Conformational Insight on WT- and Mutated-EGFR Receptor Activation and Inhibition by Epigallocatechin-3-Gallate: Over a Rational Basis for the Design of Selective Non-Small-Cell Lung Anticancer Agents


Abstract
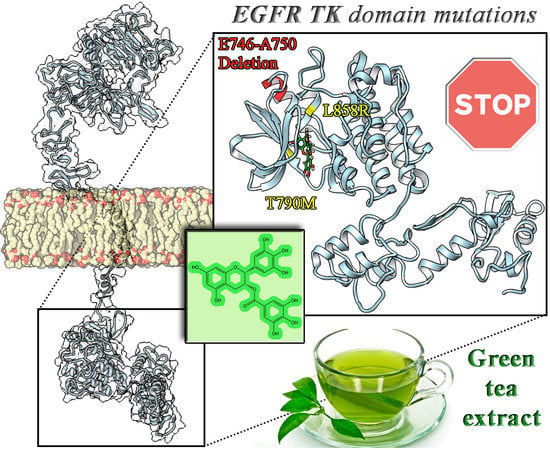
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
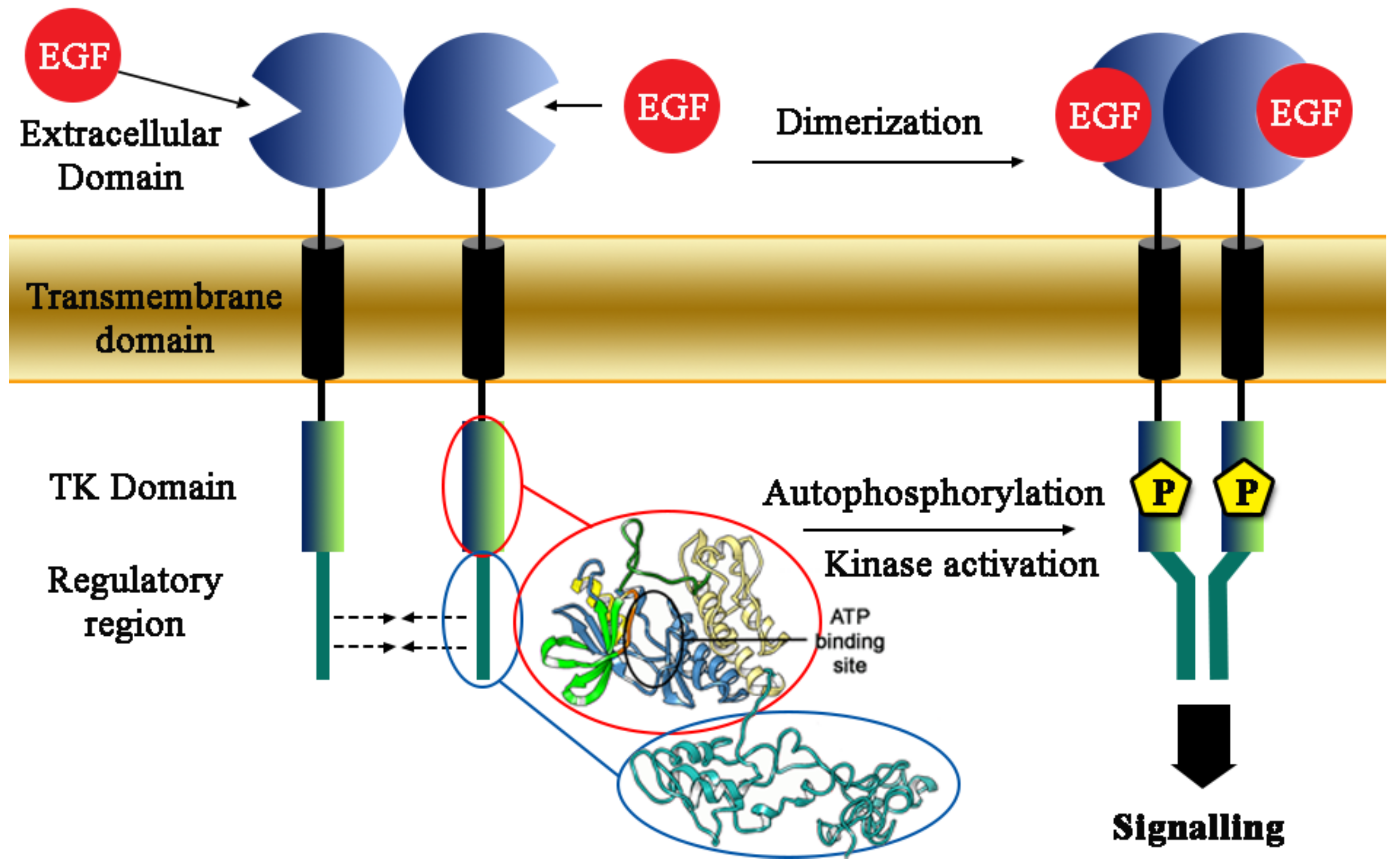
1. Introduction
2. Results and Discussion
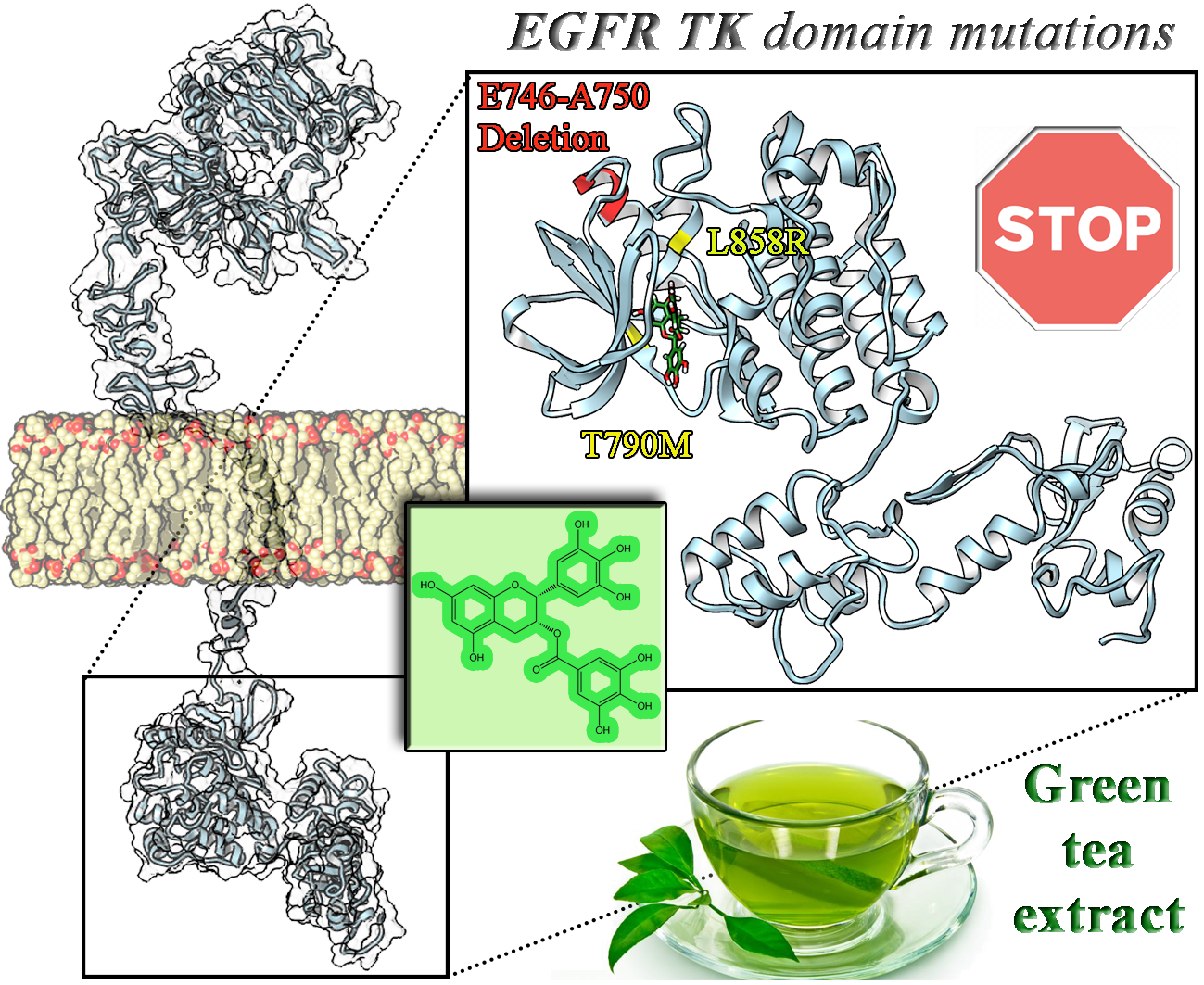
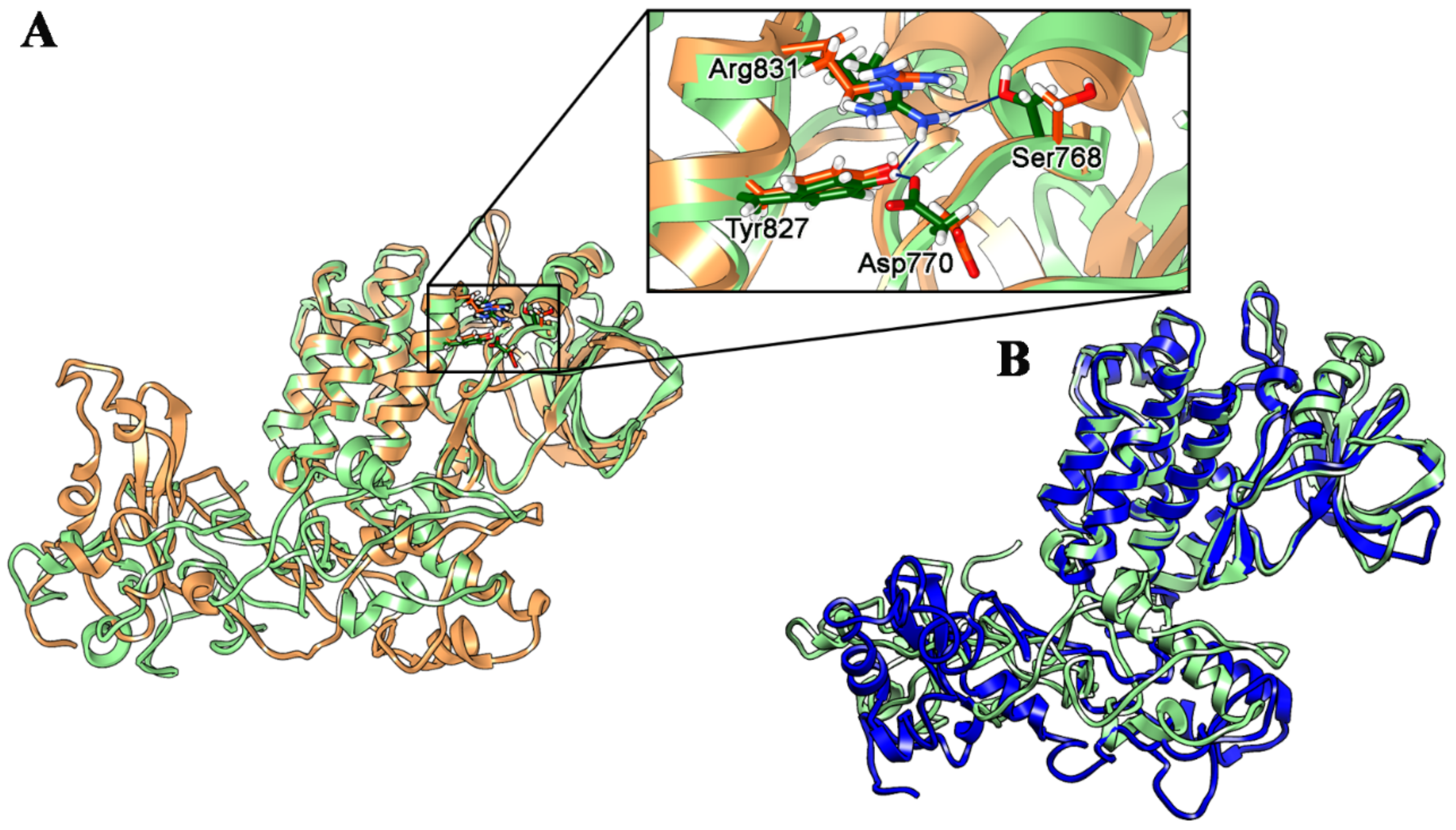
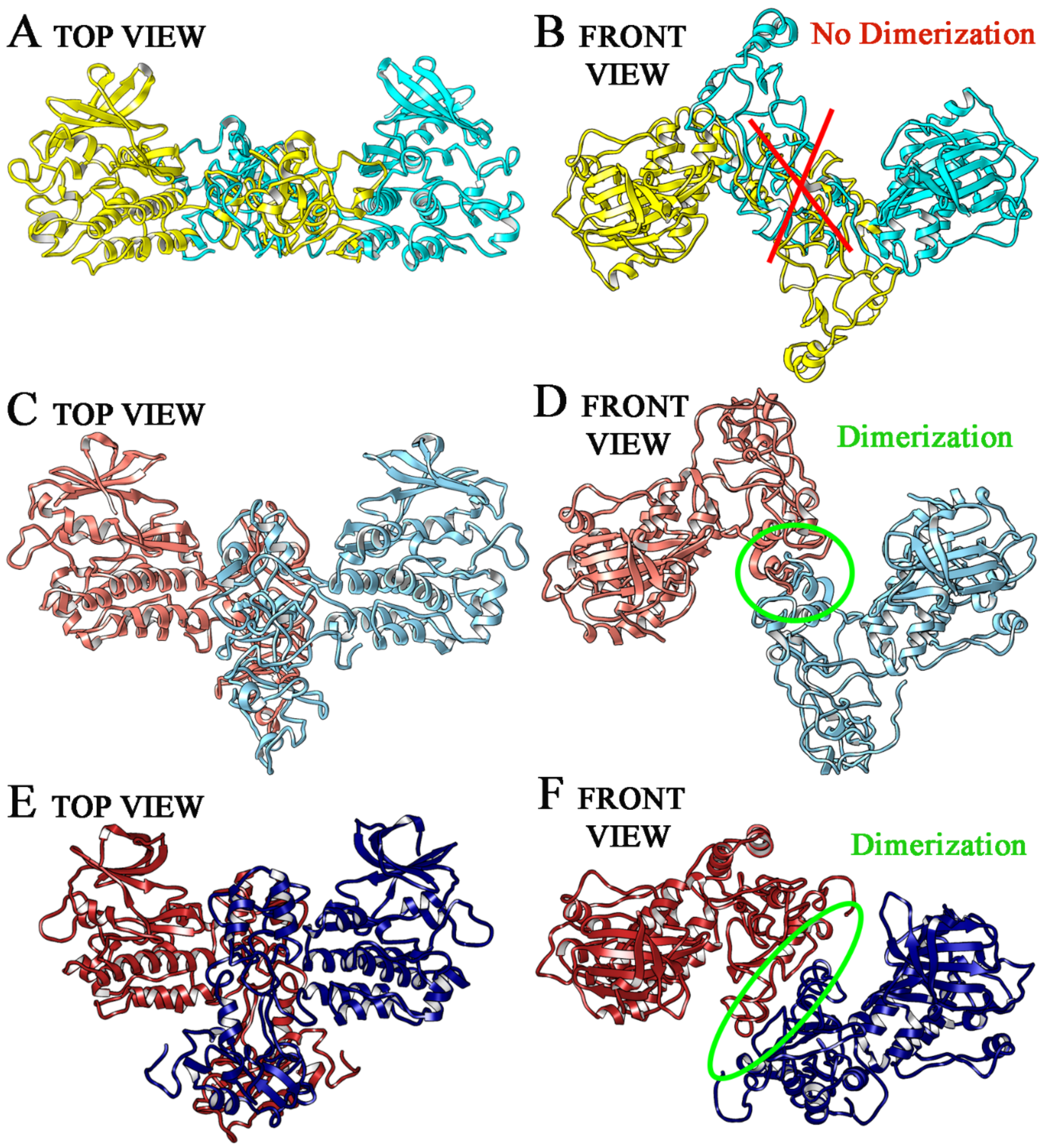
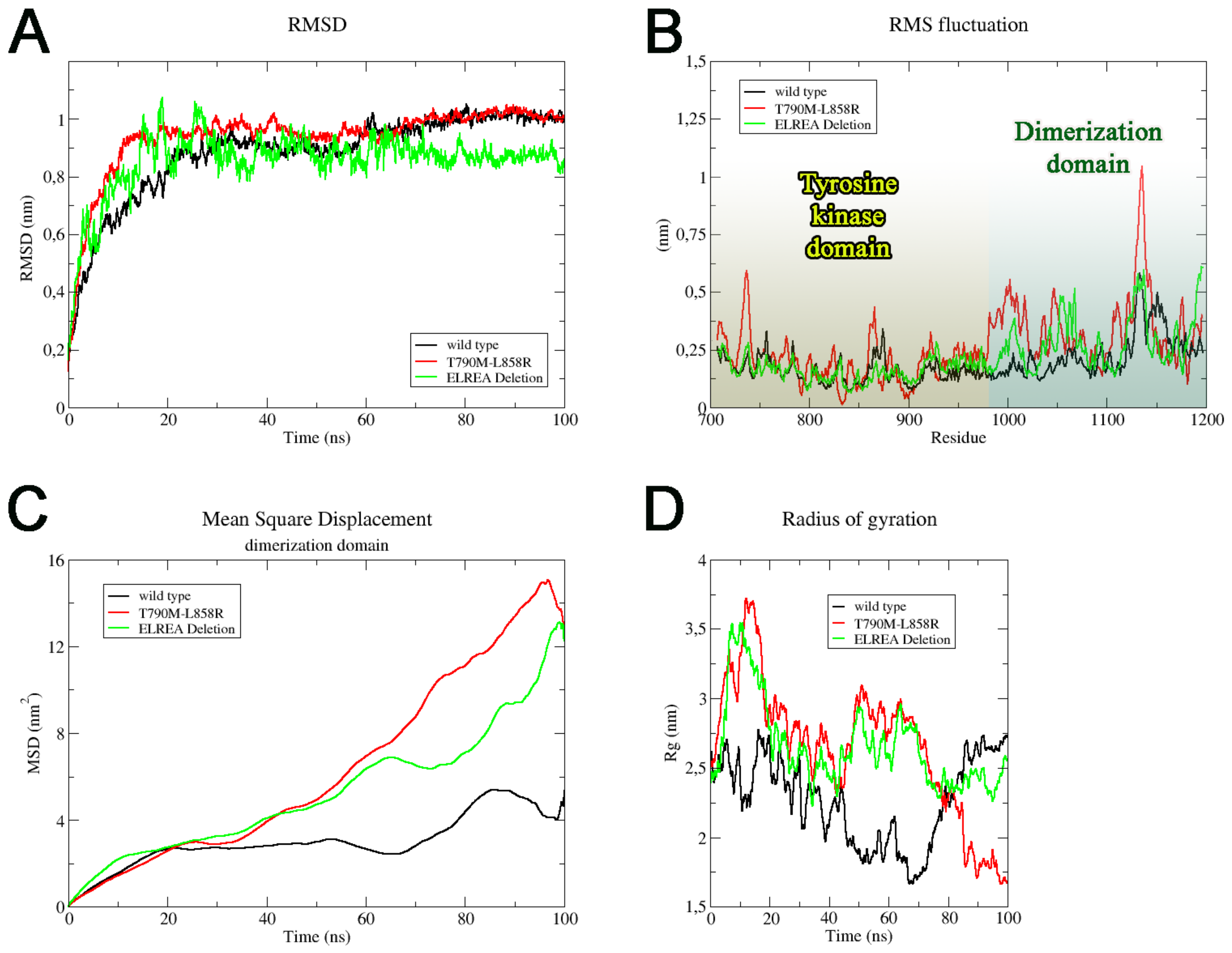
2.1. Wild Type, T790M/L858R and ELREA Deletion EGFR Differ for Conformational Mobility of the Dimerization Domain
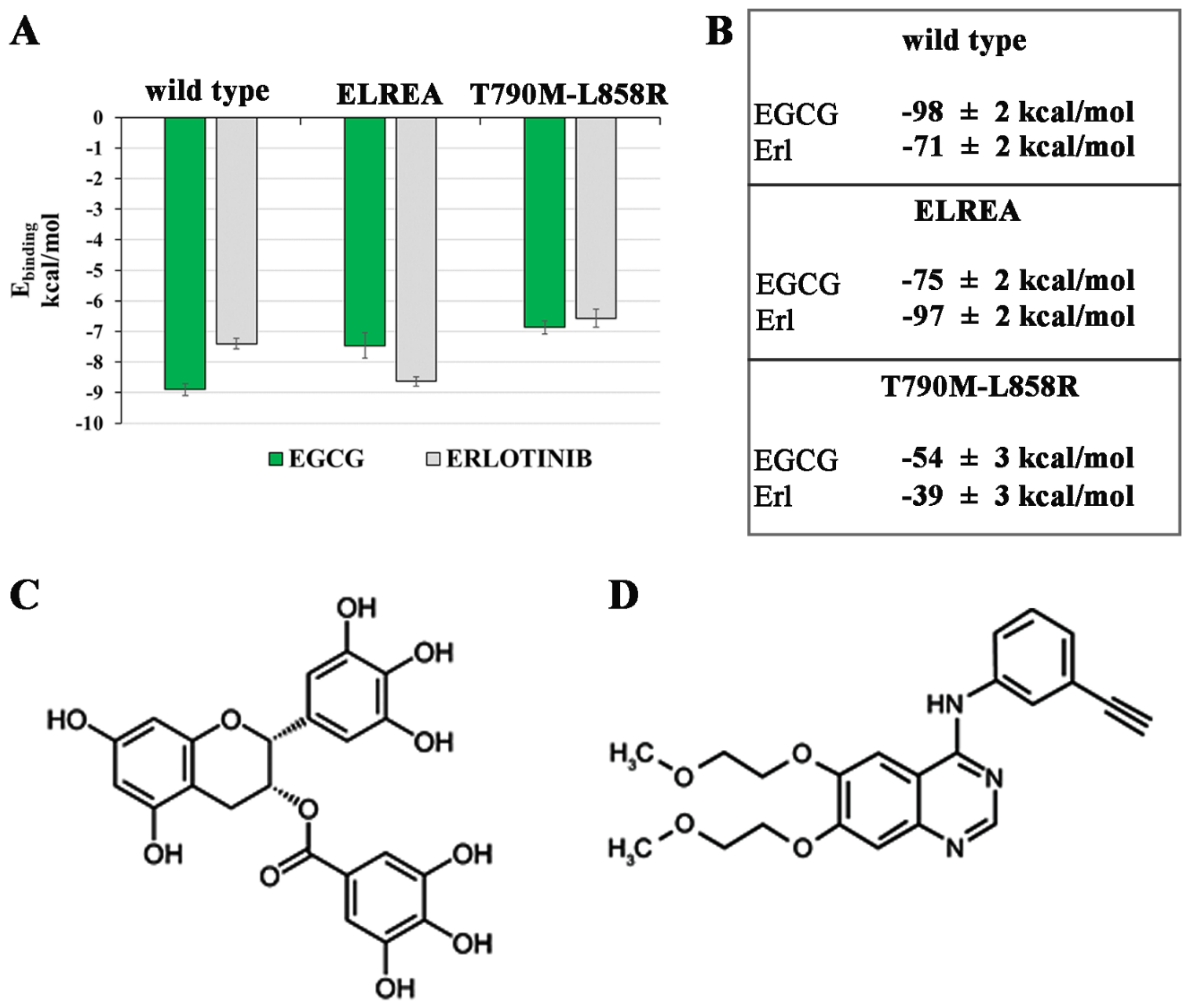
2.2. Known Inhibitors of EGFR-TK Domains Bind Differently with the Three EGCFR Receptors
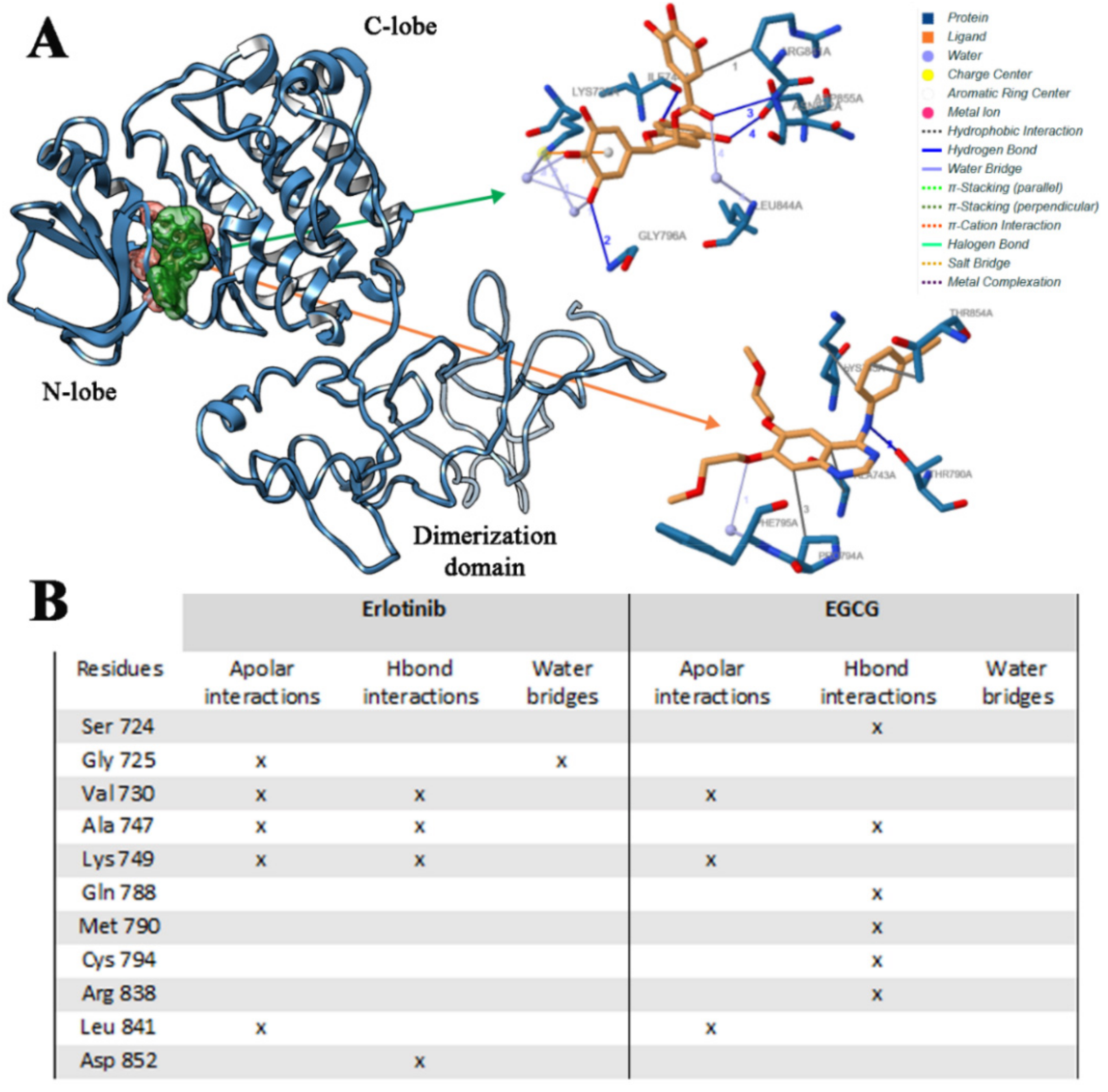
2.2.1. Erl and EGCG Binding Mode with EGFR Wild Type
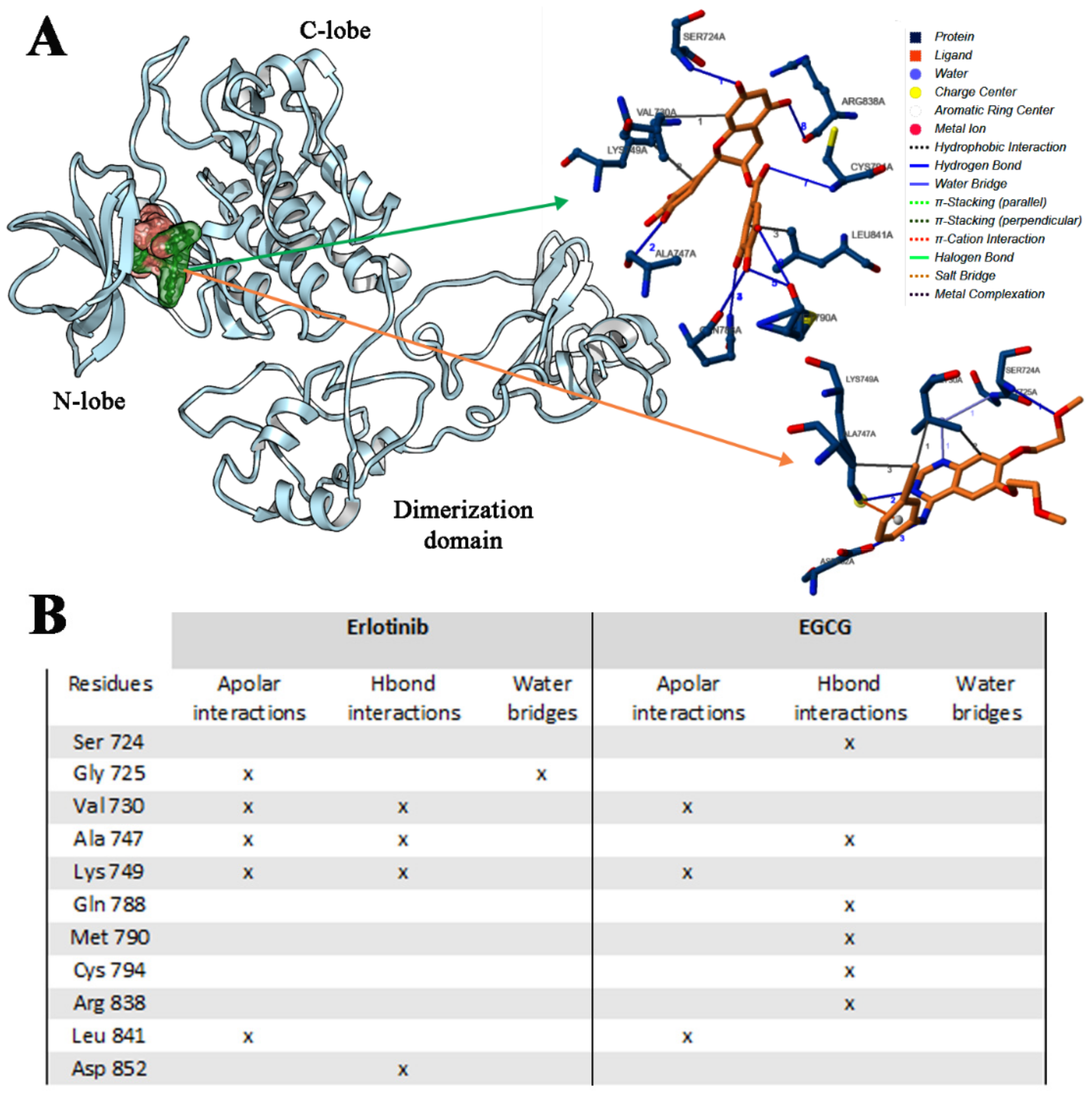
2.2.2. Erl and EGCG Binding with EGFR ELREA Deletion
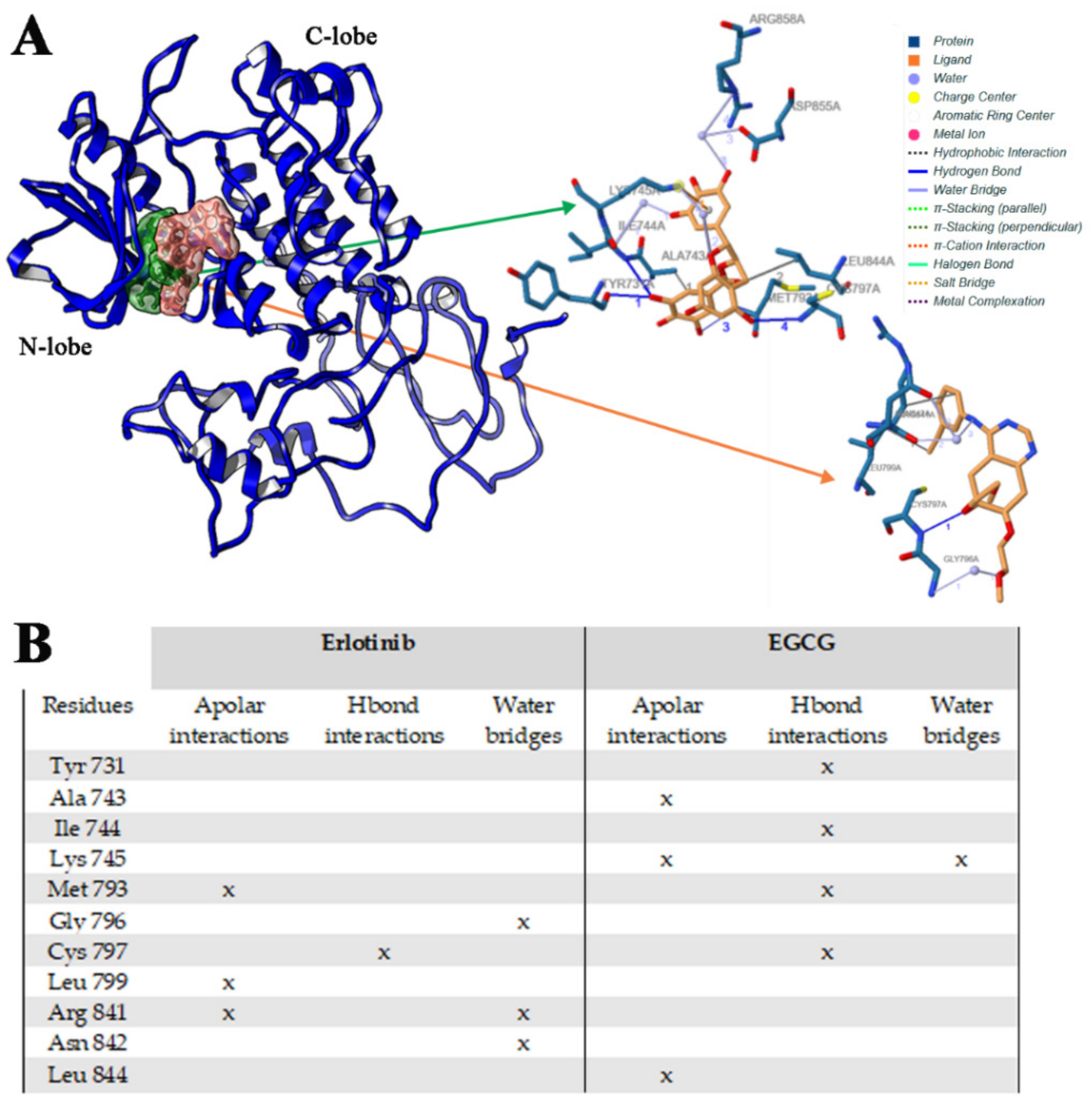
2.2.3. Erl and EGCG Binding with EGFR T790M/L858R
3. Materials and Methods
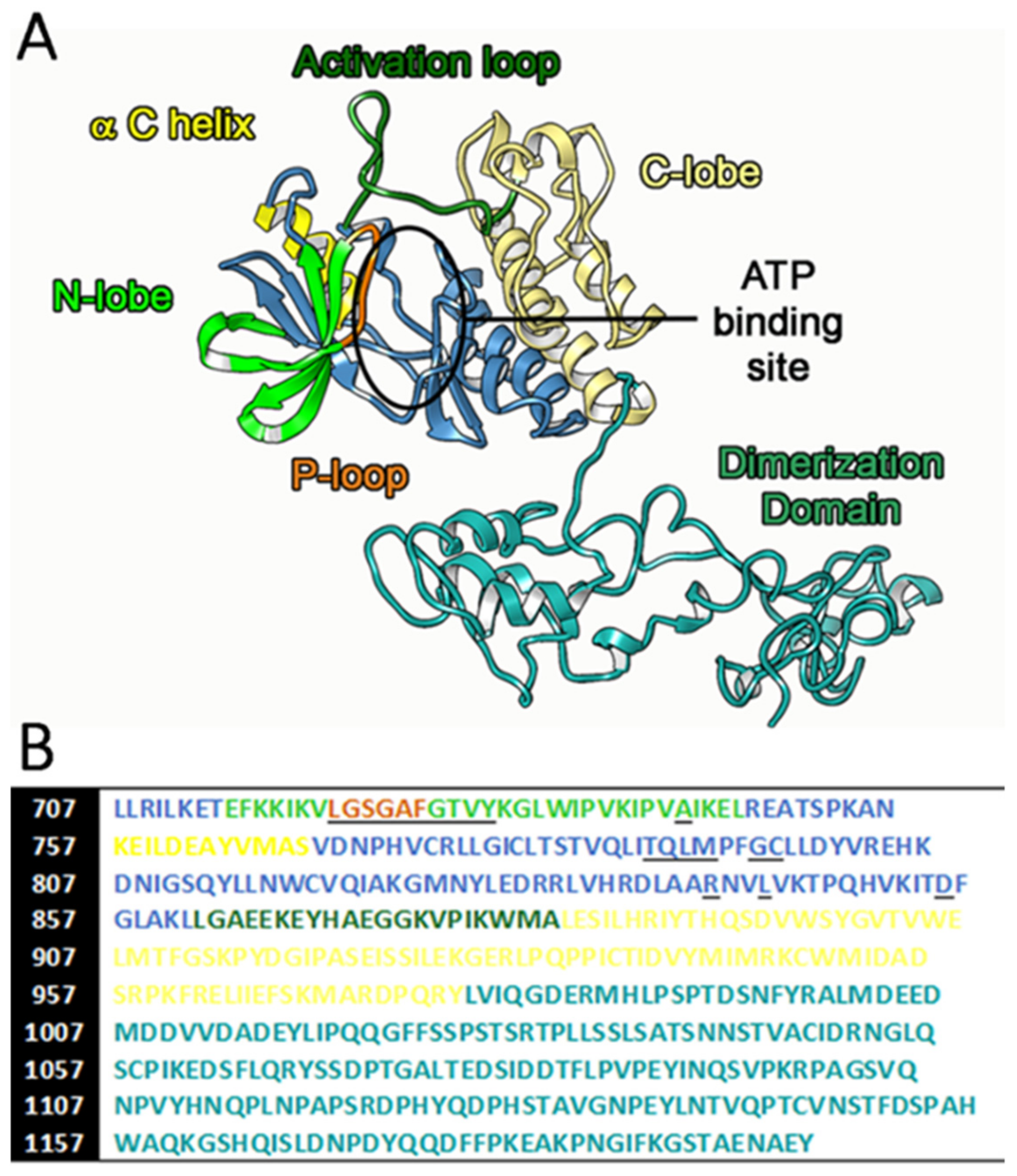
3.1. EGFR TK Cytoplasmic Domain Modeling
3.2. Inhibitors Docking to EGCFR Receptors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Longo, D.; Fauci, A.; Kasper, D.; Hauser, S.; Jameson, J.; Loscalzo, J. Harrison’s Principles of Internal Medicine, 18th ed.; McGraw-Hill Professional: New York, NY, USA, 2012. [Google Scholar]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuoka, H.; Cooper, O.; Mizutani, J.; Tong, Y.; Ren, S.G.; Bannykh, S.; Melmed, S. HER2/ErbB2 Receptor Signaling in Rat and Human Prolactinoma Cells: Strategy for Targeted Prolactinoma Therapy. Mol. Endocrinol. 2011, 25, 92–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar]
- Singh, D.; Attri, B.K.; Gill, R.K.; Bariwa, J. Review on EGFR Inhibitors: Critical Updates. Med. Chem. 2016, 16, 14. [Google Scholar]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Xu, M.J.; Johnson, D.E.; Grandis, J.R. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017, 36, 463–473. [Google Scholar] [CrossRef]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Johnson, B.E.; Jänne, P.A. Epidermal growth factor receptor mutations in patients with non-small cell lung cancer. Cancer Res. 2005, 65, 7525–7529. [Google Scholar] [CrossRef] [Green Version]
- Sutiman, N.; Tan, S.W.; Tan, E.H.; Lim, W.T.; Kanesvaran, R.; Ng, Q.S.; Jain, A.; Ang, M.K.; Tan, W.L.; Toh, C.K.; et al. EGFR Mutation Subtypes Influence Survival Outcomes following First-Line Gefitinib Therapy in Advanced Asian NSCLC Patients. J. Thorac Oncol. 2017, 12, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetti, S.; Molina, M.A.; Queralt, C.; de Aguirre, I.; Mayo, C.; Bertran-Alamillo, J.; Sanchez, J.J.; Gonzalez-Larriba, J.L.; Jimenez, U.; Isla, D.; et al. Detection of EGFR mutations with mutation-specific antibodies in stage IV non-small-cell lung cancer. J. Transl. Med. 2010, 18, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, N.; Nicolson, M.; Groves, R.W.; Steele, J.; Eaby, B.; Dunlop, J.; Mcphelim, J.; Nijjar, R.; Ukachukwu, I. Expert consensus on the management of erlotinib-associated cutaneous toxicity in the u.k. Oncologist 2009, 14, 840–847. [Google Scholar] [CrossRef]
- Minnelli, C.; Moretti, P.; Fulgenzi, G.; Mariani, P.; Laudadio, E.; Armeni, T.; Galeazzi, R.; Mobbili, G. A Poloxamer-407 modified liposome encapsulating epigallocatechin-3-gallate in the presence of magnesium: Characterization and protective effect against oxidative damage. Int. J. Pharm. 2018, 552, 225–234. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.N.; Sun, P.Y.; Zi, C.T.; Wang, X.J.; Sheng, J. Glucosylated Epigallocatechin Gallate (EGCG) Derivatives Combined with EGFR Tyrosine Kinase Inhibitor Overcome Resistance in EGFR T790M Mutant Lung Cancer. JSTR 2019, 18, 13474–13479. [Google Scholar]
- Bommu, U.D.; Konidala, K.K.; Pabbaraju, N.; Yeguvapalli, S. QSAR modeling, pharmacophore-based virtual screening, and ensemble docking insights into predicting potential epigallocatechin gallate (EGCG) analogues against epidermal growth factor receptor. J. Receptors Signal. Transduction 2019, 39, 18–27. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Yamada, S.; Itai, S.; Chang, Y.W.; Nakamura, T.; Yanaka, M.; Kato, Y. Elucidation of the critical epitope of an anti-EGFR monoclonal antibody EMab-134. Biochem. Biophys. Rep. 2018, 14, 54–57. [Google Scholar] [CrossRef]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, M.; Narasimhan, S.; de Heus, C.; Mance, D.; van Doorn, S.; Houben, K.; Popov-Čeleketić, D.; Damman, R.; Katrukha, E.A.; Jain, P.; et al. EGFR Dynamics Change during Activation in Native Membranes as Revealed by NMR. Cell 2016, 167, 1241–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purba, E.R.; Saita, E.I.; Maruyama, I.N. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Hackshaw, A.; Feng, Q.; Fu, X.; Zhang, Y.; Mao, C.; Tang, J. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: A meta-analysis. Int. J. Cancer 2017, 15, 2805–2819. [Google Scholar] [CrossRef]
- Yang, J.J.; Zhou, Q.; Yan, H.H.; Zhang, X.C.; Chen, H.J.; Tu, H.Y.; Wang, Z.; Xu, C.R.; Su, J.; Wang, B.C.; et al. A phase III randomised controlled trial of erlotinib vs. gefitinib in advanced non-small cell lung cancer with EGFR mutations. Br. J. Cancer 2017, 116, 568–574. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, K.M. A structure-based view of Epidermal Growth Factor Receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [Green Version]
- Milik, S.N.; Lasheen, D.S.; Serya, D.S.L.; Aboudiz, K.A.M. How to train your inhibitor: Design strategies to overcome resistance to Epidermal Growth Factor Receptor inhibitors. Eur. J. Med. Chem. 2017, 142, 131–151. [Google Scholar] [CrossRef] [Green Version]
- Eck, M.J.; Yun, C.H. Structural and mechanistic underprinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 2010, 1804, 559–566. [Google Scholar] [CrossRef] [Green Version]
- Lecointre, C.; Simon, V.; Kerneur, C.; Allemand, F.; Fournet, A.; Montarras, I.; Pons, J.L.; Gelin, M.; Brignatz, C.; Urbach, S.; et al. Dimerization of the Pragmin Pseudo-Kinase Regulates Protein Tyrosine Phosphorylation. Structure 2018, 26, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R. Open Source Drug Discovery Consortium, Lynn, A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Jorge, S.E.D.C.; Kobayashi, S.S.; Costa, D.B. Epidermal growth factor receptor (EGFR) mutations in lung cancer: Preclinical and clinical data. Braz. J. Med. Biol. Res. 2014, 47, 929–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazieh, A.R.; Sudairy, R.A.; Abu-Shraie, N.; Suwairi, W.A.; Ferwana, M.; Murad, M.H. Erlotinib in wild type epidermal growth factor receptor non-small cell lung cancer: A systematic review. Ann. Thorac. Med. 2013, 8, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Yu, P.; Ye, L.; Zhang, J.; Wang, H.; Zou, F.; Tian, J.; Kurihara, H. PCC0208027, a novel tyrosine kinase inhibitor, inhibits tumor growth of NSCLC by targeting EGFR and HER2 aberrations. Sci. Rep. 2019, 9, 5692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.C.; Li, C.; Gao, F.; Xu, Y.; Jiang, Z.B.; Liu, J.X.; Jin, L.Y. Epigallocatechin gallate inhibits the growth of human lung cancer by directly targeting the EGFR signaling pathway. Oncol. Rep. 2014, 31, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Fracasso, P.M.; Bukowski, R.M.; Lynch, T.J.; Munster, P.N.; Shapiro, G.I.; Jänne, P.A.; Eder, J.P.; Naughton, M.J.; Ellis, M.J.; et al. A phase I study with neratinib (HKI-272), an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin. Cancer Res. 2009, 1, 2552–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Hellmann, M.D.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Poor response to erlotinib in patients with tumors containing baseline EGFR T790M mutations found by routine clinical molecular testing. Ann. Oncol. 2014, 2, 423–428. [Google Scholar] [CrossRef]
- Whang, Z.; Longo, P.A.; Tarrant, M.K.; Kim, K.; Head, S.; Leahy, D.J.; Cole, P.A. Mechanistic Insights into the Activation of Oncogenic Forms of EGF Receptor. Nat. Struct. Mol. Biol. 2011, 18, 1388–1393. [Google Scholar] [CrossRef]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.N.; Zhao, M.M.; Wu, D.D.; Chen, Y.; Wang, Y.; Zhu, J.H.; Cai, W.J.; Zhu, Y.Z.; Zhu, Y.C. Hydrogen Sulfide Targets EGFR Cys797/Cys798 Residues to Induce Na+ /K+ -ATPase Endocytosis and Inhibition in Renal Tubular Epithelial Cells and Increase Sodium Excretion in Chronic Salt-Loaded Rats. Antioxid. Redox Signal 2014, 21, 2061–2082. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, E.; de Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.E.; Lee, G.R.; Heo, L.; Lee, H.; Seok, C. Prediction of Protein Structure and Interaction by GALAXY Protein Modeling Programs. Bio Design 2014, 2, 1. [Google Scholar]
- Dagan-Wiener, A.; Nissim, I.; Ben Abu, N.; Borgonovo, G.; Bassoli, A.; Niv, M.Y. Bitter or not? BitterPredict, a tool for predicting taste from chemical structure. Sci. Rep. 2017, 7, 12074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceroni, A.; Passerini, A.; Vullo, A.; Frasconi, P. DISULFIND: A disulfide bonding state and cysteine connectivity prediction server. Nucleic Acids Res. 2006, 34, 177–181. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Sali, A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics 2003, 19, 2500–2501. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-levelparallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.C. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. 1985, 31, 1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Ray, J.R. Elastic constants and statistical ensembles in molecular dynamics. Comput. Phys. Rep. 1988, 8, 109–151. [Google Scholar] [CrossRef]
- Galeazzi, R.; Laudadio, E.; Falconi, E.; Massaccesi, L.; Ercolani, L.; Mobbili, G.; Minnelli, C.; Scirè, A.; Cianfruglia, L.; Armeni, T. Protein-protein interactions of human glyoxalase II: Findings of a reliable docking protocol. Org. Biomol. Chem. 2018, 16, 5167–5177. [Google Scholar] [CrossRef] [PubMed]
- Laudadio, E.; Minnelli, C.; Amici, A.; Massaccesi, L.; Mobbili, G.; Galeazzi, R. Liposomal formulations for an efficient encapsulation of epigallocatechin-3-gallate: An in-silico/experimental approach. Molecules 2018, 23, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. ELSEVIER VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 3, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Sanner, M.F.; Duncan, B.S.; Carrillo, C.J.; Olson, A.J. Integrating computation and visualization for biomolecular analysis: An example using python and AVS. Pac. Symp. Biocomput. 1999, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Den Otter, W.K.; Briels, W.J. The calculation of free-energy differences by constrained molecular-dynamics simulations. J. Chem. Phys. 1998, 109, 4139. [Google Scholar] [CrossRef]
- Laudadio, E.; Cedraro, N.; Mangiaterra, G.; Citterio, B.; Mobbili, G.; Minnelli, C.; Bizzaro, D.; Biavasco, F.; Galeazzi, R. Natural Alkaloid Berberine Activity against Pseudomonas aeruginosa MexXY-Mediated Aminoglycoside Resistance: In Silico and in Vitro Studies. J. Nat. Prod. 2019, 82, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Gabbianelli, R.; Carloni, M.; Marmocchi, F.; Nasuti, C.; Fedeli, D.; Laudadio, E.; Massaccesi, L.; Galeazzi, R. Permethrin and its metabolites affect Cu/Zn superoxide conformation: Fluorescence and in silico evidences. Mol. BioSyst. 2015, 11, 208. [Google Scholar]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minnelli, C.; Laudadio, E.; Mobbili, G.; Galeazzi, R. Conformational Insight on WT- and Mutated-EGFR Receptor Activation and Inhibition by Epigallocatechin-3-Gallate: Over a Rational Basis for the Design of Selective Non-Small-Cell Lung Anticancer Agents. Int. J. Mol. Sci. 2020, 21, 1721. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051721
Minnelli C, Laudadio E, Mobbili G, Galeazzi R. Conformational Insight on WT- and Mutated-EGFR Receptor Activation and Inhibition by Epigallocatechin-3-Gallate: Over a Rational Basis for the Design of Selective Non-Small-Cell Lung Anticancer Agents. International Journal of Molecular Sciences. 2020; 21(5):1721. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051721
Chicago/Turabian StyleMinnelli, Cristina, Emiliano Laudadio, Giovanna Mobbili, and Roberta Galeazzi. 2020. "Conformational Insight on WT- and Mutated-EGFR Receptor Activation and Inhibition by Epigallocatechin-3-Gallate: Over a Rational Basis for the Design of Selective Non-Small-Cell Lung Anticancer Agents" International Journal of Molecular Sciences 21, no. 5: 1721. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051721