2-like1, acts as a repressor to negatively regulate jasmonate signaling in Arabidopsis. Plant. Cell 2013, 25, 1641–1656. [Google Scholar] [CrossRef] [Green Version]
- Fan, M.; Bai, M.-Y.; Kim, J.-G.; Wang, T.; Oh, E.; Chen, L.; Park, C.H.; Son, S.-H.; Kim, S.-K.; Mudgett, M.B. The bHLH transcription factor hbi1 mediates the trade-off between growth and pathogen-associated molecular pattern–triggered immunity in arabidopsis. Plant. Cell 2014, 26, 828–841. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Tai, H.; Li, S.; Gao, W.; Zhao, M.; Xie, C.; Li, W.X. B hlh 122 is important for drought and osmotic stress resistance in Arabidopsis and in the repression of aba catabolism. N. Phytol. 2014, 201, 1192–1204. [Google Scholar] [CrossRef]
- Gao, C.; Ju, Z.; Li, S.; Zuo, J.; Fu, D.; Tian, H.; Luo, Y.; Zhu, B. Deciphering ascorbic acid regulatory pathways in ripening tomato fruit using a weighted gene correlation network analysis approach. J. Integr. Plant. Biol. 2013, 55, 1080–1091. [Google Scholar] [CrossRef]
- Bai, Y.; Dougherty, L.; Cheng, L.; Zhong, G.-Y.; Xu, K. Uncovering co-expression gene network modules regulating fruit acidity in diverse apples. BMC Genom. 2015, 16, 612. [Google Scholar] [CrossRef] [Green Version]
- Bush, L.P.; Wilkinson, H.H.; Schardl, C.L. Bioprotective alkaloids of grass-fungal endophyte symbioses. Plant. Physiol. 1997, 114, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Yu, M.; Zhang, B.; Liu, X.; Zhang, J. Comparative nutritional characteristics of the three major chinese dendrobium species with different growth years. PLoS ONE 2019, 14, e0222666. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. Orthomcl: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Dewey, C.N.; Li, B. Rsem: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar]
- Robinson, M.D.; Mccarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Klopfenstein, D.; Pedersen, B.; Flick, P.; Sato, K.; Ramirez, F.; Yunes, J.; Mungall, C. Goatools: Tools for gene ontology. Zenodo 2015. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG orthology (ko) as a controlled vocabulary. Bioinformatics 2005, 21, 3787. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. David: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. Kobas 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, T.F.; Ghazalpour, A.; Aten, J.E.; Drake, T.A.; Lusis, A.J.; Horvath, S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm. Genome 2007, 18, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Dong, J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module Colors | Node Number |
|---|---|
| Blue | 562 |
| Brown | 892 |
| Cyan | 1132 |
| darkgreen | 1016 |
| darkolivegreen | 362 |
| darkorange | 665 |
| floralwhite | 184 |
| greenyellow | 440 |
| lightcyan | 180 |
| lightgreen | 178 |
| lightpink4 | 40 |
| lightsteelblue1 | 193 |
| paleturquoise | 470 |
| plum2 | 57 |
| royalblue | 275 |
| saddlebrown | 179 |
| salmon4 | 1015 |
| sienna3 | 109 |
| yellowgreen | 107 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Zhang, B.; Tang, X.; Zhang, J.; Lin, J. Comparative Transcriptome Analysis of Different Dendrobium Species Reveals Active Ingredients-Related Genes and Pathways. Int. J. Mol. Sci. 2020, 21, 861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030861
Yuan Y, Zhang B, Tang X, Zhang J, Lin J. Comparative Transcriptome Analysis of Different Dendrobium Species Reveals Active Ingredients-Related Genes and Pathways. International Journal of Molecular Sciences. 2020; 21(3):861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030861
Chicago/Turabian StyleYuan, Yingdan, Bo Zhang, Xinggang Tang, Jinchi Zhang, and Jie Lin. 2020. "Comparative Transcriptome Analysis of Different Dendrobium Species Reveals Active Ingredients-Related Genes and Pathways" International Journal of Molecular Sciences 21, no. 3: 861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030861