Role of HMGB1 in an Animal Model of Vascular Cognitive Impairment Induced by Chronic Cerebral Hypoperfusion

 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
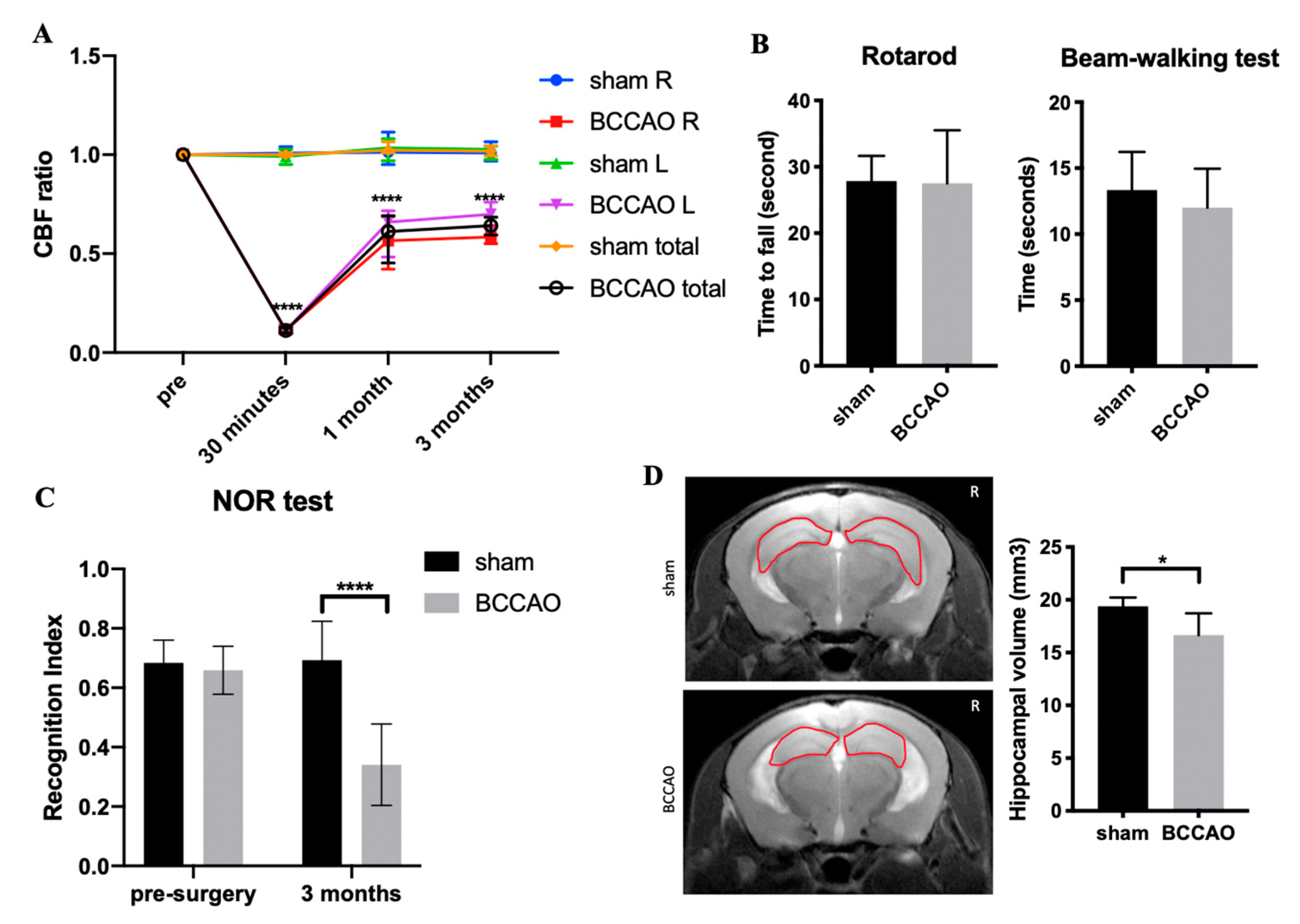
2.1. Survival Rate and CBF Values
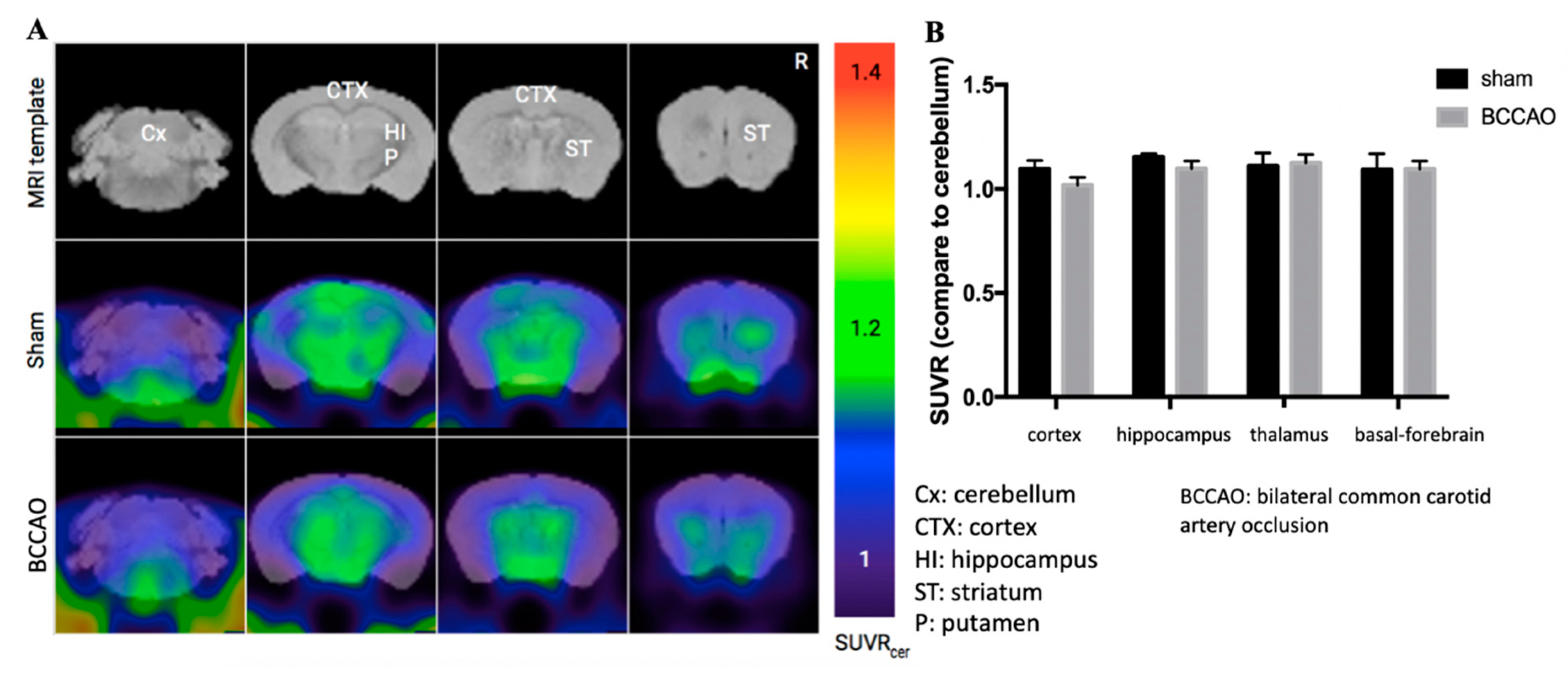
2.2. CCH Induced Hippocampal Atrophy and Memory Decline but Did Not Cause Behavioral Alterations in Motor Coordination
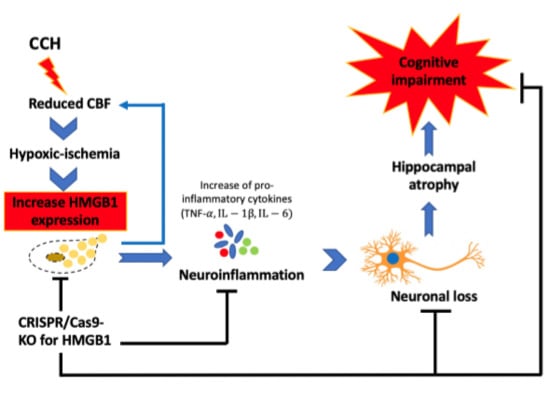
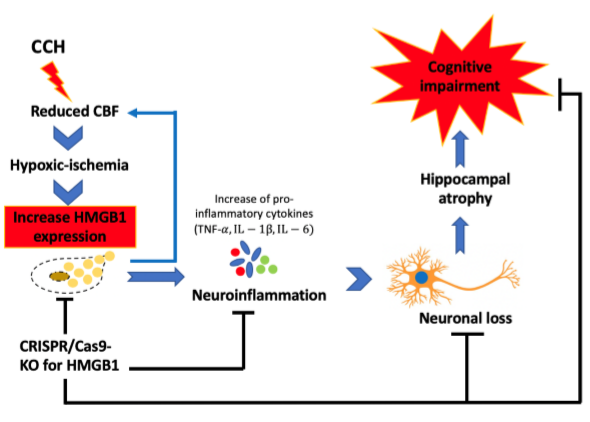
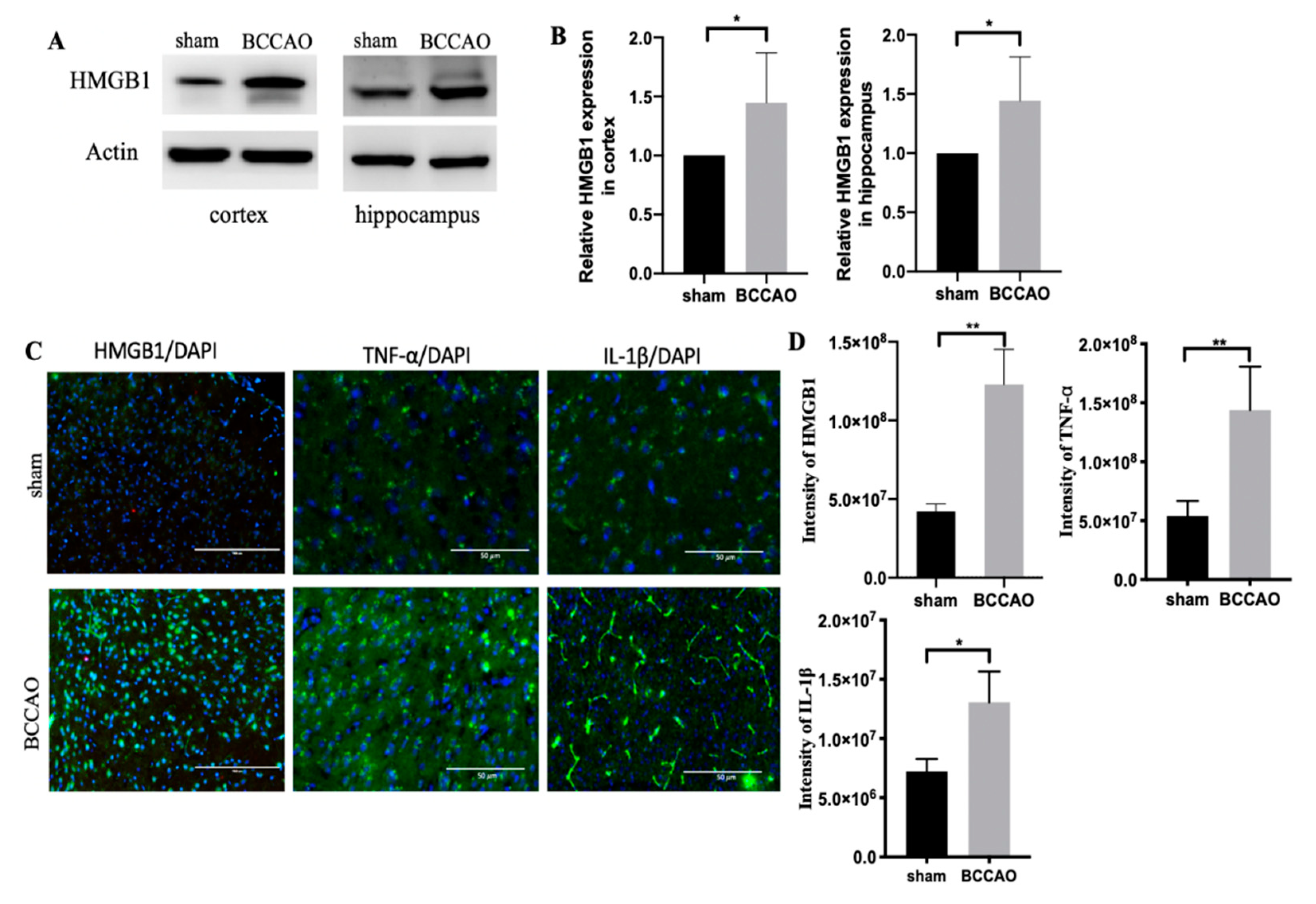
2.3. CCH Increased the Expression of HMGB1 and Its Proinflammatory Cytokines at 3 Months
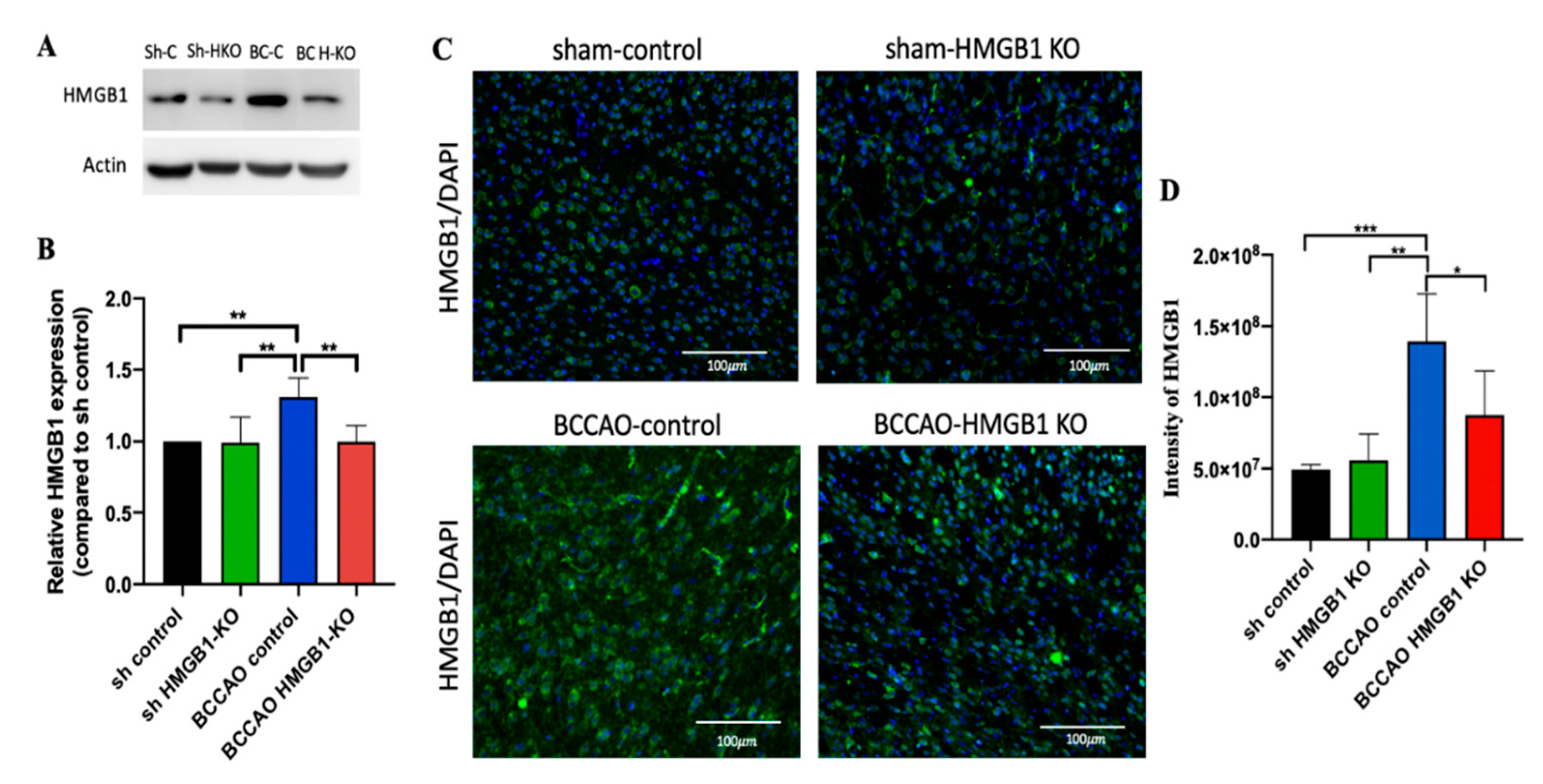
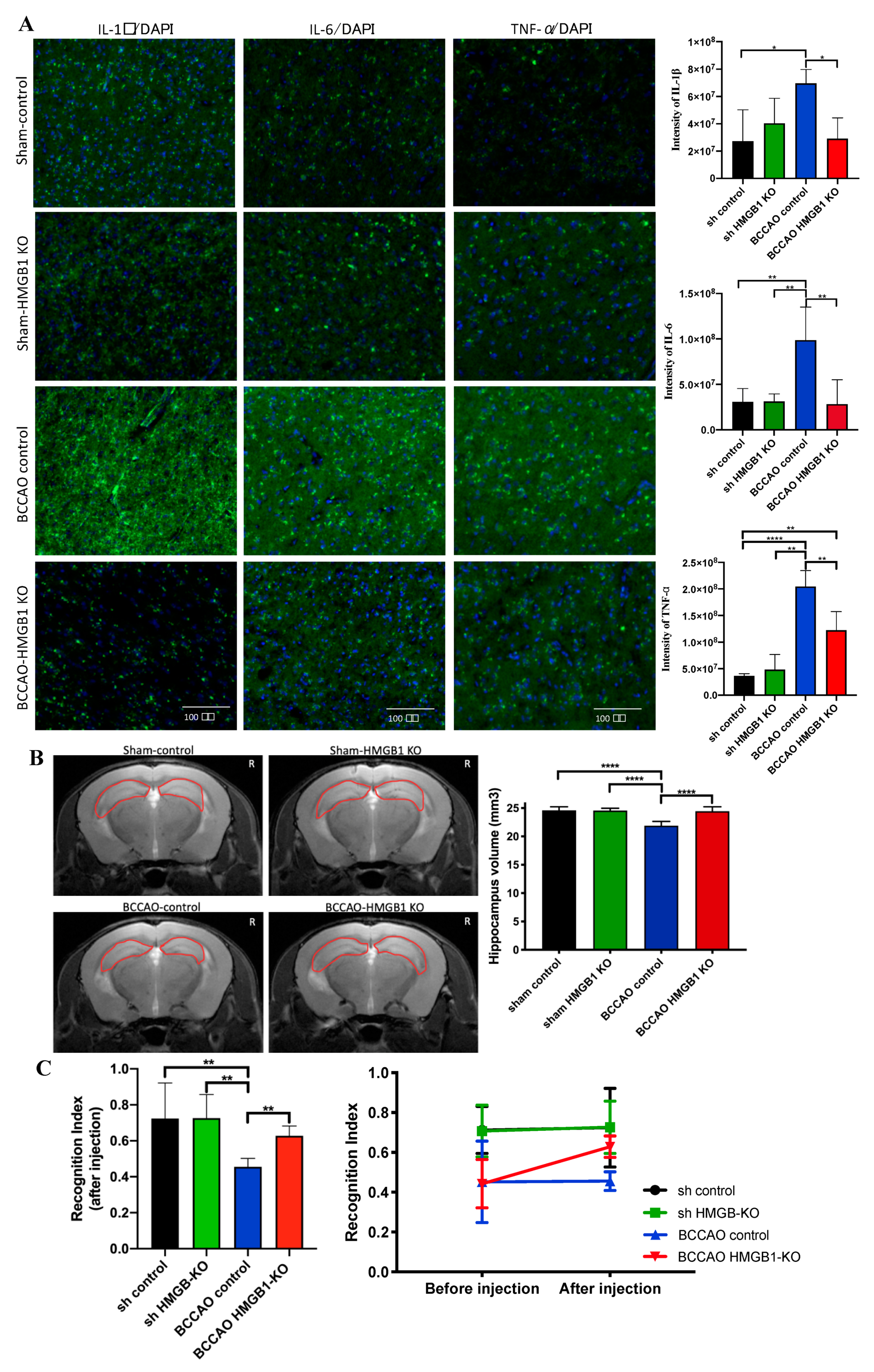
2.4. Administration of CRISPR/Cas9-KO of HMGB1 in BCCAO Mice Reduces the Expression of HMGB1 and Its Proinflammatory Cytokines, Attenuates Hippocampal Atrophy, and Improves Cognitive Decline
3. Discussion
4. Materials and Methods
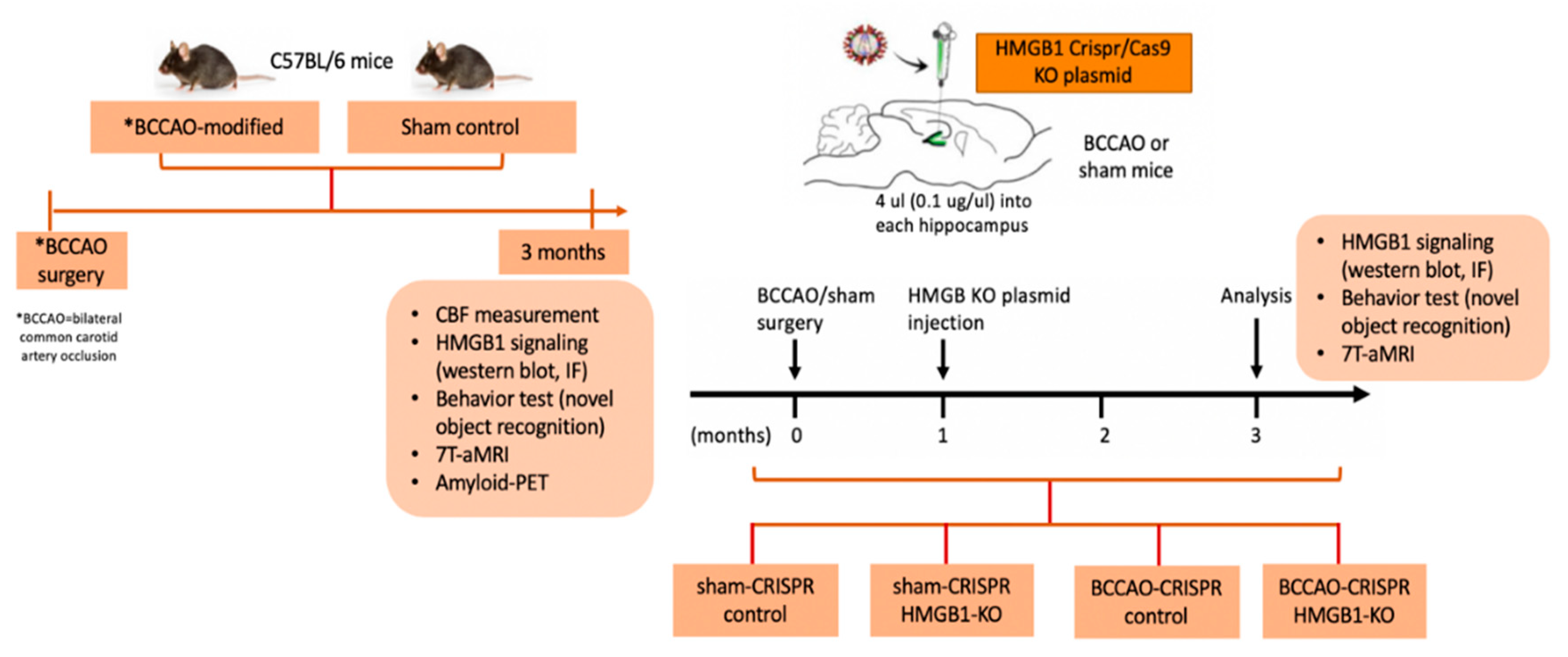
4.1. Animals and Study Design
4.2. Model Establishment of CCH through Modified BCCAO Surgery
4.3. Injection of HMGB1 CRISPR/Cas9-KO Plasmid
4.4. Novel Object Recognition
4.5. Motor Function Test
4.6. 7T-aMRI (7Tesla-Animal MRI)
4.7. Amyloid Positron Imaging Tomography Scanning
4.8. Western Blot
4.9. Immunostaining
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dichgans, M.; Leys, D. Vascular Cognitive Impairment. Circ. Res. 2017, 120, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; De Silva, T.M.; Chen, J.; Faraci, F.M. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ. Res. 2017, 120, 449–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncombe, J.; Kitamura, A.; Hase, Y.; Ihara, M.; Kalaria, R.N.; Horsburgh, K. Chronic cerebral hypoperfusion: A key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 2451–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Gu, J.-H.; Dai, C.-L.; Liu, Q.; Iqbal, K.; Liu, F.; Gong, C.-X. Chronic cerebral hypoperfusion causes decrease of O-GlcNAcylation, hyperphosphorylation of tau and behavioral deficits in mice. Front. Aging Neurosci. 2014, 6, 10. [Google Scholar] [CrossRef]
- Shibata, M.; Ohtani, R.; Ihara, M.; Tomimoto, H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 2004, 35, 2598–2603. [Google Scholar] [CrossRef] [Green Version]
- Farkas, E.; Luiten, P.G.; Bari, F. Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res. Rev. 2007, 54, 162–180. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, A.; Saito, S.; Maki, T.; Oishi, N.; Ayaki, T.; Hattori, Y.; Yamamoto, Y.; Urushitani, M.; Kalaria, R.N.; Fukuyama, H. Gradual cerebral hypoperfusion in spontaneously hypertensive rats induces slowly evolving white matter abnormalities and impairs working memory. J. Cereb. Blood Flow Metab. 2016, 36, 1592–1602. [Google Scholar] [CrossRef] [Green Version]
- Farkas, E.; Institóris, Á.; Domoki, F.; Mihály, A.; Bari, F. The effect of pre-and posttreatment with diazoxide on the early phase of chronic cerebral hypoperfusion in the rat. Brain Res. 2006, 1087, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Institoris, A.; Farkas, E.; Berczi, S.; Sule, Z.; Bari, F. Effects of cyclooxygenase (COX) inhibition on memory impairment and hippocampal damage in the early period of cerebral hypoperfusion in rats. Eur. J. Pharmacol. 2007, 574, 29–38. [Google Scholar] [CrossRef]
- Washida, K.; Hattori, Y.; Ihara, M. Animal Models of Chronic Cerebral Hypoperfusion: From Mouse to Primate. Int. J. Mol. Sci. 2019, 20, 6176. [Google Scholar] [CrossRef] [Green Version]
- Hattori, Y.; Enmi, J.-I.; Iguchi, S.; Saito, S.; Yamamoto, Y.; Nagatsuka, K.; Iida, H.; Ihara, M. Substantial reduction of parenchymal cerebral blood flow in mice with bilateral common carotid artery stenosis. Sci. Rep. 2016, 6, 32179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, Y.; Enmi, J.-I.; Iguchi, S.; Saito, S.; Yamamoto, Y.; Tsuji, M.; Nagatsuka, K.; Kalaria, R.N.; Iida, H.; Ihara, M. Gradual carotid artery stenosis in mice closely replicates hypoperfusive vascular dementia in humans. J. Am. Heart Assoc. 2016, 5, e002757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thong-Asa, W.; Tilokskulchai, K. Neuronal damage of the dorsal hippocampus induced by long-term right common carotid artery occlusion in rats. Iran. J. Basic Med Sci. 2014, 17, 220. [Google Scholar] [PubMed]
- Arundine, M.; Tymianski, M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 2003, 34, 325–337. [Google Scholar] [CrossRef]
- Martin, L.J.; Al-Abdulla, N.A.; Brambrink, A.M.; Kirsch, J.R.; Sieber, F.E.; Portera-Cailliau, C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res. Bull. 1998, 46, 281–309. [Google Scholar] [CrossRef]
- Dong, Y.-F.; Kataoka, K.; Toyama, K.; Sueta, D.; Koibuchi, N.; Yamamoto, E.; Yata, K.; Tomimoto, H.; Ogawa, H.; Kim-Mitsuyama, S. Attenuation of brain damage and cognitive impairment by direct renin inhibition in mice with chronic cerebral hypoperfusion. Hypertension 2011, 58, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Zeh, H.J., III; Lotze, M.T. High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal. 2011, 14, 1315–1335. [Google Scholar] [CrossRef] [Green Version]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331. [Google Scholar] [CrossRef]
- De Souza, A.; Westra, J.; Limburg, P.; Bijl, M.; Kallenberg, C. HMGB1 in vascular diseases: Its role in vascular inflammation and atherosclerosis. Autoimmun. Rev. 2012, 11, 909–917. [Google Scholar] [CrossRef]
- Fujita, K.; Motoki, K.; Tagawa, K.; Chen, X.; Hama, H.; Nakajima, K.; Homma, H.; Tamura, T.; Watanabe, H.; Katsuno, M. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci. Rep. 2016, 6, 31895. [Google Scholar] [CrossRef] [Green Version]
- Festoff, B.W.; Sajja, R.K.; van Dreden, P.; Cucullo, L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hei, Y.; Chen, R.; Yi, X.; Long, Q.; Gao, D.; Liu, W. HMGB1 neutralization attenuates hippocampal neuronal death and cognitive impairment in rats with chronic cerebral hypoperfusion via suppressing inflammatory responses and oxidative stress. Neuroscience 2018, 383, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Manns, J.R.; Eichenbaum, H. A cognitive map for object memory in the hippocampus. Learn. Mem. 2009, 16, 616–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driscoll, I.; Hamilton, D.A.; Petropoulos, H.; Yeo, R.A.; Brooks, W.M.; Baumgartner, R.N.; Sutherland, R.J. The aging hippocampus: Cognitive, biochemical and structural findings. Cereb. Cortex 2003, 13, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012, 13, 93–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denninger, J.K.; Smith, B.M.; Kirby, E.D. Novel Object Recognition and Object Location Behavioral Testing in Mice on a Budget. J. Vis. Exp. 2018, e58593. [Google Scholar] [CrossRef]
- Mueller, S.G.; Schuff, N.; Yaffe, K.; Madison, C.; Miller, B.; Weiner, M.W. Hippocampal atrophy patterns in mild cognitive impairment and Alzheimer’s disease. Hum. Brain Mapp. 2010, 31, 1339–1347. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Yamashita, T.; Zhai, Y.; Nakano, Y.; Morihara, R.; Li, X.; Tian, F.; Liu, X.; Huang, Y.; Shi, X. Acceleration of NLRP3 inflammasome by chronic cerebral hypoperfusion in Alzheimer’s disease model mouse. Neurosci. Res. 2019, 143, 61–70. [Google Scholar] [CrossRef]
- Ihara, M.; Tomimoto, H. Lessons from a mouse model characterizing features of vascular cognitive impairment with white matter changes. J. Aging Res. 2011, 2011, 978761. [Google Scholar] [CrossRef]
- Wang, L.; Du, Y.; Wang, K.; Xu, G.; Luo, S.; He, G. Chronic cerebral hypoperfusion induces memory deficits and facilitates Aβ generation in C57BL/6J mice. Exp. Neurol. 2016, 283, 353–364. [Google Scholar] [CrossRef]
- Ashok, A.; Rai, N.K.; Raza, W.; Pandey, R.; Bandyopadhyay, S. Chronic cerebral hypoperfusion-induced impairment of Aβ clearance requires HB-EGF-dependent sequential activation of HIF1α and MMP9. Neurobiol. Dis. 2016, 95, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Salvadores, N.; Searcy, J.L.; Holland, P.R.; Horsburgh, K. Chronic cerebral hypoperfusion alters amyloid-β peptide pools leading to cerebral amyloid angiopathy, microinfarcts and haemorrhages in Tg-SwDI mice. Clin. Sci. 2017, 131, 2109–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannai, T.; Iwata, A.; Mano, T.; Ohtomo, R.; Ohtomo, G.; Hashimoto, T.; Iwatsubo, T.; Toda, T. Chronic cerebral hypoperfusion increases amyloid plaques by accelerating amyloid beta aggregation in app/ps1 transgenic mice. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, P693–P694. [Google Scholar] [CrossRef]
- Du, S.-Q.; Wang, X.-R.; Xiao, L.-Y.; Tu, J.-F.; Zhu, W.; He, T.; Liu, C.-Z. Molecular mechanisms of vascular dementia: What can be learned from animal models of chronic cerebral hypoperfusion? Mol. Neurobiol. 2017, 54, 3670–3682. [Google Scholar] [CrossRef]
- Neto, C.J.B.F.; Paganelli, R.A.; Benetoli, A.; Lima, K.C.M.; Milani, H. Permanent, 3-stage, 4-vessel occlusion as a model of chronic and progressive brain hypoperfusion in rats: A neurohistological and behavioral analysis. Behav. Brain Res. 2005, 160, 312–322. [Google Scholar] [CrossRef]
- Kitaguchi, H.; Tomimoto, H.; Ihara, M.; Shibata, M.; Uemura, K.; Kalaria, R.N.; Kihara, T.; Asada-Utsugi, M.; Kinoshita, A.; Takahashi, R. Chronic cerebral hypoperfusion accelerates amyloid β deposition in APPSwInd transgenic mice. Brain Res. 2009, 1294, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Yoshizaki, K.; Adachi, K.; Kataoka, S.; Watanabe, A.; Tabira, T.; Takahashi, K.; Wakita, H. Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp. Neurol. 2008, 210, 585–591. [Google Scholar] [CrossRef]
- Cohen, S.J.; Munchow, A.H.; Rios, L.M.; Zhang, G.; Ásgeirsdóttir, H.N.; Stackman, R.W., Jr. The rodent hippocampus is essential for nonspatial object memory. Curr. Biol. 2013, 23, 1685–1690. [Google Scholar] [CrossRef] [Green Version]
- Kinnavane, L.; Amin, E.; Olarte-Sánchez, C.M.; Aggleton, J.P. Detecting and discriminating novel objects: The impact of perirhinal cortex disconnection on hippocampal activity patterns. Hippocampus 2016, 26, 1393–1413. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Kitagawa, K.; Matsushita, K.; Mabuchi, T.; Yagita, Y.; Yanagihara, T.; Matsumoto, M. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: Selective neuronal death in the murine transient forebrain ischemia. Brain Res. 1997, 752, 209–218. [Google Scholar] [CrossRef]
- Wahul, A.B.; Joshi, P.C.; Kumar, A.; Chakravarty, S. Transient global cerebral ischemia differentially affects cortex, striatum and hippocampus in Bilateral Common Carotid Arterial occlusion (BCCAo) mouse model. J. Chem. Neuroanat. 2018, 92, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Somredngan, S.; Thong-asa, W. Neurological Changes in Vulnerable Brain Areas of Chronic Cerebral Hypoperfusion Mice. Ann. Neurosci. 2017, 24, 233–242. [Google Scholar] [PubMed]
- Gooch, J.; Wilcock, D.M. Animal models of vascular cognitive impairment and dementia (VCID). Cell. Mol. Neurobiol. 2016, 36, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hou, T.; Wang, W.; Luo, Y.; Yan, F.; Jia, J. The effect of chronic cerebral hypoperfusion on amyloid-β metabolism in a transgenic mouse model of Alzheimer’s disease (PS1V97L). J. Alzheimer’s Dis. 2018, 62, 1609–1621. [Google Scholar] [CrossRef]
- Miyamoto, O.; Auer, R. Hypoxia, hyperoxia, ischemia, and brain necrosis. Neurology 2000, 54, 362. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Yang, H.; Harris, H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin. Immunol. 2018, 38, 40–48. [Google Scholar] [CrossRef]
- Bennett, S.A.; Tenniswood, M.; Chen, J.-H.; Davidson, C.M.; Keyes, M.T.; Fortin, T.; Pappas, B.A. Chronic cerebral hypoperfusion elicits neuronal apoptosis and behavioral impairment. Neuroreport 1998, 9, 161–166. [Google Scholar] [CrossRef]
- Kim, J.-B.; Choi, J.S.; Yu, Y.-M.; Nam, K.; Piao, C.-S.; Kim, S.-W.; Lee, M.-H.; Han, P.-L.; Park, J.-S.; Lee, J.-K. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 2006, 26, 6413–6421. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Mishima, K.; Nozako, M.; Hazekawa, M.; Mishima, S.; Fujioka, M.; Orito, K.; Egashira, N.; Iwasaki, K.; Fujiwara, M. Delayed treatment with minocycline ameliorates neurologic impairment through activated microglia expressing a high-mobility group box1–inhibiting mechanism. Stroke 2008, 39, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A. The HMGB1 receptor RAGE mediates ischemic brain damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Ko, P.W.; Lee, H.W.; Jeong, J.Y.; Lee, M.G.; Kim, J.H.; Lee, W.H.; Yu, R.; Oh, W.J.; Suk, K. Astrocyte-derived lipocalin-2 mediates hippocampal damage and cognitive deficits in experimental models of vascular dementia. Glia 2017, 65, 1471–1490. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, J.; Shang, Y.; Zhao, L.; Wang, M.; Shi, J.; Li, S. HMGB1-TLR4 axis plays a regulatory role in the pathogenesis of mesial temporal lobe epilepsy in immature rat model and children via the p38MAPK signaling pathway. Neurochem. Res. 2017, 42, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Speetzen, L.J.; Endres, M.; Kunz, A. Bilateral common carotid artery occlusion as an adequate preconditioning stimulus to induce early ischemic tolerance to focal cerebral ischemia. JoVE (J. Vis. Exp.) 2013, e4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Nan, D.; He, Q.; Yang, L.; Guo, H. Astrocyte activation and capillary remodeling in modified bilateral common carotid artery occlusion mice. Microcirculation 2017, 24, e12366. [Google Scholar] [CrossRef]
- Venkat, P.; Chopp, M.; Chen, J. Models and mechanisms of vascular dementia. Exp. Neurol. 2015, 272, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Jiwa, N.S.; Garrard, P.; Hainsworth, A.H. Experimental models of vascular dementia and vascular cognitive impairment: A systematic review. J. Neurochem. 2010, 115, 814–828. [Google Scholar] [CrossRef]
- Wang, M.; Iliff, J.J.; Liao, Y.; Chen, M.J.; Shinseki, M.S.; Venkataraman, A.; Cheung, J.; Wang, W.; Nedergaard, M. Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. J. Neurosci. 2012, 32, 17948–17960. [Google Scholar] [CrossRef]
- Luong, T.N.; Carlisle, H.J.; Southwell, A.; Patterson, P.H. Assessment of motor balance and coordination in mice using the balance beam. JoVE (J. Vis. Exp.) 2011, e2376. [Google Scholar] [CrossRef] [Green Version]
- Tung, V.W.; Burton, T.J.; Dababneh, E.; Quail, S.L.; Camp, A.J. Behavioral assessment of the aging mouse vestibular system. JoVE (J. Vis. Exp.) 2014, e51605. [Google Scholar] [CrossRef] [Green Version]
- Franklin, K.B.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates; Academic Press: New York, NY, USA, 2008; Volume 3. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidyanti, A.N.; Hsieh, J.-Y.; Lin, K.-J.; Fang, Y.-C.; Setyopranoto, I.; Hu, C.-J. Role of HMGB1 in an Animal Model of Vascular Cognitive Impairment Induced by Chronic Cerebral Hypoperfusion. Int. J. Mol. Sci. 2020, 21, 2176. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062176
Vidyanti AN, Hsieh J-Y, Lin K-J, Fang Y-C, Setyopranoto I, Hu C-J. Role of HMGB1 in an Animal Model of Vascular Cognitive Impairment Induced by Chronic Cerebral Hypoperfusion. International Journal of Molecular Sciences. 2020; 21(6):2176. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062176
Chicago/Turabian StyleVidyanti, Amelia Nur, Jia-Yu Hsieh, Kun-Ju Lin, Yao-Ching Fang, Ismail Setyopranoto, and Chaur-Jong Hu. 2020. "Role of HMGB1 in an Animal Model of Vascular Cognitive Impairment Induced by Chronic Cerebral Hypoperfusion" International Journal of Molecular Sciences 21, no. 6: 2176. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062176