Clinical Course and Electron Microscopic Findings in Lymphocytes of Patients with DRAM2-Associated Retinopathy

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results
2.1. Genetic Studies
2.2. DRAM2 Variants
2.3. In Silico Molecular Genetic Analysis
2.4. EYS Variant
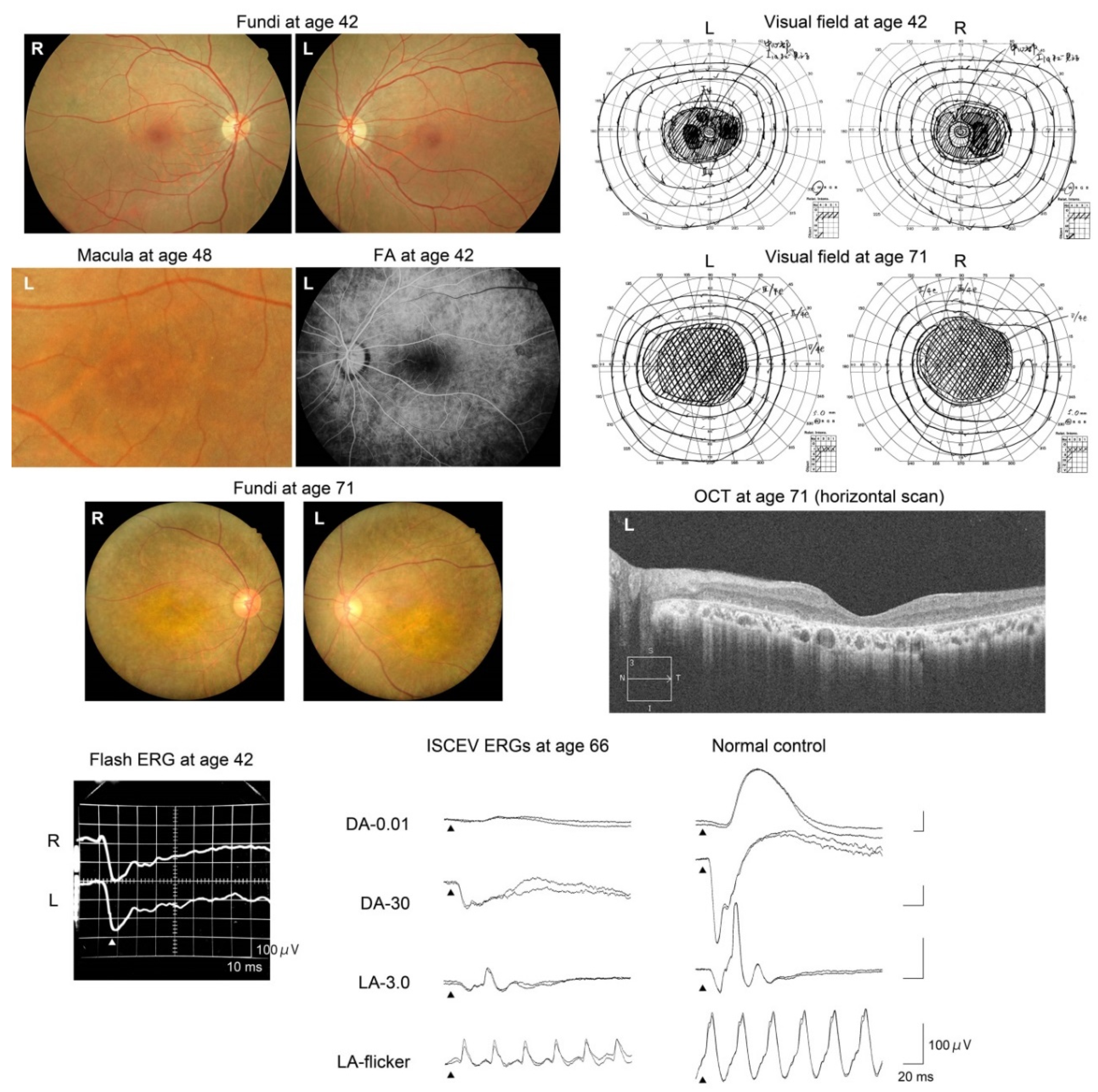
2.5. Clinical Course of Patients
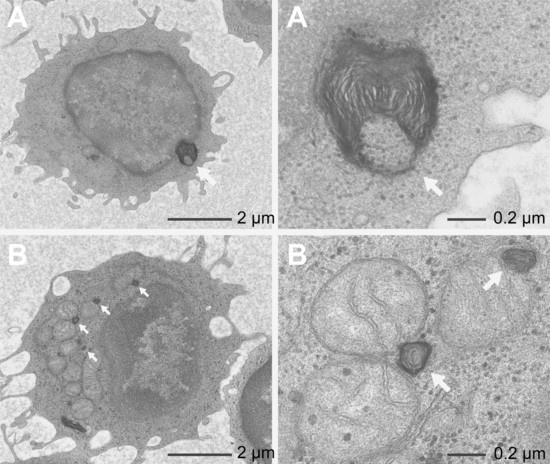
2.6. Transmission Electron Microscopy (TEM)
3. Discussion
4. Participants and Methods
4.1. Ethics Statement
4.2. Genetic Studies
4.3. Clinical Studies
4.4. Transmission Electron Microscopy (TEM)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RPE | retinal pigment epithelium |
| TEM | transmission electron microscopy |
| WES | whole exome sequence |
| HAF | high allele frequency |
| FAF | fundus autofluorescence |
| OCT | optical coherence tomography |
| ERG | electroretinogram |
| ISCEV | International Society for Clinical Electrophysiology of Vision |
References
- O’Prey, J.; Skommer, J.; Wilkinson, S.; Ryan, K.M. Analysis of DRAM-related proteins reveals evolutionarily conserved and divergent roles in the control of autophagy. Cell Cycle 2009, 8, 2260–2265. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Her, S.; Kim, M.; Jang, I.S.; Park, J. The expression of damage-regulated autophagy modulator 2 (DRAM2) contributes to autophagy induction. Mol. Biol. Rep. 2012, 39, 1087–1093. [Google Scholar] [CrossRef]
- Park, S.M.; Kim, K.; Lee, E.J.; Kim, B.K.; Lee, T.J.; Seo, T.; Jang, I.S.; Lee, S.H.; Kim, S.; Lee, J.H.; et al. Reduced expression of DRAM2/TMEM77 in tumor cells interferes with cell death. Biochem. Biophys. Res. Commun. 2009, 390, 1340–1344. [Google Scholar] [CrossRef]
- DRAM2. DNA damage regulated autophagy modulator 2 [Homo sapiens (human)]. NCBI (National Center for Biotechnology Information, U.S. National Library of Medicine). Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/gene/128338 (accessed on 7 February 2020).
- El-Asrag, M.E.; Sergouniotis, P.I.; McKibbin, M.; Plagnol, V.; Sheridan, E.; Waseem, N.; Abdelhamed, Z.; McKeefry, D.; Van Schil, K.; Poulter, J.A.; et al. Biallelic mutations in the autophagy regulator DRAM2 cause retinal dystrophy with early macular involvement. Am. J. Hum. Genet. 2015, 96, 948–954. [Google Scholar] [CrossRef] [Green Version]
- Sergouniotis, P.I.; McKibbin, M.; Robson, A.G.; Bolz, H.J.; De Baere, E.; Müller, P.L.; Heller, R.; El-Asrag, M.E.; Van Schil, K.; Plagnol, V.; et al. Disease expression in autosomal recessive retinal dystrophy associated with mutations in the DRAM2 gene. Invest. Ophthalmol. Vis. Sci. 2015, 56, 8083–8090. [Google Scholar]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef]
- Abad-Morales, V.; Burés-Jelstrup, A.; Navarro, R.; Ruiz-Nogales, S.; Méndez-Vendrell, P.; Corcóstegui, B.; Pomares, E. Characterization of the cone-rod dystrophy retinal phenotype caused by novel homozygous DRAM2 mutations. Exp. Eye Res. 2019, 187, 107752. [Google Scholar] [CrossRef]
- gnomAD. Genome Aggregation Database. Available online: https://gnomad.broadinstitute.org/ (accessed on 7 February 2020).
- Human Genetic Variation Database (HGVD). Available online: http://www.genome.med.kyoto-u.ac.jp/SnpDB/ (accessed on 7 February 2020).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Maeda, A.; Yoshida, A.; Kawai, K.; Arai, Y.; Akiba, R.; Inaba, A.; Takagi, S.; Fujiki, R.; Hirami, Y.; Kurimoto, Y.; et al. Development of a molecular diagnostic test for retinitis pigmentosa in the Japanese population. Jpn. J. Ophthalmol. 2018, 62, 451–457. [Google Scholar]
- Nikopoulos, K.; Cisarova, K.; Quinodoz, M.; Koskiniemi-Kuendig, H.; Miyake, N.; Farinelli, P.; Ur Rehman, A.; Khan, M.I.; Prunotto, A.; Akiyama, M.; et al. A frequent variant in the Japanese population determines quasi-Mendelian inheritance of rare retinal ciliopathy. Nat. Commun. 2019, 10, 2884. [Google Scholar] [CrossRef] [Green Version]
- Abd El-Aziz, M.M.; Barragan, I.; O’Driscoll, C.A.; Goodstadt, L.; Prigmore, E.; Borrego, S.; Mena, M.; Pieras, J.I.; El-Ashry, M.F.; Abu Safieh, L.; et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat. Genet. 2008, 40, 1285–1287. [Google Scholar] [CrossRef] [Green Version]
- Collin, R.W.J.; Littink, K.W.; Klevering, B.J.; van den Born, L.I.; Koenekoop, R.K.; Zonneveld, M.N.; Blokland, E.A.W.; Strom, T.M.; Hoyng, C.B.; den Hollander, A.I.; et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am. J. Hum. Genet. 2008, 83, 594–603. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Lee, M.S. Autophagy-a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 2013, 288, 7506–7518. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Perusek, L.; Maeda, A. Autophagy in light-induced retinal damage. Exp. Eye Res. 2016, 144, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Perusek, L.; Sahu, B.; Parmar, T.; Maeno, H.; Arai, E.; Le, Y.Z.; Subauste, C.S.; Chen, Y.; Palczewski, K.; Maeda, A. Di-retinoid-pyridinium-ethanolamine (A2E) accumulation and the maintenance of the visual cycle are independent of Atg7-mediated autophagy in the retinal pigmented epithelium. J. Biol. Chem. 2015, 290, 29035–29044. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Vinberg, F.; Schottler, F.; Doggett, T.A.; Kefalov, V.J.; Ferguson, T.A. Autophagy supports color vision. Autophagy 2015, 11, 1821–1832. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Doggett, T.A.; Sene, A.; Apte, R.S.; Ferguson, T.A. Autophagy supports survival and phototransduction protein levels in rod photoreceptors. Cell Death Differ. 2015, 22, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.M. Therapeutic targeting of autophagy in disease: Biology and pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef] [Green Version]
- Marmor, M.F.; Kellner, U.; Lai, T.Y.Y.; Melles, R.B.; Mieler, W.F. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology 2016, 123, 1386–1394. [Google Scholar] [CrossRef] [Green Version]
- Kint, J.A. Fabry’s disease: Alpha-galactosidase deficiency. Science 1970, 167, 1268–1269. [Google Scholar] [CrossRef]
- Fischer, E.G.; Moore, M.J.; Lager, D.J. Fabry disease: A morphologic study of 11 cases. Mod. Pathol. 2006, 19, 1295–1301. [Google Scholar] [CrossRef]
- Japan Eye Genetics Consortium (JEGC). Rare/Intractable Disease Project of Japan. Japan Agency for Medical Research and Development (AMED). Available online: http://www.jegc.org/ (accessed on 7 February 2020).
- RetNetTM. Retinal Information Network. Available online: https://sph.uth.edu/retnet/ (accessed on 7 February 2020).
- Fujinami, K.; Kameya, S.; Kikuchi, S.; Ueno, S.; Kondo, M.; Hayashi, T.; Shinoda, K.; Machida, S.; Kuniyoshi, K.; Kawamura, Y.; et al. Novel RP1L1 variants and genotype-photoreceptor microstructural phenotype associations in cohort of Japanese patients with occult macular dystrophy. Invest Ophthalmol Vis. Sci. 2016, 57, 4837–4846. [Google Scholar] [CrossRef] [Green Version]
- jMorp-Japanese Multi Omics Reference Panel. Available online: https://jmorp.megabank.tohoku.ac.jp/202001/downloads/legacy/ (accessed on 7 February 2020).
- Integrative Genomics Viewer. Available online: http://www.broadinstitute.org/igv/ (accessed on 7 February 2020).
- SIFT (Sorting Intolerant From Tolerant). Available online: https://sift.bii.a-star.edu.sg/ (accessed on 7 February 2020).
- PROVEAN (Protein Variation Effect Analyzer). Available online: http://provean.jcvi.org/index.php (accessed on 7 February 2020).
- PolyPhen-2. Prediction of functional effects of human nsSNPs. Available online: http://genetics.bwh.harvard.edu/pph2/ (accessed on 7 February 2020).
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef]
- Kutsuma, T.; Katagiri, S.; Hayashi, T.; Yoshitake, K.; Iejima, D.; Gekka, T.; Kohzaki, K.; Mizobuchi, K.; Baba, Y.; Terauchi, R.; et al. Novel biallelic loss-of-function KCNV2 variants in cone dystrophy with supernormal rod responses. Doc. Ophthalmol. 2019, 138, 229–239. [Google Scholar] [CrossRef]
- Katagiri, S.; Hosono, K.; Hayashi, T.; Kurata, K.; Mizobuchi, K.; Matsuura, T.; Yoshitake, K.; Iwata, T.; Nakano, T.; Hotta, Y. Early onset flecked retinal dystrophy associated with new compound heterozygous RPE65 variants. Mol. Vis. 2018, 24, 286–296. [Google Scholar]
- Kuniyoshi, K.; Hayashi, T.; Sakuramoto, H.; Mishima, H.; Tsuneoka, H.; Tsunoda, K.; Iwata, T.; Shimomura, Y. New truncation mutation of the NR2E3 gene in a Japanese patient with enhanced S-cone syndrome. Jpn. J. Ophthalmol. 2016, 60, 476–485. [Google Scholar] [CrossRef]
- Anderson, D.R. A method of preparing peripheral leucocytes for electron microscopy. J. Ultrastruct. Res. 1965, 13, 263–268. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient # | Sex | Age (years) | Follow-Up Period (years) | Visual Acuity (Decimal) | Refractive Error | Others | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Initial | Final | Initial (age; years) | Intermediate (age; years) | Final (age; years) | ||||||
| 1 Jikei-176-1241 | F | 38 | 43 | 5 | Right | 0.3 (38) | -- | 0.2 (43) | S−1.0D | Phakia No cataract |
| Left | 0.6 (38) | -- | 0.3 (43) | S−0.75D | ||||||
| 2 Kinki-12-1022 | M | 42 | 71 | 29 | Right | 0.2 (42) | 0.03 (55) | H.M. (71) | S−4.0D=C−1.25 | IOL implantation at age 55 in both eyes |
| Left | 0.09 (42) | 0.02 (55) | H.M. (71) | S−4.0D | ||||||
| 3 Kinki-69-1159 | F | 42 | 71 | 29 | Right | 1.2 (42) | 0.01 (55) | H.M. (71) | S+0.5D=C−0.25D | Nuclear cataract in both eyes |
| Left | 1.2 (42) | H.M. (55) | H.M. (71) | S+0.25D | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuniyoshi, K.; Hayashi, T.; Kameya, S.; Katagiri, S.; Mizobuchi, K.; Tachibana, T.; Kubota, D.; Sakuramoto, H.; Tsunoda, K.; Fujinami, K.; et al. Clinical Course and Electron Microscopic Findings in Lymphocytes of Patients with DRAM2-Associated Retinopathy. Int. J. Mol. Sci. 2020, 21, 1331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041331
Kuniyoshi K, Hayashi T, Kameya S, Katagiri S, Mizobuchi K, Tachibana T, Kubota D, Sakuramoto H, Tsunoda K, Fujinami K, et al. Clinical Course and Electron Microscopic Findings in Lymphocytes of Patients with DRAM2-Associated Retinopathy. International Journal of Molecular Sciences. 2020; 21(4):1331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041331
Chicago/Turabian StyleKuniyoshi, Kazuki, Takaaki Hayashi, Shuhei Kameya, Satoshi Katagiri, Kei Mizobuchi, Toshiaki Tachibana, Daiki Kubota, Hiroyuki Sakuramoto, Kazushige Tsunoda, Kaoru Fujinami, and et al. 2020. "Clinical Course and Electron Microscopic Findings in Lymphocytes of Patients with DRAM2-Associated Retinopathy" International Journal of Molecular Sciences 21, no. 4: 1331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041331