Correlative Light and Electron Microscopy (CLEM) Analysis of Nuclear Reorganization Induced by Clustered DNA Damage Upon Charged Particle Irradiation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
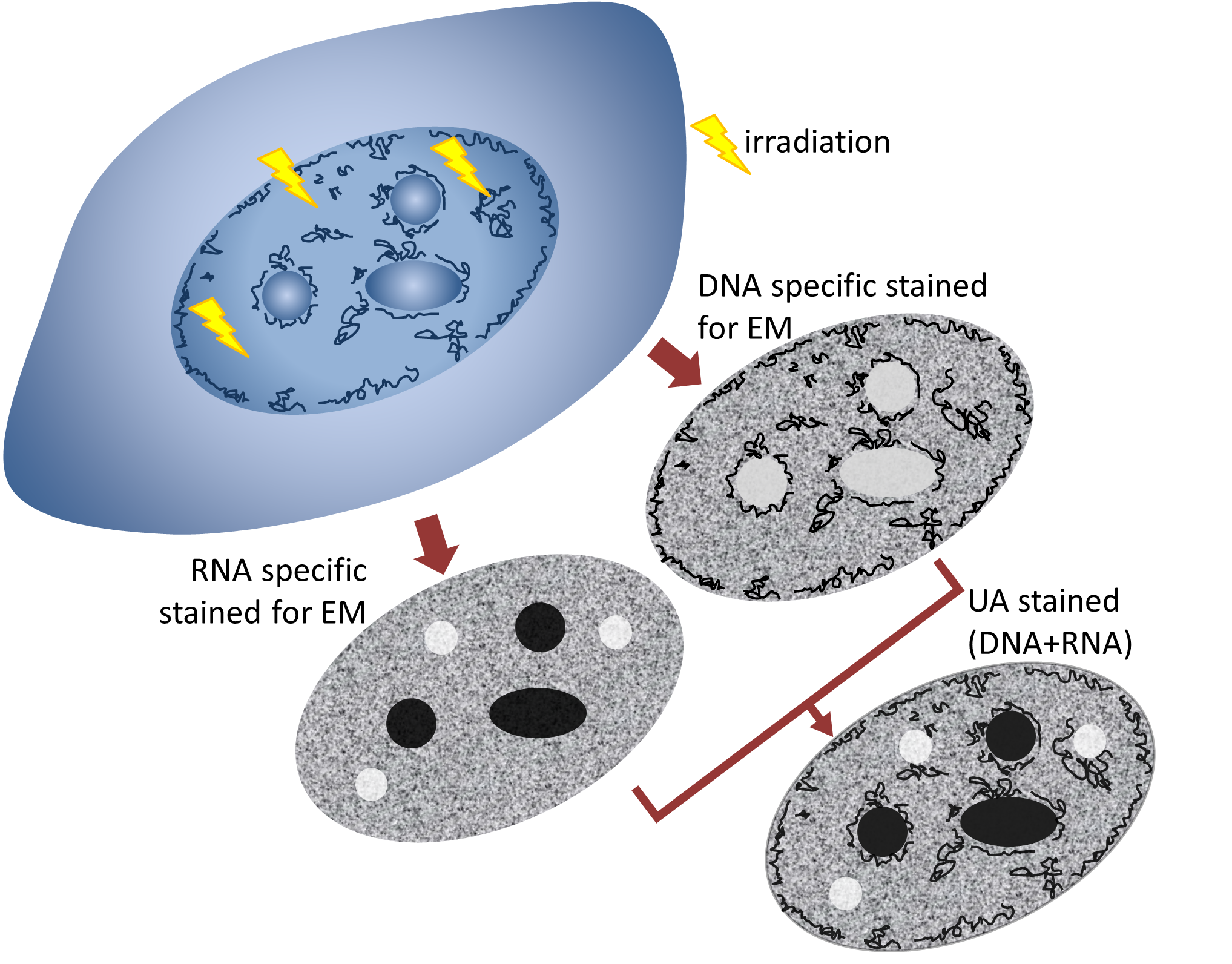
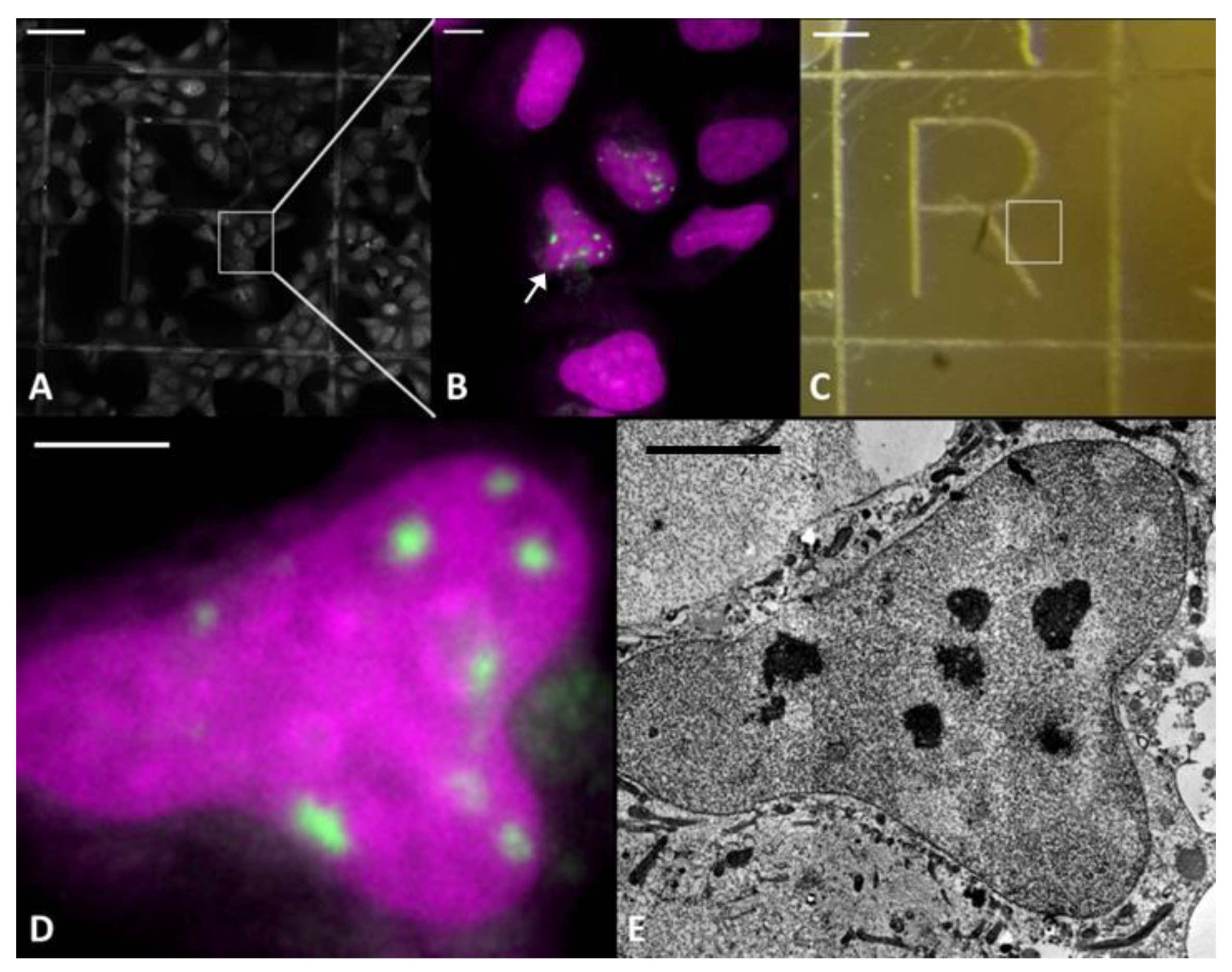
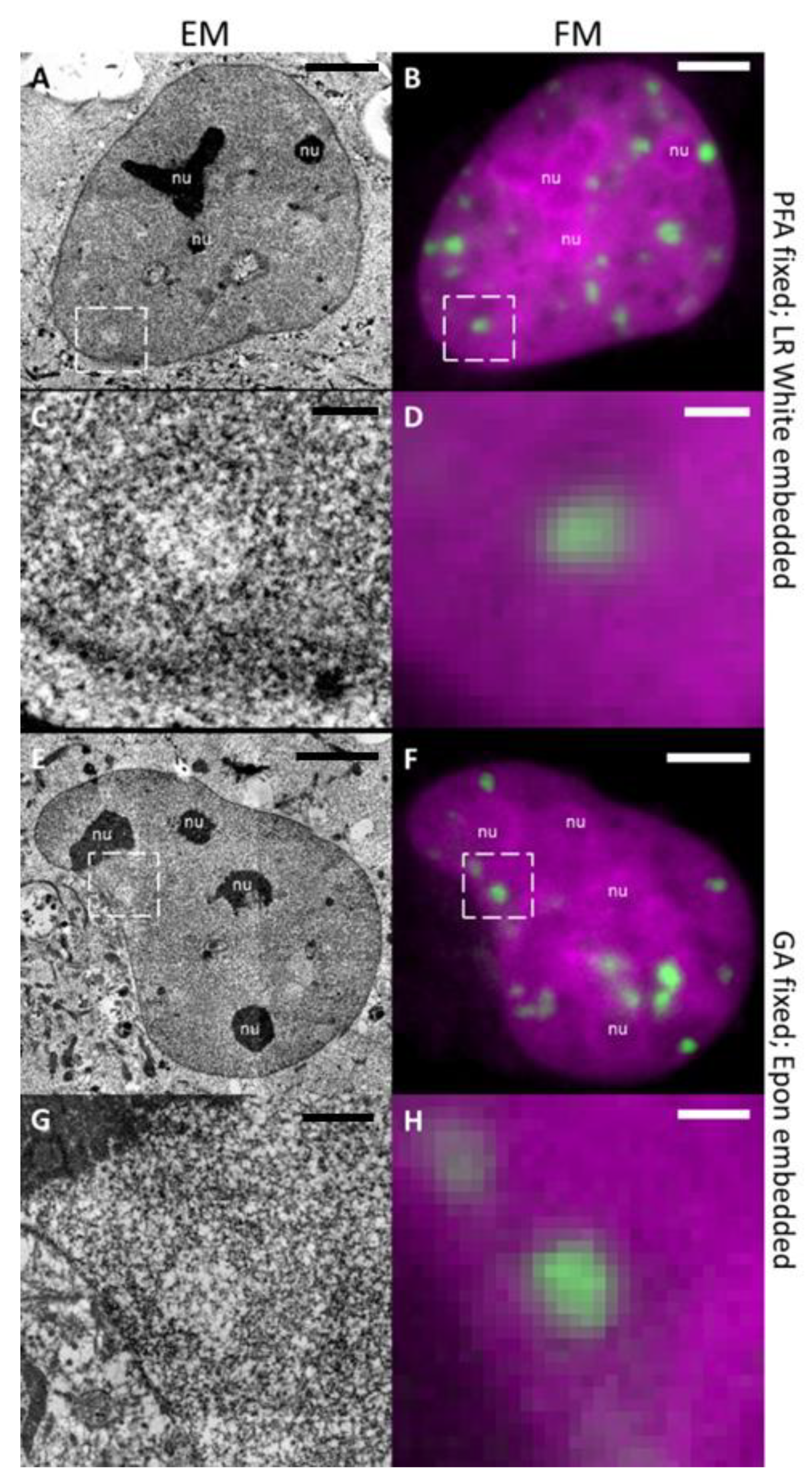
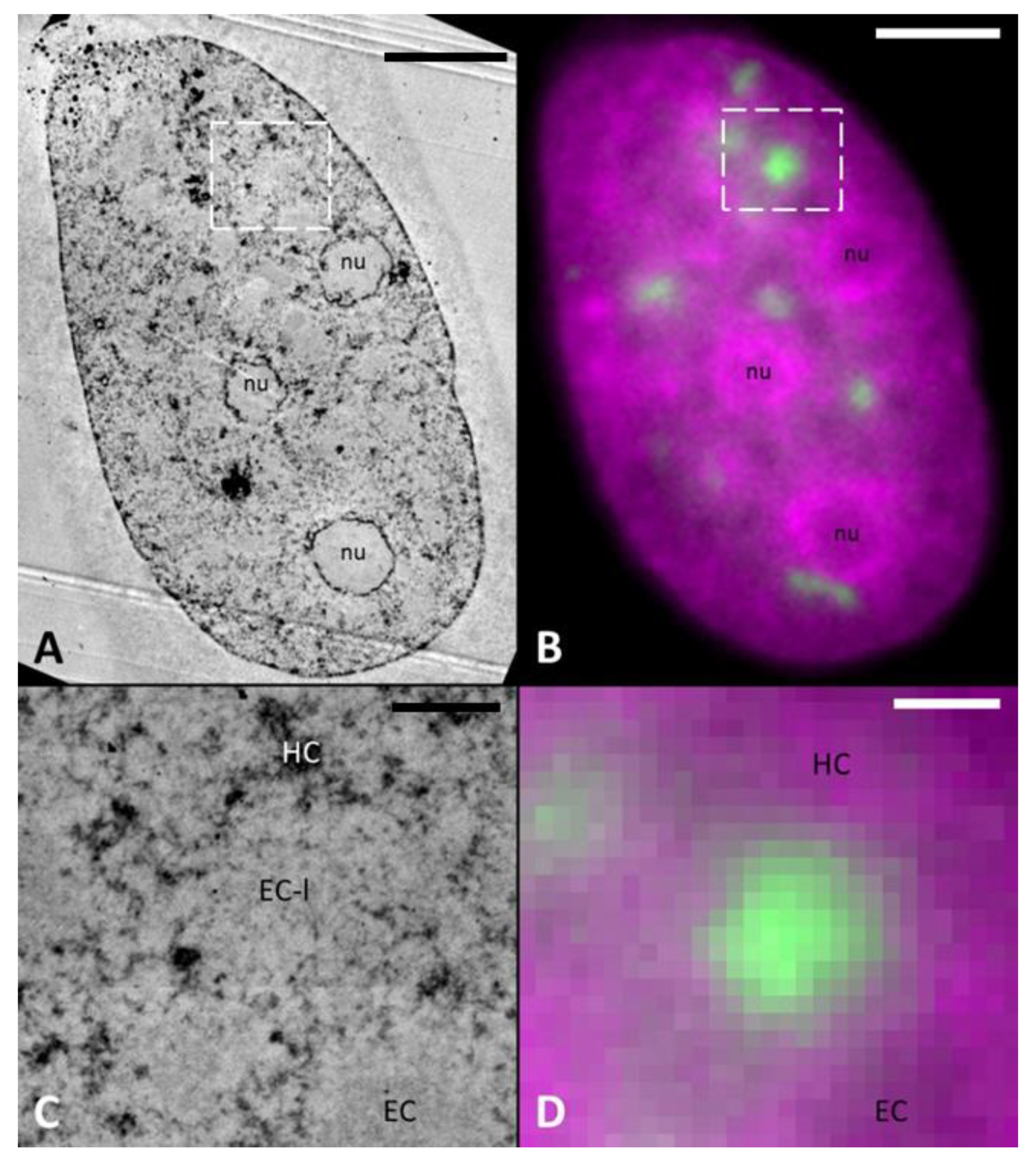
2.1. LDA Phenotype is Fixation and Embedding Independent
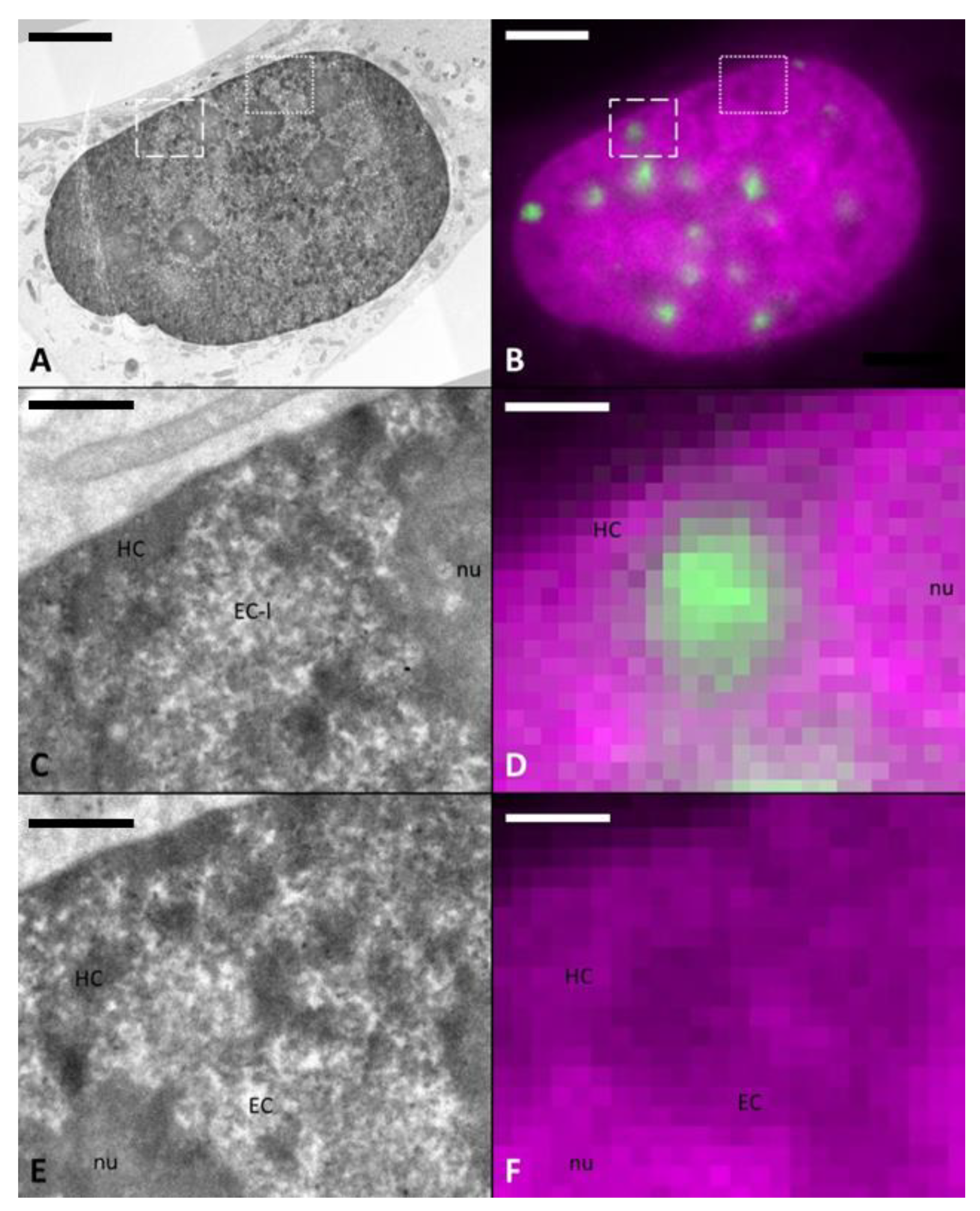
2.2. DNA-Specific Stains Reveal EC-Like Areas within DNA-Repair Foci, but No LDA Pattern
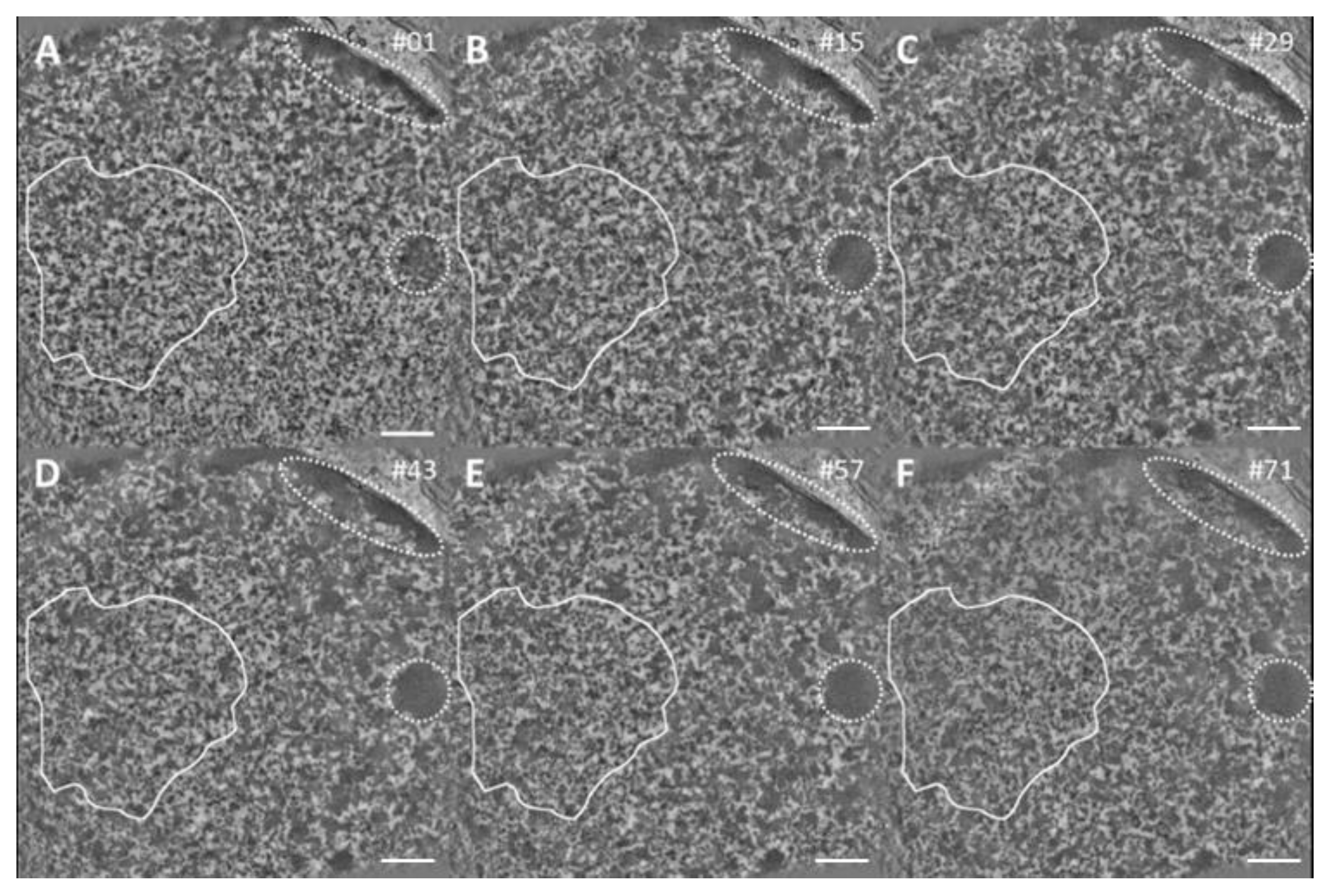
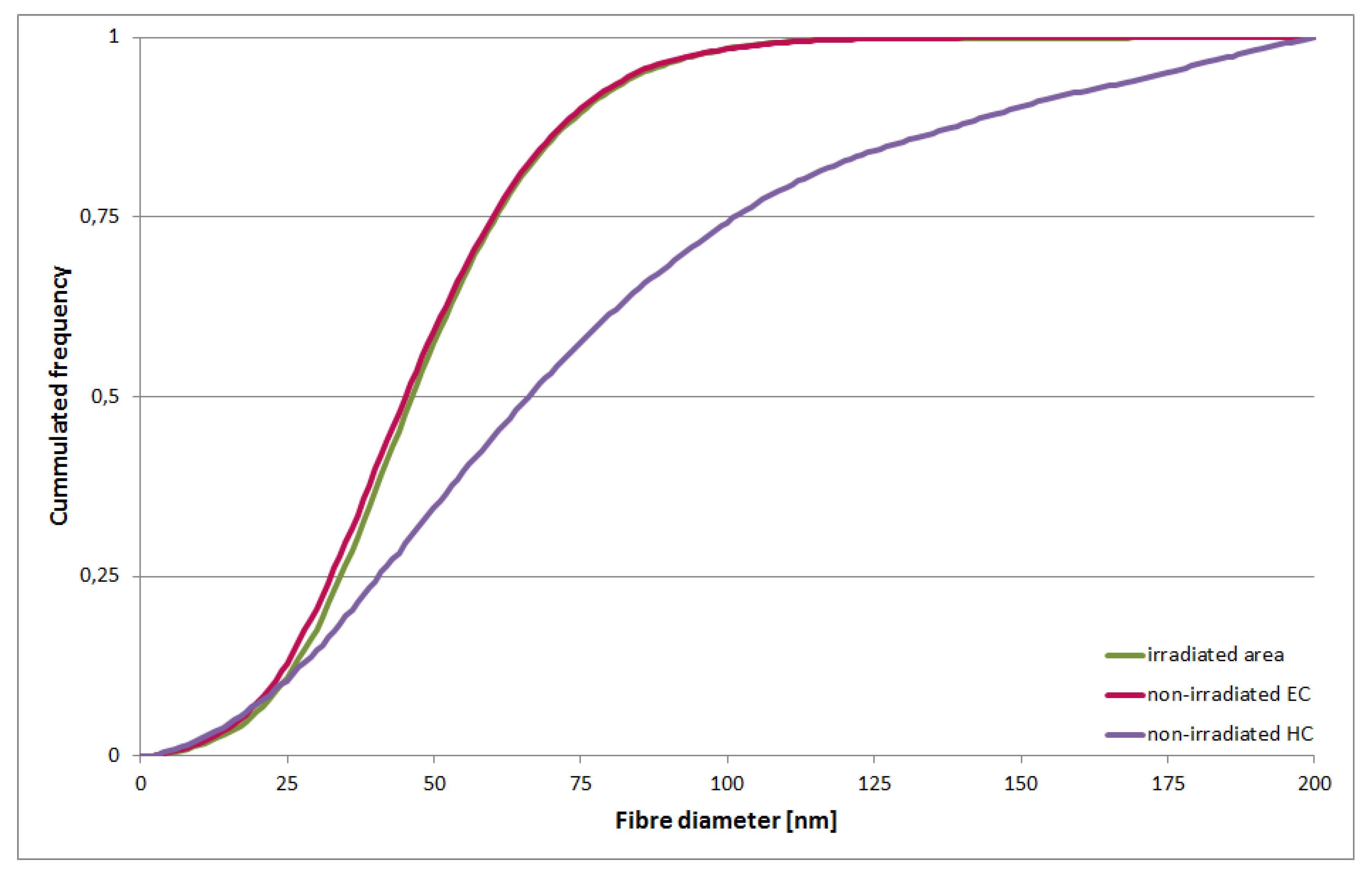
2.3. Tomographic Analysis of Chromatin Organisation within DNA Repair Foci
2.4. Reduced RNA Density within DNA Repair Regions
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Irradiation, and Fixation
4.2. Fluorescence Microscopy
4.3. Electron Microscopy
4.3.1. Embedding and Cutting for EM
4.3.2. ChromEMT Staining
4.3.3. Staining with Osmium Ammine B (OA-B)
4.3.4. RNA-Specific Staining with Terbium-Citrate (Tb)
4.3.5. Transmission EM
4.3.6. EM Tomography
4.4. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CLEM | correlative light and electron microscopy |
| LDA | low density area |
| PcG | polycomb group protein |
| HP1 | heterochromatin protein 1 |
| TAD | topologically associated domain |
| DSB | double-strand break |
| UA | uranyl acetate |
| TEM | transmission electron microscope |
| EM | electron microscope |
| OA-B | osmium ammine B |
| IRIF | ionizing radiation induced focus |
| PFA | (para) formaldehyde |
| GA | glutaraldehyde |
| 53BP1 | p53-binding protein 1 |
| GFP | green fluorescent protein |
| FM | fluorescence microscope |
| LET | linear energy transfer |
| EC | euchromatin |
| EC-l | euchromatin-like |
| HC | heterochromatin |
| DCR | decondensed chromatin region |
| PDDF | persistent DNA damage focus |
| FLIM | fluorescence lifetime imaging microscopy |
| DAB | 3,3′-Diaminobenzidine |
| LLPS | liquate-liquate phase separation |
| SCB | sodium cacodylate buffer |
| PBS-/- | phosphate buffer saline without calcium and magnesium |
| Tb | terbium-citrate |
| FCS | fetal calf serum |
| DMEM | Dulbecco’s Modified Eagle Medium |
| ROI | region of interest |
References
- Zhou, K.; Gaullier, G.; Luger, K. Nucleosome structure and dynamics are coming of age. Nat. Struct. Mol. Biol. 2019, 26, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.C.; Connolly, M.; McDonald, C.J.; Pan, A.; Pryamkova, A.; Ray, K.; Seidel, E.; Tamura, S.; Rogge, R.; Maeshima, K. The 10-nm chromatin fiber and its relationship to interphase chromosome organization. Biochem. Soc. Trans. 2018, 46, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheutin, T.; Cavalli, G. The multiscale effects of polycomb mechanisms on 3D chromatin folding. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of genome folding into topologically associating domains. Sci. Adv. 2019, 5, eaaw1668. [Google Scholar] [CrossRef] [Green Version]
- Cremer, T.; Cremer, M.; Hübner, B.; Silahtaroglu, A.; Hendzel, M.; Lanctôt, C.; Strickfaden, H.; Cremer, C. The Interchromatin Compartment Participates in the Structural and Functional Organization of the Cell Nucleus. Bioessays 2020, 42, 1900132. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, A.A.; Jeggo, P.; Löbrich, M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair (Amst.) 2010, 9, 1273–1282. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jeggo, P.A. The heterochromatic barrier to DNA double strand break repair: How to get the entry visa. Int. J. Mol. Sci. 2012, 13, 11844–11860. [Google Scholar] [CrossRef] [Green Version]
- Fortuny, A.; Polo, S.E. The response to DNA damage in heterochromatin domains. Chromosoma 2018, 127, 291–300. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Lemaître, C.; Soutoglou, E. Double strand break (DSB) repair in heterochromatin and heterochromatin proteins in DSB repair. DNA Repair (Amst.) 2014, 19, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Marteijn, J.A.; Vermeulen, W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin 2012, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.-O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef] [PubMed]
- Ryu, T.; Spatola, B.; Delabaere, L.; Bowlin, K.; Hopp, H.; Kunitake, R.; Karpen, G.H.; Chiolo, I. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 2015, 17, 1401–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chagin, V.O.; Reinhart, B.; Becker, A.; Mortusewicz, O.; Jost, K.L.; Rapp, A.; Leonhardt, H.; Cardoso, M.C. Processive DNA synthesis is associated with localized decompaction of constitutive heterochromatin at the sites of DNA replication and repair. Nucleus 2019, 10, 231–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.; Lukásová, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [Green Version]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin compaction protects genomic DNA from radiation damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef] [Green Version]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Müller, I.; Merk, B.; Voss, K.O.; Averbeck, N.; Jakob, B.; Durante, M.; Taucher-Scholz, G. Species conserved DNA damage response at the inactive human X chromosome. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2013, 756, 30–36. [Google Scholar] [CrossRef]
- Abdollahi, E.; Taucher-Scholz, G.; Durante, M.; Jakob, B. Upgrading the GSI beamline microscope with a confocal fluorescence lifetime scanner to monitor charged particle induced chromatin decondensation in living cells. Nucl. Instrum. Methods Phys. Res. B 2015, 365, 626–630. [Google Scholar] [CrossRef]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA damage concentrated in particle trajectories causes persistent large-scale rearrangements in chromatin architecture. Radiother. Oncol. 2018, 129, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019, 38, e101379. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 2017, 357, eaag0025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Nin, G.H.; Biggiogera, M.; Echeverría, O.M. Activation of osmium ammine by SO2-generating chemicals for EM Feulgen-type staining of DNA. Eur. J. Histochem. 1995, 39, 101–106. [Google Scholar] [PubMed]
- Jakob, B.; Splinter, J.; Durante, M.; Taucher-Scholz, G. Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc. Natl. Acad. Sci. USA 2009, 106, 3172–3177. [Google Scholar] [CrossRef] [Green Version]
- Jakob, B.; Dubiak-Szepietowska, M.; Janiel, E.; Schmidt, A.; Durante, M.; Taucher-Scholz, G. Differential repair protein recruitment at sites of clustered and isolated DNA double-strand breaks produced by high-energy heavy ions. Sci. Rep. 2020, 10, 1443. [Google Scholar] [CrossRef]
- Lorat, Y.; Timm, S.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered double-strand breaks in heterochromatin perturb DNA repair after high linear energy transfer irradiation. Radiother. Oncol. 2016, 121, 154–161. [Google Scholar] [CrossRef]
- Smith, P.J.; Blunt, N.; Wiltshire, M.; Hoy, T.; Teesdale-Spittle, P.; Craven, M.R.; Watson, J.V.; Amos, W.B.; Errington, R.J.; Patterson, L.H. Characteristics of a novel deep red/infrared fluorescent cell-permeant DNA probe, DRAQ5, in intact human cells analyzed by flow cytometry, confocal and multiphoton microscopy. Cytometry 2000, 40, 280–291. [Google Scholar] [CrossRef]
- Heiß, M.; Fischer, B.E.; Jakob, B.; Fournier, C.; Becker, G.; Taucher-Scholz, G. Targeted irradiation of mammalian cells using a heavy-ion microprobe. Radiat. Res. 2006, 165, 231–239. [Google Scholar] [CrossRef]
- Gautier, A. Ultrastructural localization of DNA in ultrathin tissue sections. Int. Rev. Cytol. 1976, 44, 113–191. [Google Scholar] [CrossRef] [PubMed]
- Biggiogera, M.; Fakan, S. Fine structural specific visualization of RNA on ultrathin sections. J. Histochem. Cytochem. 1998, 46, 389–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata-Garrido, J.; Casafont, I.; Tapia, O.; Berciano, M.T.; Lafarga, M. Neuronal accumulation of unrepaired DNA in a novel specific chromatin domain: Structural, molecular and transcriptional characterization. Acta Neuropathol. Commun. 2016, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, K.; Eltsov, M.; Laemmli, U.K. Chromosome structure: Improved immunolabeling for electron microscopy. Chromosoma 2005, 114, 365–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fussner, E.; Ching, R.W.; Bazett-Jones, D.P. Living without 30 nm chromatin fibers. Trends Biochem. Sci. 2011, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, K.; Hihara, S.; Eltsov, M. Chromatin structure: Does the 30-nm fibre exist In Vivo? Curr. Opin. Cell Biol. 2010, 22, 291–297. [Google Scholar] [CrossRef]
- Olins, A.L.; Moyer, B.A.; Kim, S.H.; Allison, D.P. Synthesis of a more stable osmium ammine electron-dense DNA stain. J. Histochem. Cytochem. 1989, 37, 395–398. [Google Scholar] [CrossRef] [Green Version]
- Bernhard, W. A new staining procedure for electron microscopical cytology. J. Ultrastruct. Res. 1969, 27, 250–265. [Google Scholar] [CrossRef]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Strickfaden, H.; Xu, Z.Z.; Hendzel, M.J. Visualization of miniSOG Tagged DNA Repair Proteins in Combination with Electron Spectroscopic Imaging (ESI). J. Vis. Exp. 2015, 103, e52893. [Google Scholar] [CrossRef] [Green Version]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legartová, S.; Sehnalová, P.; Malyšková, B.; Küntziger, T.; Collas, P.; Cmarko, D.; Raška, I.; Sorokin, D.V.; Kozubek, S.; Bártová, E. Localized Movement and Levels of 53BP1 Protein Are Changed by γ-irradiation in PML Deficient Cells. J. Cell. Biochem. 2016, 117, 2583–2596. [Google Scholar] [CrossRef] [PubMed]
- Meers, C.; Keskin, H.; Storici, F. DNA repair by RNA: Templated, or not templated, that is the question. DNA Repair (Amst.) 2016, 44, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, A.; Tapryal, N.; Venkova, T.; Horikoshi, N.; Pandita, R.K.; Sarker, A.H.; Sarkar, P.S.; Hazra, T.K. Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes. Nat. Commun. 2016, 7, 13049. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Kruhlak, M.; Crouch, E.E.; Orlov, M.; Montaño, C.; Gorski, S.A.; Nussenzweig, A.; Misteli, T.; Phair, R.D.; Casellas, R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 2007, 447, 730–734. [Google Scholar] [CrossRef]
- Manfrini, N.; Clerici, M.; Wery, M.; Colombo, C.V.; Descrimes, M.; Morillon, A.; Di d’Adda Fagagna, F.; Longhese, M.P. Resection is responsible for loss of transcription around a double-strand break in Saccharomyces cerevisiae. Elife 2015, 4, e08942. [Google Scholar] [CrossRef]
- Averbeck, N.B.; Ringel, O.; Herrlitz, M.; Jakob, B.; Durante, M.; Taucher-Scholz, G. DNA end resection is needed for the repair of complex lesions in G1-phase human cells. Cell Cycle 2014, 13, 2509–2516. [Google Scholar] [CrossRef] [Green Version]
- Pryde, F.; Khalili, S.; Robertson, K.; Selfridge, J.; Ritchie, A.-M.; Melton, D.W.; Jullien, D.; Adachi, Y. 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J. Cell Sci. 2005, 118, 2043–2055. [Google Scholar] [CrossRef] [Green Version]
- Pessina, F.; Giavazzi, F.; Yin, Y.; Gioia, U.; Vitelli, V.; Galbiati, A.; Barozzi, S.; Garre, M.; Oldani, A.; Flaus, A.; et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol. 2019, 21, 1286–1299. [Google Scholar] [CrossRef]
- Michelini, F.; Pitchiaya, S.; Vitelli, V.; Sharma, S.; Gioia, U.; Pessina, F.; Cabrini, M.; Wang, Y.; Capozzo, I.; Iannelli, F.; et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 2017, 19, 1400–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, A.S.; Hawley, B.R.; Wilczynska, A.; Bushell, M. The roles of RNA in DNA double-strand break repair. Br. J. Cancer 2020, 122, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-H.; Hariharan, A.; Bastianello, G.; Toyama, Y.; Shivashankar, G.V.; Foiani, M.; Sheetz, M.P. DNA damage causes rapid accumulation of phosphoinositides for ATR signaling. Nat. Commun. 2017, 8, 2118. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, G. Fine Structure Immunocytochemistry; Springer: Berlin/Heidelberg, Germany, 1993; pp. 26–80. ISBN 978-3-642-77095-1. [Google Scholar]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005, 152, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Luther, P.K.; Lawrence, M.C.; Crowther, R.A. A method for monitoring the collapse of plastic sections as a function of electron dose. Ultramicroscopy 1988, 24, 7–18. [Google Scholar] [CrossRef]
- Braunfeld, M.B.; Koster, A.J.; Sedat, J.W.; Agard, D.A. Cryo automated electron tomography: Towards high-resolution reconstructions of plastic-embedded structures. J. Microsc. 1994, 174, 75–84. [Google Scholar] [CrossRef]
- Kremer, J.R.; Mastronarde, D.N.; McIntosh, J.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996, 116, 71–76. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonnemacher, S.; Eltsov, M.; Jakob, B. Correlative Light and Electron Microscopy (CLEM) Analysis of Nuclear Reorganization Induced by Clustered DNA Damage Upon Charged Particle Irradiation. Int. J. Mol. Sci. 2020, 21, 1911. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061911
Tonnemacher S, Eltsov M, Jakob B. Correlative Light and Electron Microscopy (CLEM) Analysis of Nuclear Reorganization Induced by Clustered DNA Damage Upon Charged Particle Irradiation. International Journal of Molecular Sciences. 2020; 21(6):1911. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061911
Chicago/Turabian StyleTonnemacher, Susanne, Mikhail Eltsov, and Burkhard Jakob. 2020. "Correlative Light and Electron Microscopy (CLEM) Analysis of Nuclear Reorganization Induced by Clustered DNA Damage Upon Charged Particle Irradiation" International Journal of Molecular Sciences 21, no. 6: 1911. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061911