Disruption of Exocytosis in Sympathoadrenal Chromaffin Cells from Mouse Models of Neurodegenerative Diseases

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Central and Peripheral Control of the Stress Fight-or-Flight Response
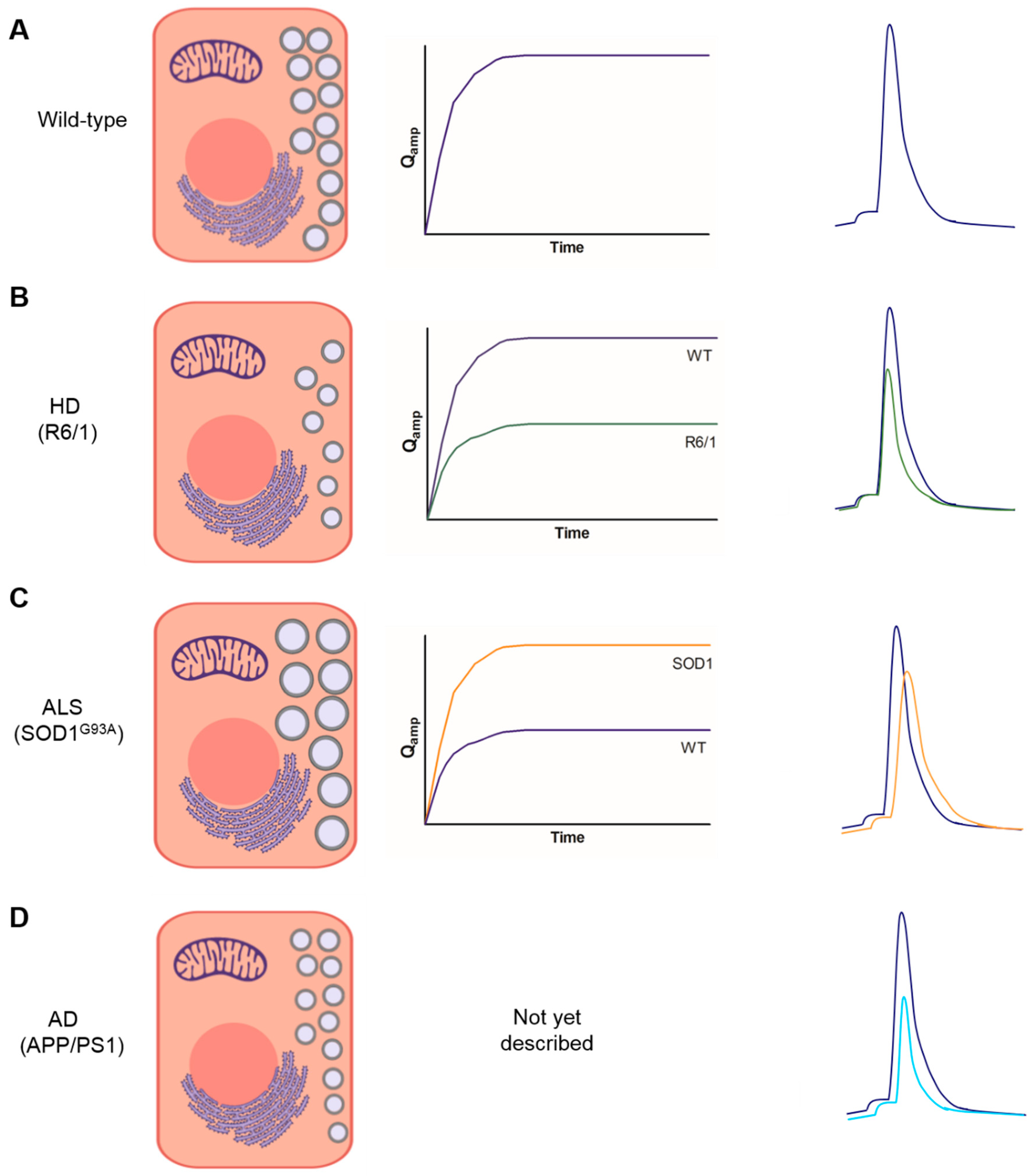
3. Bulk Release of Catecholamines from Chromaffin Cells of Mouse Models of NDDs
4. Kinetics of Exocytotic Fusion Pore, Pore Expansion, and Closure in CCs from Mouse Models of NDDs
5. Changes in Ion Currents and [Ca2+]c Signaling
6. Some Pathological Proteins Associated with NDDs Are Also Expressed in CCs to Modify Exocytosis
7. A Hypothesis on The Implication of The Sympathoadrenal Axis in The Pathogenesis of NDDs
8. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Smith-Dijak, A.I.; Sepers, M.D.; Raymond, L.A. Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 2019, 150, 346–365. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Manzano, R.; Vaz, R.; Osta, R.; Brites, D. Synaptic Failure: Focus in an Integrative View of ALS. Brain Plast. 2016, 1, 159–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tampellini, D. Synaptic activity and Alzheimer’s disease: A critical update. Front. Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, A.; Shen, J. Presenilins in synaptic function and disease. Trends Mol. Med. 2011, 17, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wu, B.; Beglopoulos, V.; Wines-Samuelson, M.; Zhang, D.; Dragatsis, I.; Südhof, T.C.; Shen, J. Presenilins are essential for regulating neurotransmitter release. Nature 2009, 460, 632–636. [Google Scholar] [CrossRef] [Green Version]
- Atwood, C.S.; Obrenovich, M.E.; Liu, T.; Chan, H.; Perry, G.; Smith, M.A.; Martins, R.N. Amyloid-beta: A chameleon walking in two worlds: A review of the trophic and toxic properties of amyloid-beta. Brain Res. Brain Res. Rev. 2003, 43, 1–16. [Google Scholar] [CrossRef]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Von Lewinski, F.; Keller, B.U. Ca2+, mitochondria and selective motoneuron vulnerability: Implications for ALS. Trends Neurosci. 2005, 28, 494–500. [Google Scholar] [CrossRef]
- Johnson, M.A.; Rajan, V.; Miller, C.E.; Wightman, R.M. Dopamine release is severely compromised in the R6/2 mouse model of Huntington’s disease. J. Neurochem. 2006, 97, 737–746. [Google Scholar] [CrossRef]
- Nicniocaill, B.; Haraldsson, B.; Hansson, O.; O’Connor, W.T.; Brundin, P. Altered striatal amino acid neurotransmitter release monitored using microdialysis in R6/1 Huntington transgenic mice. Eur. J. Neurosci. 2001, 13, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Cannon, W.B. Organization for Physiological Homeostasis. Physiol. Rev. 1929, 9, 399–431. [Google Scholar] [CrossRef]
- Hervonen, A.; Partanen, S.; Vaalasti, A.; Partanen, M.; Kanerva, L.; Alho, H. The distribution and endocrine nature of the abdominal paraganglia of adult man. Am. J. Anat. 1978, 153, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Coupland, R.E.; Pyper, A.S.; Hopwood, D. A Method for Differentiating between Noradrenaline- and Adrenaline-storing Cells in the Light and Electron Microscope. Nature 1964, 201, 1240–1242. [Google Scholar] [CrossRef]
- Hillarp, N.-Å.; Lagerstedt, S.; Nilson, B. The Isolation of n Granular Fraction from the Suprarenal Medulla, Containing the Symyathomimetic Catechol Amines 1. Acta Physiol. Scand. 1953, 29, 251–263. [Google Scholar] [CrossRef]
- Moro, M.A.; Garcia, A.G.; Langley, O.K. Characterization of two chromaffin cell populations isolated from bovine adrenal medulla. J. Neurochem. 1991, 57, 363–369. [Google Scholar] [CrossRef]
- Jansen, A.S.; Nguyen, X.V.; Karpitskiy, V.; Mettenleiter, T.C.; Loewy, A.D. Central command neurons of the sympathetic nervous system: Basis of the fight-or-flight response. Science 1995, 270, 644–646. [Google Scholar] [CrossRef]
- Bereiter, D.A.; Engeland, W.C.; Gann, D.S. Adrenal secretion of epinephrine after stimulation of trigeminal nucleus caudalis depends on stimulus pattern. Neuroendocrinology 1987, 45, 54–61. [Google Scholar] [CrossRef]
- Lindgren, P.; Rosen, A.; Uvnas, B. The release of catechols from the adrenal medulla on activation of the bulbar part of the sympathetic vasodilator outflow in cats. Acta Physiol. Scand. 1959, 47, 233–242. [Google Scholar] [CrossRef]
- Matsui, H. Effect of myelencephalic stimulation on the secretion of noradrenaline and adrenaline of the adrenal gland in the cat. Tohoku J. Exp. Med. 1965, 87, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Folkow, B.; Von Euler, U.S. Selective Activation of Noradrenaline and Adrenaline Producing Cells in the Cat’s Adrenal Gland by Hypothalamic Stimulation. Circ. Res. 1954, 2, 191–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, R.L.; Culberson, J.L.; Carmichael, S.W. Influence of hypothalamic stimulation on the secretion of adrenal medullary catecholamines. J. Auton. Nerv. Syst. 1983, 8, 89–96. [Google Scholar] [CrossRef]
- Von Euler, U.S.; Folkow, B. The effect of stimulation of autonomic areas in the cerebral cortex upon the adrenaline and noradrenaline secretion from the adrenal gland in the cat. Acta Physiol. Scand. 1958, 42, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Anderson, C.R.; Southwell, B.R.; McAllen, R.M. Distinct preganglionic neurons innervate noradrenaline and adrenaline cells in the cat adrenal medulla. Neuroscience 1996, 70, 825–832. [Google Scholar] [CrossRef]
- Morrison, S.F.; Cao, W.H. Different adrenal sympathetic preganglionic neurons regulate epinephrine and norepinephrine secretion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1763–R1775. [Google Scholar] [CrossRef] [Green Version]
- Wightman, R.M.; Jankowski, J.A.; Kennedy, R.T.; Kawagoe, K.T.; Schroeder, T.J.; Leszczyszyn, D.J.; Near, J.A.; Diliberto, E.J.; Viveros, O.H. Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc. Natl. Acad. Sci. USA 1991, 88, 10754–10758. [Google Scholar] [CrossRef] [Green Version]
- De Diego, A.M.G.; García, A.G. Altered exocytosis in chromaffin cells from mouse models of neurodegenerative diseases. Acta Physiol. (Oxf) 2018, 224, e13090. [Google Scholar] [CrossRef]
- Neher, E. Vesicle pools and Ca2+ microdomains: New tools for understanding their roles in neurotransmitter release. Neuron 1998, 20, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Ramírez, C.; Baraibar, A.M.; Nanclares, C.; Méndez-López, I.; Gómez, A.; Muñoz, M.P.; de Diego, A.M.G.; Gandía, L.; Casarejos, M.J.; García, A.G. Altered excitability and exocytosis in chromaffin cells from the R6/1 mouse model of Huntington’s disease is linked to over-expression of mutated huntingtin. J. Neurochem. 2018, 147, 454–476. [Google Scholar] [CrossRef]
- Calvo-Gallardo, E.; de Pascual, R.; Fernández-Morales, J.-C.; Arranz-Tagarro, J.-A.; Maroto, M.; Nanclares, C.; Gandía, L.; de Diego, A.M.G.; Padín, J.-F.; García, A.G. Depressed excitability and ion currents linked to slow exocytotic fusion pore in chromaffin cells of the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Am. J. Physiol. Cell Physiol. 2015, 308, C1–C19. [Google Scholar] [CrossRef] [Green Version]
- Chow, R.H.; von Rüden, L.; Neher, E. Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature 1992, 356, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Almers, W. Exocytosis. Annu. Rev. Physiol. 1990, 52, 607–624. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Hartung, K.; Langosch, D.; Rehm, H.; Bamberg, E.; Franke, W.W.; Betz, H. Identification of synaptophysin as a hexameric channel protein of the synaptic vesicle membrane. Science 1988, 242, 1050–1053. [Google Scholar] [CrossRef] [PubMed]
- De Diego, A.M.; Lorrio, S.; Calvo-Gallardo, E.; García, A.G. Smaller quantal size and faster kinetics of single exocytotic events in chromaffin cells from the APP/PS1 mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2012, 428, 482–486. [Google Scholar] [CrossRef]
- García, A.G.; García-De-Diego, A.M.; Gandía, L.; Borges, R.; García-Sancho, J. Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol. Rev. 2006, 86, 1093–1131. [Google Scholar] [CrossRef] [Green Version]
- Fumimura, Y.; Ikemura, M.; Saito, Y.; Sengoku, R.; Kanemaru, K.; Sawabe, M.; Arai, T.; Ito, G.; Iwatsubo, T.; Fukayama, M.; et al. Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in lewy body disease. J. Neuropathol. Exp. Neurol. 2007, 66, 354–362. [Google Scholar] [CrossRef] [Green Version]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.; Shen, J.; Zhang, Y.; Li, S.-H.; Li, X.-J.; Li, H. Immunohistochemical localization of huntingtin-associated protein 1 in endocrine system of the rat. J. Histochem. Cytochem. 2005, 53, 1517–1524. [Google Scholar] [CrossRef]
- Mackenzie, K.D.; Duffield, M.D.; Peiris, H.; Phillips, L.; Zanin, M.P.; Teo, E.H.; Zhou, X.-F.; Keating, D.J. Huntingtin-associated protein 1 regulates exocytosis, vesicle docking, readily releasable pool size and fusion pore stability in mouse chromaffin cells. J. Physiol. (Lond.) 2014, 592, 1505–1518. [Google Scholar] [CrossRef] [Green Version]
- Selye, H. Stress in Health and Disease; Elsevier Science: Saint Louis, MI, USA, 1976; ISBN 978-1-4831-9221-5. [Google Scholar]
- Carbone, E.; Borges, R.; Eiden, L.E.; García, A.G.; Hernández-Cruz, A. Chromaffin Cells of the Adrenal Medulla: Physiology, Pharmacology, and Disease. Compr. Physiol. 2019, 9, 1443–1502. [Google Scholar] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Diego, A.M.G.; Ortega-Cruz, D.; García, A.G. Disruption of Exocytosis in Sympathoadrenal Chromaffin Cells from Mouse Models of Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 1946. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061946
de Diego AMG, Ortega-Cruz D, García AG. Disruption of Exocytosis in Sympathoadrenal Chromaffin Cells from Mouse Models of Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(6):1946. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061946
Chicago/Turabian Stylede Diego, Antonio M. G., Diana Ortega-Cruz, and Antonio G. García. 2020. "Disruption of Exocytosis in Sympathoadrenal Chromaffin Cells from Mouse Models of Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 6: 1946. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061946