Engineering Prostate Cancer from Induced Pluripotent Stem Cells—New Opportunities to Develop Preclinical Tools in Prostate and Prostate Cancer Studies


Abstract
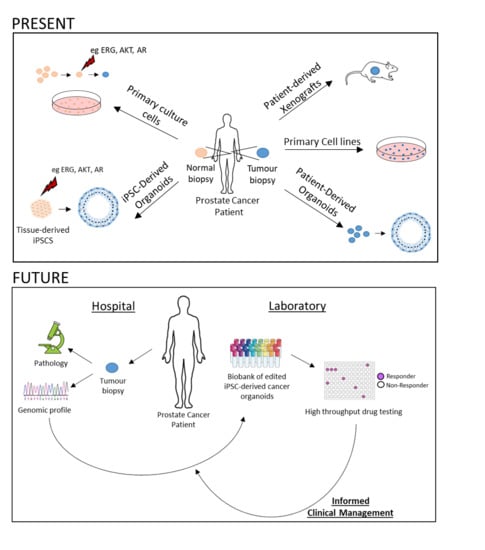
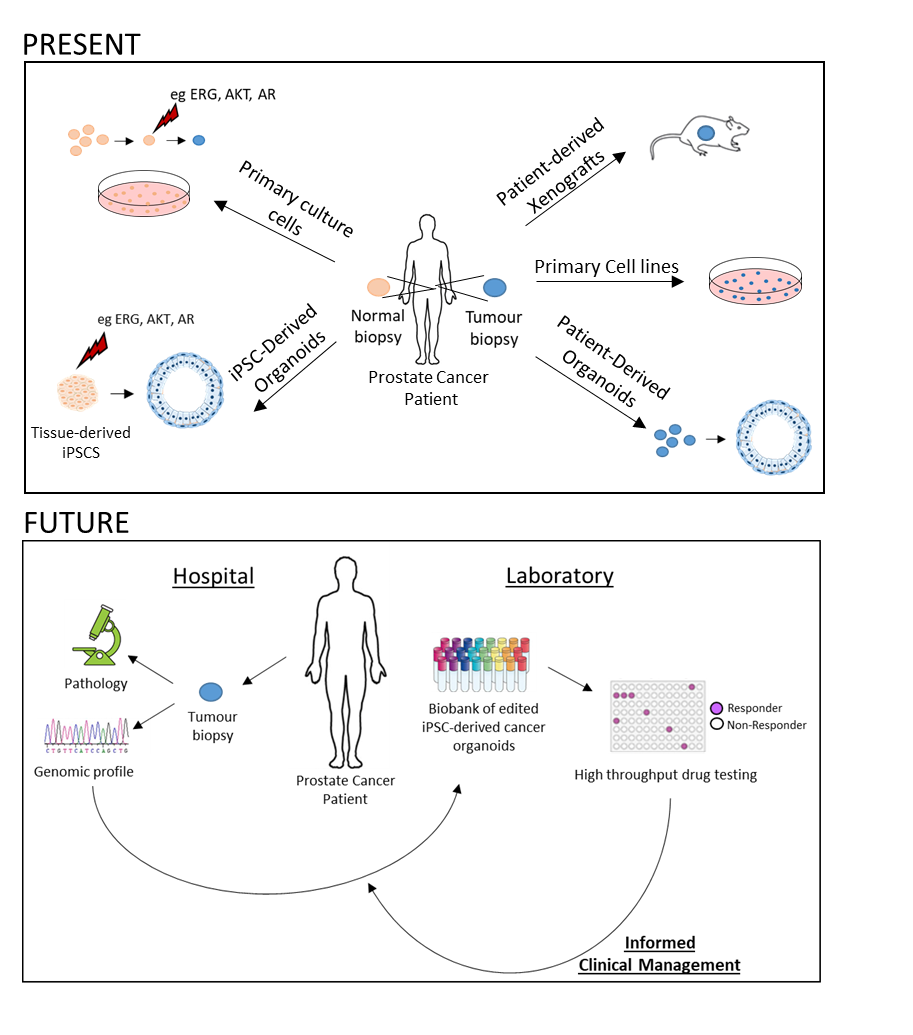
:
1. Introduction
2. Challenges in Current Preclinical Prostate Cancer Models
2.1. Cell Lines
2.2. Primary Culture Cells
2.3. Patient-Derived Xenografts (PDXs)
2.4. Patient-Derived Organoids (PDOs)
3. Human iPSCs for Disease Modelling
3.1. Human iPSC-Derived Tissue and Organ Models
3.1.1. Cerebral iDOs
3.1.2. Retinal iDOs
3.1.3. Intestine iDOs
3.1.4. Liver iDOs
3.1.5. Kidney iDOs
3.1.6. Lung iDOs
4. Emerging Approaches in Preclinical Prostate Cancer Research
4.1. Transformation of Primary Prostate Cells
4.2. Prostate iDOs
4.3. Genome Editing Technology and Precision Medicine
5. Final Remarks
Funding
Conflicts of Interest
Abbreviations
| AR | Androgen receptor |
| CRPC | Castration-resistant prostate cancer |
| CRPC-NE | Castration-resistant prostate cancer neuroendocrine |
| CF | Cystic fibrosis |
| ESC | Embryonic stem cells |
| iPSCs | Induced-pluripotent stem cells |
| iDOs | iPSC-derived organoids |
| PAM | Protospacer-adjacent motif |
| PDOs | Patient-derived organoids |
| PDXs | Patient-derived xenografts |
| PSA | Prostate-specific antigen |
| sgRNA | Single-guide RNA sequence |
| UGM | Urogenital mesenchyme |
References
- Boyd, L.K.; Mao, X.; Lu, Y.J. The complexity of prostate cancer: Genomic alterations and heterogeneity. Nat. Rev. Urol. 2012, 9, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bill-Axelson, A.; Holmberg, L.; Garmo, H.; Rider, J.R.; Taari, K.; Busch, C.; Nordling, S.; Haggman, M.; Andersson, S.O.; Spangberg, A.; et al. Radical prostatectomy or watchful waiting in early prostate cancer. N. Engl. J. Med. 2014, 370, 932–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klotz, L.; Zhang, L.; Lam, A.; Nam, R.; Mamedov, A.; Loblaw, A. Clinical results of long-term follow-up of a large, active surveillance cohort with localized prostate cancer. J. Clin. Oncol. 2010, 28, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Wilt, T.J.; Brawer, M.K.; Jones, K.M.; Barry, M.J.; Aronson, W.J.; Fox, S.; Gingrich, J.R.; Wei, J.T.; Gilhooly, P.; Grob, B.M.; et al. Radical prostatectomy versus observation for localized prostate cancer. N. Engl. J. Med. 2012, 367, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Hamdy, F.C.; Donovan, J.L.; Lane, J.A.; Mason, M.; Metcalfe, C.; Holding, P.; Davis, M.; Peters, T.J.; Turner, E.L.; Martin, R.M.; et al. 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N. Engl. J. Med. 2016, 375, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sawyers, C.L.; Scher, H.I. Targeting the androgen receptor pathway in prostate cancer. Curr. Opin. Pharm. 2008, 8, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J. Urol. 2002, 168, 9–12. [Google Scholar] [CrossRef]
- Mateo, J.; Fizazi, K.; Gillessen, S.; Heidenreich, A.; Perez-Lopez, R.; Oyen, W.J.G.; Shore, N.; Smith, M.; Sweeney, C.; Tombal, B.; et al. Managing nonmetastatic castration-resistant prostate cancer. Eur. Urol. 2019, 75, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on systemic prostate cancer therapies: Management of metastatic castration-resistant prostate cancer in the era of precision oncology. Eur. Urol. 2019, 75, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.W.; Ali, A.; Ingleby, F.C.; Hoyle, A.; Amos, C.L.; Attard, G.; Brawley, C.D.; Calvert, J.; Chowdhury, S.; Cook, A.; et al. Addition of docetaxel to hormonal therapy in low- and high-burden metastatic hormone sensitive prostate cancer: Long-term survival results from the stampede trial. Ann. Oncol. 2019, 30, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Hoyle, A.P.; Ali, A.; James, N.D.; Cook, A.; Parker, C.C.; de Bono, J.S.; Attard, G.; Chowdhury, S.; Cross, W.R.; Dearnaley, D.P.; et al. Abiraterone in “high-” and “low-risk” metastatic hormone-sensitive prostate cancer. Eur. Urol. 2019, 76, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Frame, F.M.; Noble, A.R.; Klein, S.; Walker, H.F.; Suman, R.; Kasprowicz, R.; Mann, V.M.; Simms, M.S.; Maitland, N.J. Tumor heterogeneity and therapy resistance—Implications for future treatments of prostate cancer. J. Cancer Metastasis Treat. 2017, 3, 302–314. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen receptor pathway-independent prostate cancer is sustained through fgf signaling. Cancer Cell 2017, 32, 474–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-institutional prospective study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef]
- Gibas, Z.; Becher, R.; Kawinski, E.; Horoszewicz, J.; Sandberg, A.A. A high-resolution study of chromosome changes in a human prostatic carcinoma cell line (lncap). Cancer Genet. Cytogenet. 1984, 11, 399–404. [Google Scholar] [CrossRef]
- Esquenet, M.; Swinnen, J.V.; Heyns, W.; Verhoeven, G. Lncap prostatic adenocarcinoma cells derived from low and high passage numbers display divergent responses not only to androgens but also to retinoids. J. Steroid Biochem. Mol. Biol. 1997, 62, 391–399. [Google Scholar] [CrossRef]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; Mirand, E.A.; Murphy, G.P. Lncap model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar]
- Kaighn, M.E.; Lechner, J.F.; Narayan, K.S.; Jones, L.W. Prostate carcinoma: Tissue culture cell lines. Natl. Cancer Inst. Monogr. 1978, 49, 17–21. [Google Scholar]
- Klein, K.A.; Reiter, R.E.; Redula, J.; Moradi, H.; Zhu, X.L.; Brothman, A.R.; Lamb, D.J.; Marcelli, M.; Belldegrun, A.; Witte, O.N.; et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient scid mice. Nat. Med. 1997, 3, 402–408. [Google Scholar] [CrossRef]
- Korenchuk, S.; Lehr, J.E.; McLean, L.; Lee, Y.G.; Whitney, S.; Vessella, R.; Lin, D.L.; Pienta, K.J. Vcap, a cell-based model system of human prostate cancer. In Vivo 2001, 15, 163–168. [Google Scholar]
- Mertz, K.D.; Setlur, S.R.; Dhanasekaran, S.M.; Demichelis, F.; Perner, S.; Tomlins, S.; Tchinda, J.; Laxman, B.; Vessella, R.L.; Beroukhimt, R.; et al. Molecular characterization of tmprss2-erg gene fusion in the nci-h660 prostate cancer cell line: A new perspective for an old model. Neoplasia 2007, 9, 200–IN203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navone, N.M.; Olive, M.; Ozen, M.; Davis, R.; Troncoso, P.; Tu, S.M.; Johnston, D.; Pollack, A.; Pathak, S.; von Eschenbach, A.C.; et al. Establishment of two human prostate cancer cell lines derived from a single bone metastasis. Clin. Cancer Res. 1997, 3, 2493–2500. [Google Scholar]
- Sramkoski, R.M.; Pretlow, T.G.; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.-S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A new human prostate carcinoma cell line, 22rv1. In Vitro Cell. Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (du 145). Int. J. Cancer 1978, 21, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.F.; Narayan, K.S.; Ohnuki, Y.; Babcock, M.S.; Jones, L.W.; Kaighn, M.E. Replicative epithelial cell cultures from normal human prostate gland: Brief communication2. J. Natl. Cancer Inst. 1978, 60, 797–801. [Google Scholar] [CrossRef]
- Niranjan, B.; Lawrence, M.G.; Papargiris, M.M.; Richards, M.G.; Hussain, S.; Frydenberg, M.; Pedersen, J.; Taylor, R.A.; Risbridger, G.P. Primary culture and propagation of human prostate epithelial cells. Methods Mol. Biol. 2013, 945, 365–382. [Google Scholar]
- Saifuddin, S.R.; Devlies, W.; Santaolalla, A.; Cahill, F.; George, G.; Enting, D.; Rudman, S.; Cathcart, P.; Challacombe, B.; Dasgupta, P.; et al. King’s health partners’ prostate cancer biobank (khp pcabb). BMC Cancer 2017, 17, 784. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Rhee, J.K.; Choi, H.; Kwon, A.; Kim, J.; Lee, G.D.; Jekarl, D.W.; Lee, S.; Kim, Y.; Kim, T.M. Passage-dependent accumulation of somatic mutations in mesenchymal stromal cells during in vitro culture revealed by whole genome sequencing. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, A.G.; Silva, I.B.B.; Campos-Fernandez, E.; Barcelos, L.S.; Souza, J.B.; Marangoni, K.; Goulart, L.R.; Alonso-Goulart, V. Comparative assay of 2d and 3d cell culture models: Proliferation, gene expression and anticancer drug response. Curr. Pharm. Des. 2018, 24, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Toivanen, R.; Berman, D.M.; Wang, H.; Pedersen, J.; Frydenberg, M.; Meeker, A.K.; Ellem, S.J.; Risbridger, G.P.; Taylor, R.A. Brief report: A bioassay to identify primary human prostate cancer repopulating cells. Stem Cells 2011, 29, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, J.X.; Xue, H.; Lin, D.; Dong, X.; Gout, P.W.; Gao, X.; Pang, J. Subrenal capsule grafting technology in human cancer modeling and translational cancer research. Differentiation 2016, 91, 15–19. [Google Scholar] [CrossRef]
- Lin, D.; Wyatt, A.W.; Xue, H.; Wang, Y.; Dong, X.; Haegert, A.; Wu, R.; Brahmbhatt, S.; Mo, F.; Jong, L.; et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014, 74, 1272–1283. [Google Scholar] [CrossRef] [Green Version]
- Toivanen, R.; Taylor, R.A.; Pook, D.W.; Ellem, S.J.; Risbridger, G.P. Breaking through a roadblock in prostate cancer research: An update on human model systems. J. Steroid Biochem. Mol. Biol. 2012, 131, 122–131. [Google Scholar] [CrossRef]
- Lin, D.; Bayani, J.; Wang, Y.; Sadar, M.D.; Yoshimoto, M.; Gout, P.W.; Squire, J.A.; Wang, Y. Development of metastatic and non-metastatic tumor lines from a patient’s prostate cancer specimen-identification of a small subpopulation with metastatic potential in the primary tumor. Prostate 2010, 70, 1636–1644. [Google Scholar] [CrossRef]
- Risbridger, G.P.; Toivanen, R.; Taylor, R.A. Preclinical models of prostate cancer: Patient-derived xenografts, organoids, and other explant models. Cold Spring Harb. Perspect. Med. 2018, 8, a030536. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Dong, X.; Wang, K.; Wyatt, A.W.; Crea, F.; Xue, H.; Wang, Y.; Wu, R.; Bell, R.H.; Haegert, A.; et al. Identification of dek as a potential therapeutic target for neuroendocrine prostate cancer. Oncotarget 2015, 6, 1806–1820. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5+ve stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnemann, J.R.; Miura, H.; Meixner, L.K.; Irmler, M.; Kloos, U.J.; Hirschi, B.; Bartsch, H.S.; Sass, S.; Beckers, J.; Theis, F.J.; et al. Quantification of regenerative potential in primary human mammary epithelial cells. Development 2015, 142, 3239–3251. [Google Scholar] [CrossRef] [Green Version]
- Chua, C.W.; Shibata, M.; Lei, M.; Toivanen, R.; Barlow, L.J.; Bergren, S.K.; Badani, K.K.; McKiernan, J.M.; Benson, M.C.; Hibshoosh, H.; et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat. Cell Biol. 2014, 16, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Karthaus, W.R.; Iaquinta, P.J.; Drost, J.; Gracanin, A.; Van Boxtel, R.; Wongvipat, J.; Dowling, C.M.; Gao, D.; Begthel, H.; Sachs, N.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roerink, S.F.; Sasaki, N.; Lee-Six, H.; Young, M.D.; Alexandrov, L.B.; Behjati, S.; Mitchell, T.J.; Grossmann, S.; Lightfoot, H.; Egan, D.A.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef]
- Rubin, E.H.; Gilliland, D.G. Drug development and clinical trials--the path to an approved cancer drug. Nat. Rev. Clin. Oncol. 2012, 9, 215–222. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Aronson, S.J.; Rehm, H.L. Building the foundation for genomics in precision medicine. Nature 2015, 526, 336–342. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor organoids as a pre-clinical cancer model for drug discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 2017. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 2018, 23, 882–897. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of human-induced pluripotent stem cells: From clinical trial in a dish to precision medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef]
- Hepburn, A.; Curry, E.; Moad, M.; Steele, R.; Franco, O.; Wilson, L.; Singh, P.; Crawford, S.; Gaughan, L.; Mills, I. High-throughput propagation of human prostate tissue from induced-pluripotent stem cells. bioRxiv 2019, 637876. [Google Scholar]
- Moad, M.; Pal, D.; Hepburn, A.C.; Williamson, S.C.; Wilson, L.; Lako, M.; Armstrong, L.; Hayward, S.W.; Franco, O.E.; Cates, J.M.; et al. A novel model of urinary tract differentiation, tissue regeneration, and disease: Reprogramming human prostate and bladder cells into induced pluripotent stem cells. Eur. Urol. 2013, 64, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.; Morley, M.; Hawkins, F.; McCauley, K.B.; Jean, J.C.; Heins, H.; Na, C.L.; Weaver, T.E.; Vedaie, M.; Hurley, K.; et al. Differentiation of human pluripotent stem cells into functional lung alveolar epithelial cells. Cell Stem Cell 2017, 21, 472–488. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Lee, J.S.; Leem, J.W.; Huh, Y.J.; Kim, J.Y.; Kim, H.S.; Park, I.H.; Daley, G.Q.; Hwang, D.Y.; Kim, D.W. Robust enhancement of neural differentiation from human es and ips cells regardless of their innate difference in differentiation propensity. Stem Cell Rev. Rep. 2010, 6, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.Q.; Freedman, B.S.; Bonventre, J.V. Directed differentiation of pluripotent stem cells to kidney cells. Semin. Nephrol. 2014, 34, 445–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, S.; Fujita, J.; Fujita, C.; Yamaguchi, M.; Kanaami, S.; Ohno, R.; Sakamoto, K.; Kodama, M.; Kurokawa, J.; Kanazawa, H.; et al. Efficient large-scale 2d culture system for human induced pluripotent stem cells and differentiated cardiomyocytes. Stem Cell Rep. 2017, 9, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Carpentier, A.; Cheng, X.; Block, P.D.; Zhao, Y.; Zhang, Z.; Protzer, U.; Liang, T.J. Human stem cell-derived hepatocytes as a model for hepatitis b virus infection, spreading and virus-host interactions. J. Hepatol. 2017, 66, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.L.; Chang, D.C.; Chang-Lin, S.; Lin, C.H.; Wu, D.T.; Chen, D.T.; Ying, S.Y. Mir-302 reprograms human skin cancer cells into a pluripotent es-cell-like state. RNA 2008, 14, 2115–2124. [Google Scholar] [CrossRef] [Green Version]
- Utikal, J.; Maherali, N.; Kulalert, W.; Hochedlinger, K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J. Cell Sci. 2009, 122, 3502–3510. [Google Scholar] [CrossRef] [Green Version]
- Corominas-Faja, B.; Cufi, S.; Oliveras-Ferraros, C.; Cuyas, E.; Lopez-Bonet, E.; Lupu, R.; Alarcon, T.; Vellon, L.; Iglesias, J.M.; Leis, O.; et al. Nuclear reprogramming of luminal-like breast cancer cells generates sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mtor pathway. Cell Cycle 2013, 12, 3109–3124. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, N.; Ishii, H.; Nagai, K.; Hoshino, H.; Mimori, K.; Tanaka, F.; Nagano, H.; Sekimoto, M.; Doki, Y.; Mori, M. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Carette, J.E.; Pruszak, J.; Varadarajan, M.; Blomen, V.A.; Gokhale, S.; Camargo, F.D.; Wernig, M.; Jaenisch, R.; Brummelkamp, T.R. Generation of ipscs from cultured human malignant cells. Blood 2010, 115, 4039–4042. [Google Scholar] [CrossRef] [Green Version]
- Stricker, S.H.; Feber, A.; Engstrom, P.G.; Caren, H.; Kurian, K.M.; Takashima, Y.; Watts, C.; Way, M.; Dirks, P.; Bertone, P.; et al. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev. 2013, 27, 654–669. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M.; et al. Hif induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.J. Applications of ipscs in cancer research. Biomark Insights 2015, 10, 125–131. [Google Scholar] [CrossRef]
- Ahfeldt, T.; Schinzel, R.T.; Lee, Y.K.; Hendrickson, D.; Kaplan, A.; Lum, D.H.; Camahort, R.; Xia, F.; Shay, J.; Rhee, E.P.; et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat Cell Biol. 2012, 14, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Guntur, A.R.; Nguyen, D.C.; Fakory, S.S.; Doucette, C.C.; Leech, C.; Lotana, H.; Kelley, M.; Kohli, J.; Martino, J.; et al. A renewable source of human beige adipocytes for development of therapies to treat metabolic syndrome. Cell Rep. 2018, 25, 3215–3228. [Google Scholar] [CrossRef] [Green Version]
- Diederichs, S.; Klampfleuthner, F.A.M.; Moradi, B.; Richter, W. Chondral differentiation of induced pluripotent stem cells without progression into the endochondral pathway. Front Cell Dev. Biol. 2019, 7, 270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef] [Green Version]
- Grandi, E.; Pandit, S.V.; Voigt, N.; Workman, A.J.; Dobrev, D.; Jalife, J.; Bers, D.M. Human atrial action potential and ca2+ model: Sinus rhythm and chronic atrial fibrillation. Circ. Res. 2011, 109, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Papapetrou, E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016, 22, 1392–1401. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human escs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes prpf31 retinitis pigmentosa. Nat. Commun. 2018, 9, 4234. [Google Scholar] [CrossRef] [PubMed]
- Saengwimol, D.; Rojanaporn, D.; Chaitankar, V.; Chittavanich, P.; Aroonroch, R.; Boontawon, T.; Thammachote, W.; Jinawath, N.; Hongeng, S.; Kaewkhaw, R. A three-dimensional organoid model recapitulates tumorigenic aspects and drug responses of advanced human retinoblastoma. Sci. Rep. 2018, 8, 15664. [Google Scholar] [CrossRef] [PubMed]
- Aurora, M.; Spence, J.R. Hpsc-derived lung and intestinal organoids as models of human fetal tissue. Dev. Biol. 2016, 420, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, T.; Sekine, K.; Kimura, M.; Yoshizawa, E.; Ayano, S.; Koido, M.; Funayama, S.; Nakanishi, N.; Hisai, T.; Kobayashi, T.; et al. Massive and reproducible production of liver buds entirely from human pluripotent stem cells. Cell Rep. 2017, 21, 2661–2670. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, H.; Deng, P.; Chen, W.; Guo, Y.; Tao, T.; Qin, J. In situ differentiation and generation of functional liver organoids from human ipscs in a 3d perfusable chip system. Lab Chip 2018, 18, 3606–3616. [Google Scholar] [CrossRef]
- Forbes, T.A.; Howden, S.E.; Lawlor, K.; Phipson, B.; Maksimovic, J.; Hale, L.; Wilson, S.; Quinlan, C.; Ho, G.; Holman, K.; et al. Patient-ipsc-derived kidney organoids show functional validation of a ciliopathic renal phenotype and reveal underlying pathogenetic mechanisms. Am. J. Hum. Genet. 2018, 102, 816–831. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.W.; Huang, S.X.; de Carvalho, A.; Ho, S.H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.; Tsai, Y.H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D.; et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Dye, B.R.; Ferrer-Torres, D.; Hill, D.R.; Overeem, A.W.; Shea, L.D.; Spence, J.R. Generation of lung organoids from human pluripotent stem cells in vitro. Nat. Protoc. 2019, 14, 518–540. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Kamiya, D.; Nishiyama, A.; Katayama, T.; Nozaki, S.; Kawasaki, H.; Watanabe, Y.; Mizuseki, K.; Sasai, Y. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat. Neurosci. 2005, 8, 288–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human es cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [Green Version]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef]
- Hansen, D.V.; Lui, J.H.; Parker, P.R.; Kriegstein, A.R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 2010, 464, 554–561. [Google Scholar] [CrossRef]
- Hallam, D.; Hilgen, G.; Dorgau, B.; Zhu, L.; Yu, M.; Bojic, S.; Hewitt, P.; Schmitt, M.; Uteng, M.; Kustermann, S.; et al. Human-induced pluripotent stem cells generate light responsive retinal organoids with variable and nutrient-dependent efficiency. Stem Cells 2018, 36, 1535–1551. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, S.J.; Llonch, S.; Borsch, O.; Ader, M. Transplantation of photoreceptors into the degenerative retina: Current state and future perspectives. Prog. Retin. Eye Res. 2019, 69, 1–37. [Google Scholar] [CrossRef]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Crespo, M.; Vilar, E.; Tsai, S.Y.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 2017, 23, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single lgr5+ liver stem cells induced by wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, S.; Carlos, A.R.; Sundaram, B.; Jeney, V.; Ribeiro, A.; Gozzelino, R.; Bank, C.; Gjini, E.; Braza, F.; Martins, R.; et al. Renal control of disease tolerance to malaria. Proc. Natl. Acad. Sci. USA 2019, 116, 5681–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korogi, Y.; Gotoh, S.; Ikeo, S.; Yamamoto, Y.; Sone, N.; Tamai, K.; Konishi, S.; Nagasaki, T.; Matsumoto, H.; Ito, I.; et al. In vitro disease modeling of hermansky-pudlak syndrome type 2 using human induced pluripotent stem cell-derived alveolar organoids. Stem Cell Rep. 2019, 13, 235. [Google Scholar] [CrossRef]
- Leibel, S.L.; Winquist, A.; Tseu, I.; Wang, J.; Luo, D.; Shojaie, S.; Nathan, N.; Snyder, E.; Post, M. Reversal of surfactant protein b deficiency in patient specific human induced pluripotent stem cell derived lung organoids by gene therapy. Sci. Rep. 2019, 9, 13450. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a cell of origin for human prostate cancer. Science 2010, 329, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Unno, K.; Roh, M.; Yoo, Y.A.; Al-Shraideh, Y.; Wang, L.; Nonn, L.; Abdulkadir, S.A. Modeling african american prostate adenocarcinoma by inducing defined genetic alterations in organoids. Oncotarget 2017, 8, 51264–51276. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.A.; Cowin, P.A.; Cunha, G.R.; Pera, M.; Trounson, A.O.; Pedersen, J.; Risbridger, G.P. Formation of human prostate tissue from embryonic stem cells. Nat. Methods 2006, 3, 179–181. [Google Scholar] [CrossRef]
- Robertson, J.A. Human embryonic stem cell research: Ethical and legal issues. Nat. Rev. Genet. 2001, 2, 74–78. [Google Scholar] [CrossRef]
- Halevy, T.; Urbach, A. Comparing esc and ipsc-based models for human genetic disorders. J. Clin. Med. 2014, 3, 1146–1162. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The master neural transcription factor brn2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderon-Gierszal, E.L.; Prins, G.S. Directed differentiation of human embryonic stem cells into prostate organoids in vitro and its perturbation by low-dose bisphenol a exposure. PLoS ONE 2015, 10, e0133238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moad, M.; Hannezo, E.; Buczacki, S.J.; Wilson, L.; El-Sherif, A.; Sims, D.; Pickard, R.; Wright, N.A.; Williamson, S.C.; Turnbull, D.M.; et al. Multipotent basal stem cells, maintained in localized proximal niches, support directed long-ranging epithelial flows in human prostates. Cell Rep. 2017, 20, 1609–1622. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; Clevers, H. Crispr/cas 9 genome editing and its applications in organoids. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G257–G265. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using crispr/cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of cftr by crispr/cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driehuis, E.; Clevers, H. H. Crispr-induced tmprss2-erg gene fusions in mouse prostate organoids. JSM Biotechnol. Biomed. Eng. 2017, 4, 1076. [Google Scholar]
- Demichelis, F.; Fall, K.; Perner, S.; Andren, O.; Schmidt, F.; Setlur, S.R.; Hoshida, Y.; Mosquera, J.M.; Pawitan, Y.; Lee, C.; et al. Tmprss2:Erg gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 2007, 26, 4596–4599. [Google Scholar] [CrossRef] [Green Version]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of tmprss2 and ets transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the tmprss2-erg gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Model | Advantages | Disadvantages |
|---|---|---|
| Cancer cell lines | Easy and cheap to grow; Useful for basic science; High throughput drug screening | Limited to 2D; Mutation accumulation over time; Limited number available |
| Primary cells | Derived from patients; Initial drug studies; Use for PDXs, PDOs and iPSCs | Difficult to grow; Tissue accessibility; Limited to 2D; Mutation accumulation over time |
| Patient-derived xenografts (PDXs) | Retain 3D tissue architecture; Intact endocrine system; Disease stage-specific models available | Time consuming and expensive; Low engraftment efficiencies; Mouse has deficient immunity and different microenvironment |
| Patient-derived organoids (PDOs) | Retain 3D tissue architecture; Histological and molecular resemblance to tissue of origin; Drug testing responses more accurate | At present only established from aggressive prostate cancer specimens; Low establishment rate; Lack microenvironment and immune influence |
| iPSC-derived organoids (iDOs) | Retain 3D tissue architecture; Unlimited source of iPSCs; Isogenic lines; Gene editing to introduce patient-specific mutations; High throughput drug screening; ‘avatar’ for precision medicine | Lack microenvironment and immune influence |
| Tissue/Organ | Method | Key Small Molecules | References |
|---|---|---|---|
| Brain | Self-organisation by embryoid bodies formation, and the addition of temporal small molecules | IWR1 and SB431542 | [91,92] |
| Eye | Self-organisation by embryoid bodies formation, and the addition of temporal small molecules | BMP4 and IGF1 | [93,94,95] |
| Intestine | Extracellular support matrix and culture medium supplemented with pro-intestine growth factors | Activin A, WNT3A and FGF4 | [96] |
| Liver | Co-culture of iPSCs with mesenchymal and endothelial cells followed by self-organisation by cell-to-cell contact or self-organisation by embryoid bodies formation on 3D perfusable chip | Activin-A, bFGF and HGF | [97,98] |
| Kidney | Mesoderm induction step followed by self-organisation in 3D culture | CHIR99021 and FGF9 | [99] |
| Lung | Endoderm induction, addition of temporal small molecules and culture in extracellular support matrix or transwell inserts | Activin A, Noggin, SB431542, SAG, FGF4, CHIR99021 and FGF10 | [96,100,101,102] |
| Prostate | Endoderm induction step and co-culture of iPSCs with rodent urogenital sinus mesenchyme (UGM), followed by self-organisation by cell-to-cell contact in extracellular support matrix | Activin A, EGF, R-spondin1, Noggin, and A83-01 | [69] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hepburn, A.C.; Sims, C.H.C.; Buskin, A.; Heer, R. Engineering Prostate Cancer from Induced Pluripotent Stem Cells—New Opportunities to Develop Preclinical Tools in Prostate and Prostate Cancer Studies. Int. J. Mol. Sci. 2020, 21, 905. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030905
Hepburn AC, Sims CHC, Buskin A, Heer R. Engineering Prostate Cancer from Induced Pluripotent Stem Cells—New Opportunities to Develop Preclinical Tools in Prostate and Prostate Cancer Studies. International Journal of Molecular Sciences. 2020; 21(3):905. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030905
Chicago/Turabian StyleHepburn, Anastasia C., C. H. Cole Sims, Adriana Buskin, and Rakesh Heer. 2020. "Engineering Prostate Cancer from Induced Pluripotent Stem Cells—New Opportunities to Develop Preclinical Tools in Prostate and Prostate Cancer Studies" International Journal of Molecular Sciences 21, no. 3: 905. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030905