High-Density Lipoprotein (HDL) Inhibits Serum Amyloid A (SAA)-Induced Vascular and Renal Dysfunctions in Apolipoprotein E-Deficient Mice

 , ,
, ,
Abstract
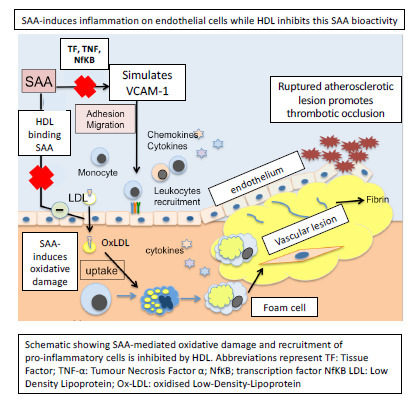
:
1. Introduction
2. Results
2.1. Evidence that HDL Inhibits the Pro-Atherogenic Activity of SAA
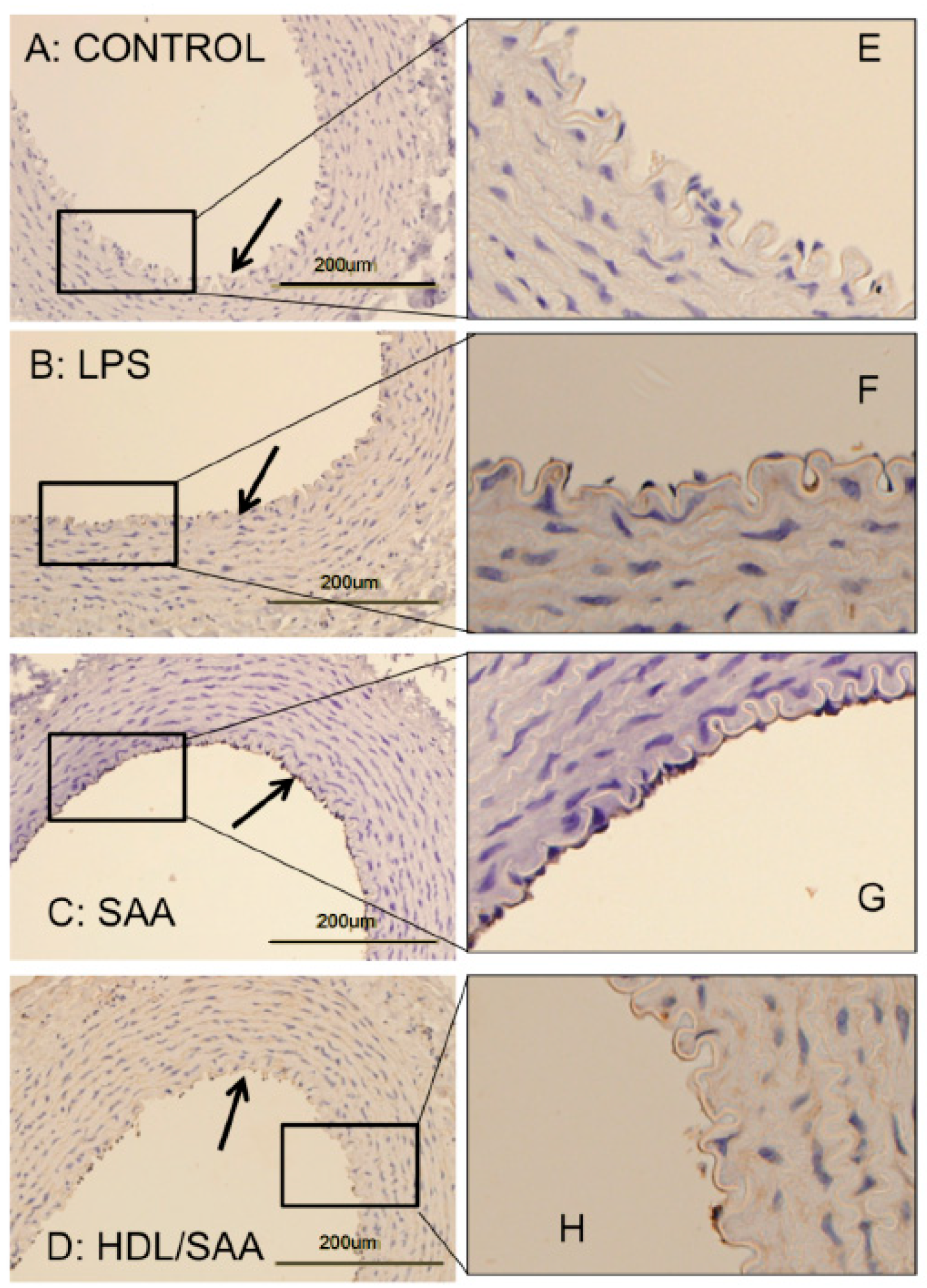
2.1.1. HDL Pretreatment Inhibits SAA-Induced Aortic Lesions
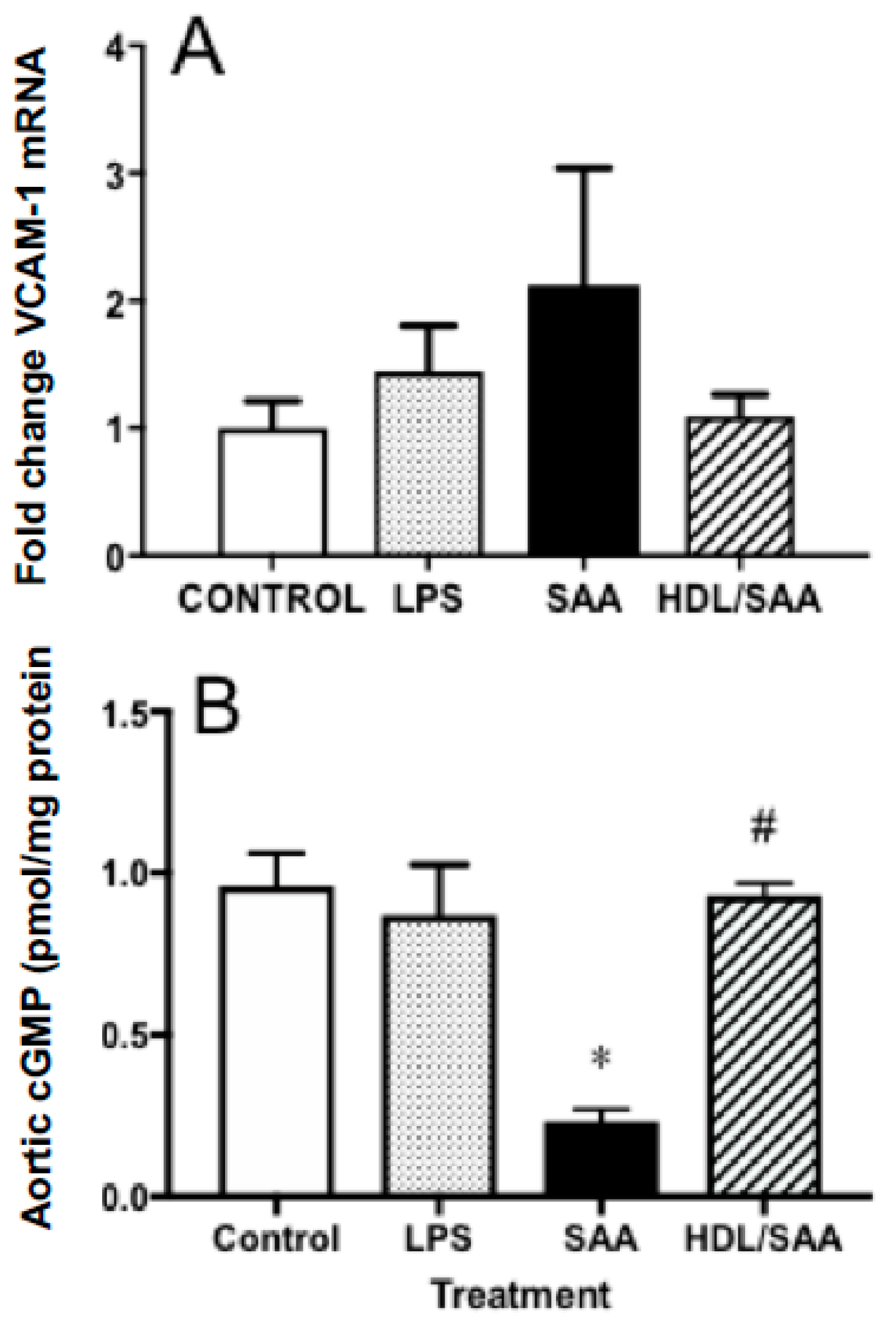
2.1.2. HDL Pretreatment Mitigates SAA-Induced Pro-Atherogenic Changes to the Vasculature
2.2. HDL Pretreatment Inhibits Oxidative Lipid Damage in the Vasculature
2.3. Analyses of Heart Tissues
2.3.1. HDL Pretreatment Mitigates SAA-Induced Pro-Atherogenic Cardiac Vasculature
2.3.2. Inflammatory Cytokines Expression
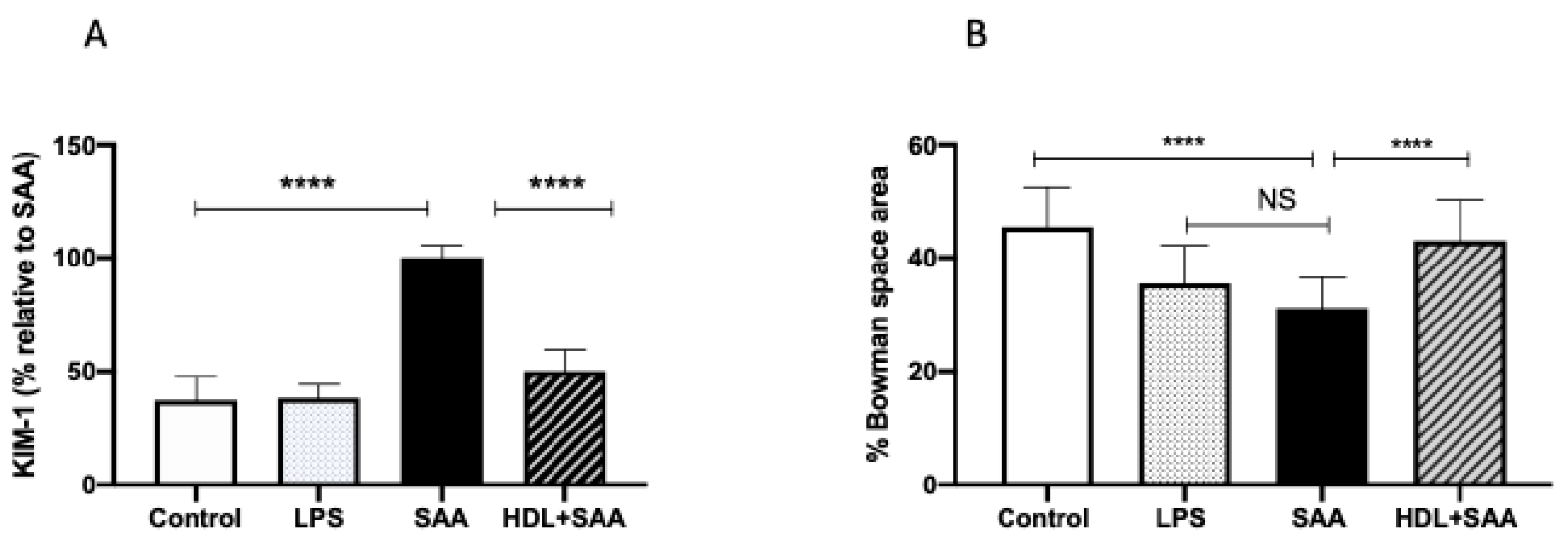
2.4. Kidney Function Studies
2.4.1. SAA Stimulates Renal Vascular Endothelium Dysfunction and HDL Mitigates These Changes
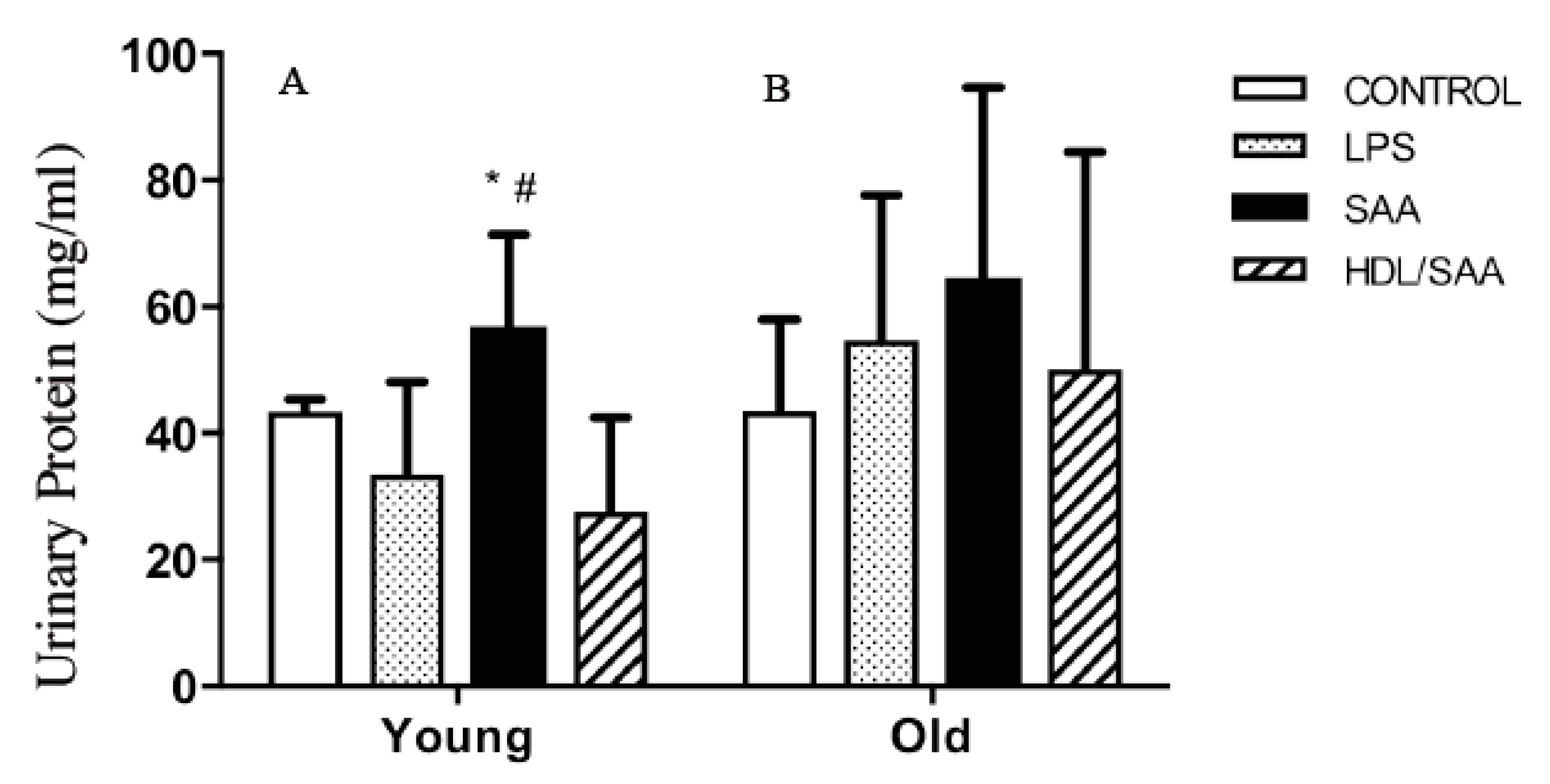
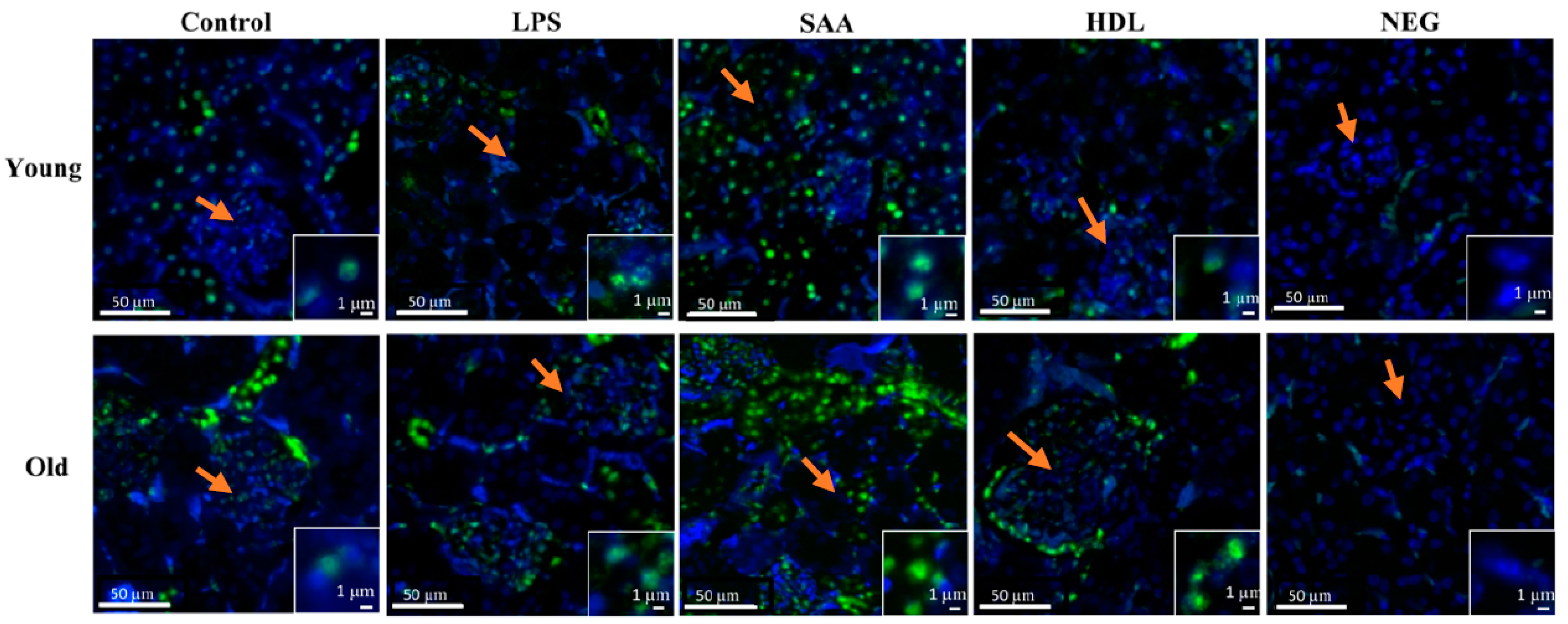
2.4.2. HDL Pretreatment Protects Renal Tissues from SAA-Induced Acute Injury
2.4.3. Human HDL Modulates SAA-Mediated Oxidative Stress in Renal Tissues
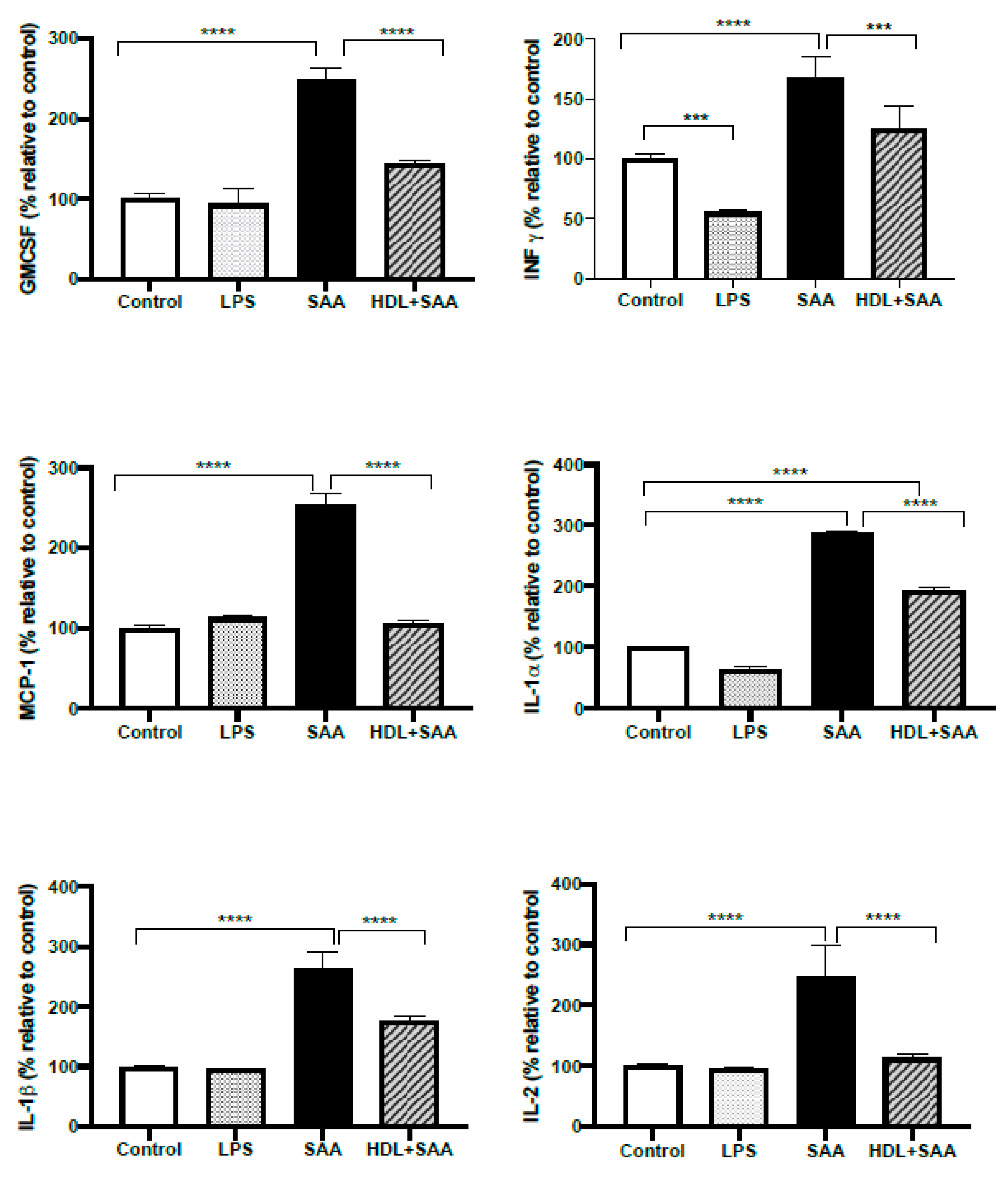
2.4.4. Pretreatment with HDL Inhibits SAA Induced Pro-Inflammatory Cytokine Stimulation
2.4.5. SAA Stimulates p-38/MAPK Activation
3. Discussion
Study Limitations
- We used a common genetically modified mouse model to assess atherosclerosis and accept that the shortcomings of this model, as identified in the statement, are a general limitation of this experimental model.
- Further validating experiments using nonrodent-based animal models (such as rabbits, pigs, and nonhuman primates) are absolutely required before the conclusions drawn from this study can be translated to human conditions where elevated SAA levels may impact on vascular and renal function.
- In the experimental design, we refrained from using a high-fat diet to accelerate atherosclerosis as the hypothesis being tested was that SAA itself plays a role in promoting pro-atherogenic factors that accelerate atherosclerosis and so a high-fat diet would interfere with this assessment.
- We used a reputable supplier of ApoE-/- mice in Australia (Animal Resources Centre (Perth, Western Australia)) that supply a range of mice for research purposes, and mice were contained in the same environment within the animal facility with the access to the same chow and water supply.
- Mice (8 weeks old) were transported to a local site for husbandry, allowed to acclimate, and then were randomly divided into four groups without internal bias.
- Data obtained using (1) analytical and (2) imaging techniques reported in this study were repeated using the same tissues at the same time for all of the treatment cohorts to gain a valid and rigorous comparison between vehicle, LPS, SAA, and HDL-intervention cohorts.
- We reported how often a given experiment was repeated to substantiate the outcome, and this was established using the nominated statistical tests with appropriate corrections.
4. Experimental Section
4.1. Materials
4.2. Methods
4.2.1. Isolation of Human HDL
4.2.2. Animals
4.2.3. Experimental Groups
- (1)
- Vehicle control group: Mice received 100 µL of sterile saline via intraperitoneal (i.p.) injection route every third day for 14 days.
- (2)
- LPS group: Mice received LPS (equivalent to 25 pg LPS/kg) via i.p. injection route every third day over 14 days. This second (positive) control was included at a slightly higher concentration of LPS determined in the SAA preparation to rule out whether biological effects induced by SAA could be attributed to the LPS contaminant in the SAA protein preparation.
- (3)
- SAA group: Mice received 100 µL of SAA (stock solution 120 μg SAA/mL, total SAA 10 μg/kg mouse/injection) via i.p. injection route every third day for 14 days.
- (4)
- HDL group: Mice designated to receive HDL supplements were pre-injected with 100 μL of stock purified human high-density lipoprotein (HDL) (freshly isolated human HDL preparations were diluted in sterile PBS to yield a stock concentration of 1 mg HDL protein/mL, total 100 μg HDL protein/per kg mouse) every third day via tail vein injection for 14 days prior to treatment with SAA (as described above in group 3). In summary, mice received HDL for two weeks prior to the administration of SAA for the ensuing two weeks.
4.2.4. Urine and Blood
4.2.5. Collection of Aorta, Heart, and Kidney Specimens
4.2.6. Gene Expression Studies
4.2.7. Analysis of Inflammatory Proteins and a Biomarker of Kidney Injury with ELISA
4.2.8. Assessment of Renal 3-Chloro-Tyrosine/Tyrosine Ratio
4.2.9. Assessment of Aortic Lipid Oxidation
4.2.10. Cyclic Guanosine Monophosphate (cGMP) Assessment
4.2.11. Immunohistochemistry (IHC) Studies
4.2.12. Immunofluorescence (IF) Studies
4.2.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cahill, P.A.; Redmond, E.M. Vascular endothelium-Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Huveneers, S.; Daemen, M.J.; Hordijk, P.L. Between Rho(k) and a hard place: The relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ. Res. 2015, 116, 895–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEneny, J.; Daniels, J.A.; McGowan, A.; Gunness, A.; Moore, K.; Stevenson, M.; Young, I.S.; Gibney, J. A Cross-Sectional Study Demonstrating Increased Serum Amyloid A Related Inflammation in High-Density Lipoproteins from Subjects with Type 1 Diabetes Mellitus and How this Association Was Augmented by Poor Glycaemic Control. J. Diabetes Res. 2015, 2015, 351601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.G.; Balasubramani, G.K.; Metes, I.D.; Levesque, M.C.; Bridges, S.L., Jr.; Moreland, L.W. Differential response of serum amyloid A to different therapies in early rheumatoid arthritis and its potential value as a disease activity biomarker. Arthritis Res. Ther. 2016, 18, 108. [Google Scholar]
- Fischer, K.; Fischer, K.; Theil, G.; Hoda, R.; Fornara, P. Serum amyloid A: A biomarker for renal cancer. Anticancer Res. 2012, 32, 1801–1804. [Google Scholar] [PubMed]
- Ren, Y.; Ren, Y.; Wang, H.; Lu, D.; Xie, X.; Chen, X.; Peng, J.; Hu, Q.; Shi, G.; Liu, S. Expression of serum amyloid A in uterine cervical cancer. Diagn. Pathol. 2014, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.C.; Jayne, C.; Thompson, J.; Wilson, P.G.; Yoder, M.H.; Webb, N.; Tannock, L.R. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J. Lipid. Res. 2015, 56, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Uhlar, C.M.; Whitehead, A.S. Serum amyloid A, the major vertebrate acute-Phase reactant. Eur. J. Biochem. 1999, 265, 501–523. [Google Scholar] [CrossRef]
- Yamada, T.; Wada, A.; Itoh, K.; Igari, J. Serum amyloid A secretion from monocytic leukaemia cell line THP-1 and cultured human peripheral monocytes. Scand. J. Immunol. 2000, 52, 7–12. [Google Scholar] [CrossRef]
- Meek, R.L.; Urieli-Shoval, S.; Benditt, E.P. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: Implications for serum amyloid A function. Proc. Natl. Acad. Sci. USA 1994, 91, 3186–3190. [Google Scholar] [CrossRef] [Green Version]
- Gotto, A.M., Jr.; Brinton, E.A. Assessing low levels of high-density lipoprotein cholesterol as a risk factor in coronary heart disease: A working group report and update. J. Am. Coll. Cardiol. 2004, 43, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chami, B.; Barrie, N.; Cai, X.; Wang, X.; Paul, M.; Morton-Chandra, R.; Sharland, A.; Dennis, J.M.; Freedman, S.B.; Witting, P.K. Serum amyloid A receptor blockade and incorporation into high-density lipoprotein modulates its pro-inflammatory and pro-Thrombotic activities on vascular endothelial cells. Int. J. Mol. Sci. 2015, 16, 11101–11124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witting, P.K.; Song, C.; Hsu, K.; Hua, S.; Parry, S.N.; Aran, R.; Geczy, C.; Freedman, S.B. The acute-Phase protein serum amyloid A induces endothelial dysfunction that is inhibited by high-Density lipoprotein. Free Radic. Biol. Med. 2011, 51, 1390–1398. [Google Scholar] [CrossRef]
- Dieter, B.P.; McPherson, S.M.; Afkarian, M.; de Boer, I.H.; Mehrotra, R.; Short, R.; Barbosa-Leiker, C.; Alicic, R.Z.; Meek, R.L.; Tuttle, K.R. Serum amyloid a and risk of death and end-Stage renal disease in diabetic kidney disease. J. Diabetes Complicat. 2016, 30, 1467–1472. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Freedman, S.B.; Witting, P.K. Serum amyloid A stimulates cultured endothelial cells to migrate and proliferate: Inhibition by the multikinase inhibitor BIBF1120. Clin. Exp. Pharmacol. Physiol. 2013, 40, 662–670. [Google Scholar] [CrossRef]
- Song, C.; Shen, Y.; Yamen, E.; Hsu, K.; Yan, W.; Witting, P.K.; Geczy, C.L.; Freedman, S.B. Serum amyloid A may potentiate prothrombotic and proinflammatory events in acute coronary syndromes. Atherosclerosis 2009, 202, 596–604. [Google Scholar] [CrossRef]
- Ashby, D.; Gamble, J.; Vadas, M.; Fidge, N.; Siggins, S.; Rye, K.; Barter, P.J. Lack of effect of serum amyloid A (SAA) on the ability of high-Density lipoproteins to inhibit endothelial cell adhesion molecule expression. Atherosclerosis 2001, 154, 113–121. [Google Scholar] [CrossRef]
- Chiba, T.; Chang, M.Y.; Wang, S.; Wight, T.N.; McMillen, T.S.; Oram, J.F.; Vaisar, T.; Heinecke, J.W.; De Beer, F.C.; De Beer, M.C.; et al. Serum amyloid A facilitates the binding of high-density lipoprotein from mice injected with lipopolysaccharide to vascular proteoglycans. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1326–1332. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.Y.; Sun, J.T.; Yang, K.; Shen, W.F.; Lu, L.; Zhang, R.Y.; Tong, X.; Liu, Y. Serum amyloid A enrichment impairs the anti-inflammatory ability of HDL from diabetic nephropathy patients. J. Diabetes Complicat. 2017, 31, 1538–1543. [Google Scholar] [CrossRef]
- Barter, P.J.; Nicholls, S.; Rye, K.A.; Anantharamaiah, G.M.; Navab, M.; Fogelman, A.M. Antiinflammatory properties of HDL. Circ. Res. 2004, 95, 764–772. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Dousset, N.; Ferretti, G.; Bacchetti, T.; Curatola, G.; Salvayre, R. Antioxidant and cytoprotective properties of high-Density lipoproteins in vascular cells. Free Radic. Biol. Med. 2006, 41, 1031–1040. [Google Scholar] [CrossRef]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-Density lipoprotein inhibits serum amyloid A-Mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Ohkawa, R.; Yoshimoto, A.; Yano, K.; Ichimura, N.; Nishimori, M.; Okubo, S.; Yatomi, Y.; Tozuka, M. Effects of serum amyloid A on the structure and antioxidant ability of high-density lipoprotein. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; de Beer, M.C.; Wroblewski, J.M.; Webb, N.R.; de Beer, F.C. SAA does not induce cytokine production in physiological conditions. Cytokine 2013, 61, 506–512. [Google Scholar]
- Rye, K.A.; Barter, P.J. Antiinflammatory actions of HDL: A new insight. Arterioscler. Thromb. Vas. Biol. 2008, 28, 1890–1891. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.Y.; Hafiane, A.; Schwertani, A.; Genest, J. High-Density Lipoproteins: Biology, Epidemiology, and Clinical Management. Can. J. Cardiol. 2017, 33, 325–333. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Antiatherogenic small, dense HDL-Guardian angel of the arterial wall? Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 144–153. [Google Scholar] [CrossRef]
- Rader, D.J.; Hovingh, G.K. HDL and cardiovascular disease. Lancet 2014, 384, 618–625. [Google Scholar] [CrossRef]
- Rader, D.J.; deGoma, E.M. Approach to the patient with extremely low HDL-Cholesterol. J. Clin. Endocrinol. Metab. 2012, 97, 3399–3407. [Google Scholar] [CrossRef] [Green Version]
- Hovingh, G.K.; de Groot, E.; van der Steeg, W.; Boekholdt, S.M.; Hutten, B.A.; Kuivenhoven, J.A.; Kastelein, J.J. Inherited disorders of HDL metabolism and atherosclerosis. Curr. Opin. Lipidol. 2005, 16, 139–145. [Google Scholar] [CrossRef]
- Santos, R.D.; Asztalos, B.F.; Martinez, L.R.; Miname, M.H.; Polisecki, E.; Schaefer, E.J. Clinical presentation, laboratory values, and coronary heart disease risk in marked high-Density lipoprotein-Deficiency states. J. Clin. Lipidol. 2008, 2, 237–247. [Google Scholar] [CrossRef]
- Oldoni, F.; Sinke, R.J.; Kuivenhoven, J.A. Mendelian disorders of high-Density lipoprotein metabolism. Circ. Res. 2014, 114, 124–142. [Google Scholar] [CrossRef] [Green Version]
- Karalis, I.; Jukema, J.W. HDL Mimetics Infusion and Regression of Atherosclerosis: Is It Still Considered a Valid Therapeutic Option? Curr. Cardiol. Rep. 2018, 20, 66. [Google Scholar] [CrossRef] [Green Version]
- Cybulsky, M.I.; Liyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Chami, B.; Hossain, F.; Hambly, T.W.; Cai, X.; Aran, R.; Fong, G.; Vellajo, A.; Martin, N.J.J.; Wang, X.; Dennis, J.M.; et al. Serum Amyloid A Stimulates Vascular and Renal Dysfunction in Apolipoprotein E-Deficient Mice Fed a Normal Chow Diet. Front. Immunol. 2019, 10, 380. [Google Scholar] [CrossRef]
- Chami, B.; Martin, N.J.J.; Dennis, J.M.; Witting, P.K. Myeloperoxidase in the inflamed colon: A novel target for treating inflammatory bowel disease. Arch. Biochem. Biophys. 2018, 645, 61–71. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-Associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Jousilahti, P.; Salomaa, V.; Rasi, V.; Vahtera, E.; Palosuo, T. The association of c-Reactive protein, serum amyloid a and fibrinogen with prevalent coronary heart disease-Baseline findings of the PAIS project. Atherosclerosis 2001, 156, 451–456. [Google Scholar] [CrossRef]
- Dong, Z.; Wu, T.; Qin, W.; An, C.; Wang, Z.; Zhang, M.; Zhang, Y.; Zhang, C.; An, F. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol. Med. 2011, 17, 1357–1364. [Google Scholar] [CrossRef]
- Krishack, P.A.; Bhanvadia, C.V.; Lukens, J.; Sontag, T.J.; De Beer, M.C.; Getz, G.S.; Reardon, C.A. Serum Amyloid A Facilitates Early Lesion Development in Ldlr(-/-) Mice. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Wang, Y.; Chen, W.; Li, W.; Wang, A.; Wong, S.; Bao, G.; Li, J.; Yang, H.; Tracey, K.J. High-Density Lipoprotein (HDL) Counter-Regulates Serum Amyloid A (SAA)-Induced sPLA2-IIE and sPLA2-V Expression in Macrophages. PLoS ONE 2016, 11, e0167468. [Google Scholar] [CrossRef] [Green Version]
- Morrow, J.D. Quantification of isoprostanes as indices of oxidant stress and the risk of atherosclerosis in humans. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Roberts, L.J. Measurement of lipid peroxidation. Free Radic. Res. 1998, 28, 659–671. [Google Scholar] [CrossRef]
- De Beer, M.C.; Wroblewski, J.M.; Noffsinger, V.P.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Ji, A.L.; Shridas, P.; Thompson, J.C.; van der Westhuyzen, D.R.; et al. Deficiency of Endogenous Acute Phase Serum Amyloid A Does Not Affect Atherosclerotic Lesions in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Nitsch, D.; Lawlor, D.A.; Patel, R.; Carson, C.; Ebrahim, S. The association of renal impairment with all-cause and cardiovascular disease mortality. Nephrol. Dial. Transpl. 2010, 25, 1191–1199. [Google Scholar] [CrossRef] [Green Version]
- Soedamah-Muthu, S.S.; Visseren, F.L.; Algra, A.; van der Graaf, Y.; Smart Study Group. The impact of Type 2 diabetes and microalbuminuria on future cardiovascular events in patients with clinically manifest vascular disease from the Second Manifestations of ARTerial disease (SMART) study. Diabet. Med. 2008, 25, 51–57. [Google Scholar] [CrossRef]
- Saulnier, P.J.; Dieter, B.P.; Tanamas, S.K.; McPherson, S.M.; Wheelock, K.M.; Knowler, W.C.; Looker, H.C.; Meek, R.L.; Nelson, R.G.; Tuttle, K.R.; et al. Association of Serum Amyloid A with Kidney Outcomes and All-Cause Mortality in American Indians with Type 2 Diabetes. Am. J. Nephrol. 2017, 46, 276–284. [Google Scholar] [CrossRef]
- Anderberg, R.J.; Meek, R.L.; Hudkins, K.L.; Cooney, S.K.; Alpers, C.E.; Leboeuf, R.C.; Tuttle, K.R. Serum amyloid A and inflammation in diabetic kidney disease and podocytes. Lab. Invest. 2015, 95, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; An, J.N.; Hwang, J.H.; Lee, H.; Lee, J.P.; Kim, S.G. p38 MAPK activity is associated with the histological degree of interstitial fibrosis in IgA nephropathy patients. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Auger-Messier, M.; Accornero, F.; Goonasekera, S.A.; Bueno, O.F.; Lorenz, J.N.; van Berlo, J.H.; Willette, R.N.; Molkentin, J.D. Unrestrained p38 MAPK activation in Dusp1/4 double-null mice induces cardiomyopathy. Circ. Res. 2013, 112, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Kokubo, S.; Sakai, N.; Furuichi, K.; Toyama, T.; Kitajima, S.; Okumura, T.; Matsushima, K.; Kaneko, S.; Wada, T. Activation of p38 mitogen-activated protein kinase promotes peritoneal fibrosis by regulating fibrocytes. Perit. Dial. Int. 2012, 32, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, S.; Makino, J.; Nagashima, A.; Takezawa, T.; Nomoto, N.; Uchihashi, K.; Matsunobu, A.; Sanai, T.; Sugihara, H.; Toda, S.; et al. Fluid flow stress affects peritoneal cell kinetics: Possible pathogenesis of peritoneal fibrosis. Perit. Dial. Int. 2011, 31, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Kolosova, I.; Nethery, D.; Kern, J.A. Role of Smad2/3 and p38 MAP kinase in TGF-beta1-induced epithelial-mesenchymal transition of pulmonary epithelial cells. J. Cell. Physiol. 2011, 226, 1248–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, T.; Yamaguchi, H.; Sato, H.; Kasai, K.; Tanaka, T.; Tanaka, N. Role of p38 mitogen-activated protein kinase pathway on renal failure in the infant rat after burn injury. Shock 2004, 21, 535–542. [Google Scholar] [CrossRef] [PubMed]
- de Beer, M.C.; Webb, N.R.; Wroblewski, J.M.; Noffsinger, V.P.; Rateri, D.L.; Ji, A.; van der Westhuyzen, D.R. de Beer, F.C. Impact of serum amyloid A on high density lipoprotein composition and levels. J. Lipid. Res. 2010, 51, 3117–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolle, M.; Huang, T.; Schuchardt, M.; Jankowski, V.; Prufer, N.; Jankowski, J.; Tietge, U.J.; Zidek, W.; van der Giet, M. High-Density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-Inflammatory-Serum amyloid A. Cardiovasc. Res. 2012, 94, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J. Clin. Investig. 2016, 126, 266–281. [Google Scholar] [CrossRef] [Green Version]
- Schuchardt, M.; Prufer, N.; Tu, Y.; Herrmann, J.; Hu, X.P.; Chebli, S.; Dahlke, K.; Zidek, W.; van der Giet, M.; Tolle, M. Dysfunctional high-Density lipoprotein activates toll-like receptors via serum amyloid A in vascular smooth muscle cells. Sci. Rep. 2019, 9, 3421. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.; Song, C.; Geczy, C.L.; Freedman, S.B.; Witting, P.K. A role for acute-Phase serum amyloid A and high-Density lipoprotein in oxidative stress, endothelial dysfunction and atherosclerosis. Redox Rep. 2009, 14, 187–196. [Google Scholar] [CrossRef]
- Frame, N.M.; Jayaraman, S.; Gantz, D.L.; Gursky, O. Serum amyloid A self-Assembles with phospholipids to form stable protein-Rich nanoparticles with a distinct structure: A hypothetical function of SAA as a “molecular mop” in immune response. J. Struct. Biol. 2017, 200, 293–302. [Google Scholar] [CrossRef]
- Daugherty, A.; Tall, A.R.; Falk, E.; Daemen, M.J.A.P.; Fisher, E.A.; Garcia-Cardena, G.; Lusis, A.J.; Owens, A.P., 3rd; Rosenfeld, M.E.; Virmani, R. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e131–e157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Song, C.; Endoh, I.; Goyette, J.; Jessup, W.; Freedman, S.B.; McNeil, H.P.; Geczy, C.L. Serum amyloid A induces monocyte tissue factor. J. Immunol. 2007, 178, 1852–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, C.M.; Vaisar, T.; Hoofnagle, A.N. Isolating and Quantifying Plasma HDL Proteins by Sequential Density Gradient Ultracentrifugation and Targeted Proteomics. Methods Mol. Biol. 2016, 1410, 105–120. [Google Scholar] [PubMed] [Green Version]
- Witting, P.K.; Mohr, D.; Stocker, R. Assessment of prooxidant activity of vitamin E in human low-density lipoprotein and plasma. Methods Enzymol. 1999, 299, 362–375. [Google Scholar] [PubMed]
- Baggiolini, M.; Walz, A.; Kunkel, S.L. Neutrophil-Activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Investig. 1989, 84, 1045–1049. [Google Scholar] [CrossRef]
- Talib, J.; Pattison, D.I.; Harmer, J.A.; Celermajer, D.S.; Davies, M.J. High plasma thiocyanate levels modulate protein damage induced by myeloperoxidase and perturb measurement of 3-Chlorotyrosine. Free Radic. Biol. Med. 2012, 53, 20–29. [Google Scholar] [CrossRef]
- Shanu, A.; Groebler, L.; Kim, H.B.; Wood, S.; Weekley, C.M.; Aitken, J.B.; Harris, H.H.; Witting, P.K. Selenium inhibits renal oxidation and inflammation but not acute kidney injury in an animal model of rhabdomyolysis. Antioxid. Redox. Signal. 2013, 18, 756–769. [Google Scholar] [CrossRef] [Green Version]
- Chami, B.; Jeong, G.; Varda, A.; Maw, A.M.; Kim, H.B.; Fong, G.M.; Simone, M.; Rayner, B.S.; Wang, X.S.; Dennis, J.M.; et al. The nitroxide 4-methoxy TEMPO inhibits neutrophil-stimulated kinase activation in H9c2 cardiomyocytes. Arch. Biochem. Biophys. 2017, 629, 19–35. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse | NCIB Accession Number |
|---|---|---|---|
| TNF | 5′-ATGAGCACTGAAAGCATGATCC-3′ | 5′-GAGGGCTGATTAGAGAGAGGTC-3′ | NM_000594.4 |
| NFκB | 5′-CTGGAAGCACGAATGACAGA-3′ | 5′-TGAGGTCCATCTCCTTGGTC-3′ | NM_001319226.2 |
| VCAM-1 | 5’CCACAAGGCTACATGAGGGT-3’ | 5’-CAGTGTGGATGTAGCCCCTT-3’ | NM_012889.1 |
| VEGF | 5′-TTTCTTGCGCTTTCGTTTTT-3′ | 5′-CCCACTGAGGAGTCCAACAT-3′ | NM_001025366.3 |
| ACTB | 5′-CATGTACGTTGCTATCCAGG-3′ | 5′-CTCCTTAATGTCACGCACGAT-3′ | NM_001101.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, X.; Ahmad, G.; Hossain, F.; Liu, Y.; Wang, X.; Dennis, J.; Freedman, B.; Witting, P.K. High-Density Lipoprotein (HDL) Inhibits Serum Amyloid A (SAA)-Induced Vascular and Renal Dysfunctions in Apolipoprotein E-Deficient Mice. Int. J. Mol. Sci. 2020, 21, 1316. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041316
Cai X, Ahmad G, Hossain F, Liu Y, Wang X, Dennis J, Freedman B, Witting PK. High-Density Lipoprotein (HDL) Inhibits Serum Amyloid A (SAA)-Induced Vascular and Renal Dysfunctions in Apolipoprotein E-Deficient Mice. International Journal of Molecular Sciences. 2020; 21(4):1316. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041316
Chicago/Turabian StyleCai, Xiaoping, Gulfam Ahmad, Farjaneh Hossain, Yuyang Liu, XiaoSuo Wang, Joanne Dennis, Ben Freedman, and Paul K. Witting. 2020. "High-Density Lipoprotein (HDL) Inhibits Serum Amyloid A (SAA)-Induced Vascular and Renal Dysfunctions in Apolipoprotein E-Deficient Mice" International Journal of Molecular Sciences 21, no. 4: 1316. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041316