Fli-1 Activation through Targeted Promoter Activity Regulation Using a Novel 3’, 5’-diprenylated Chalcone Inhibits Growth and Metastasis of Prostate Cancer Cells

 ,
,
Abstract
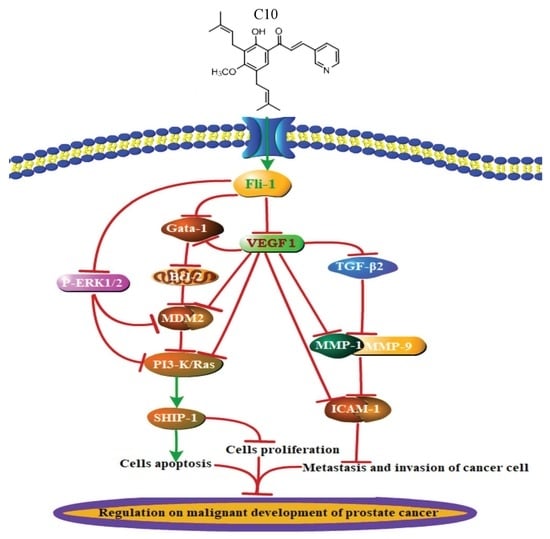
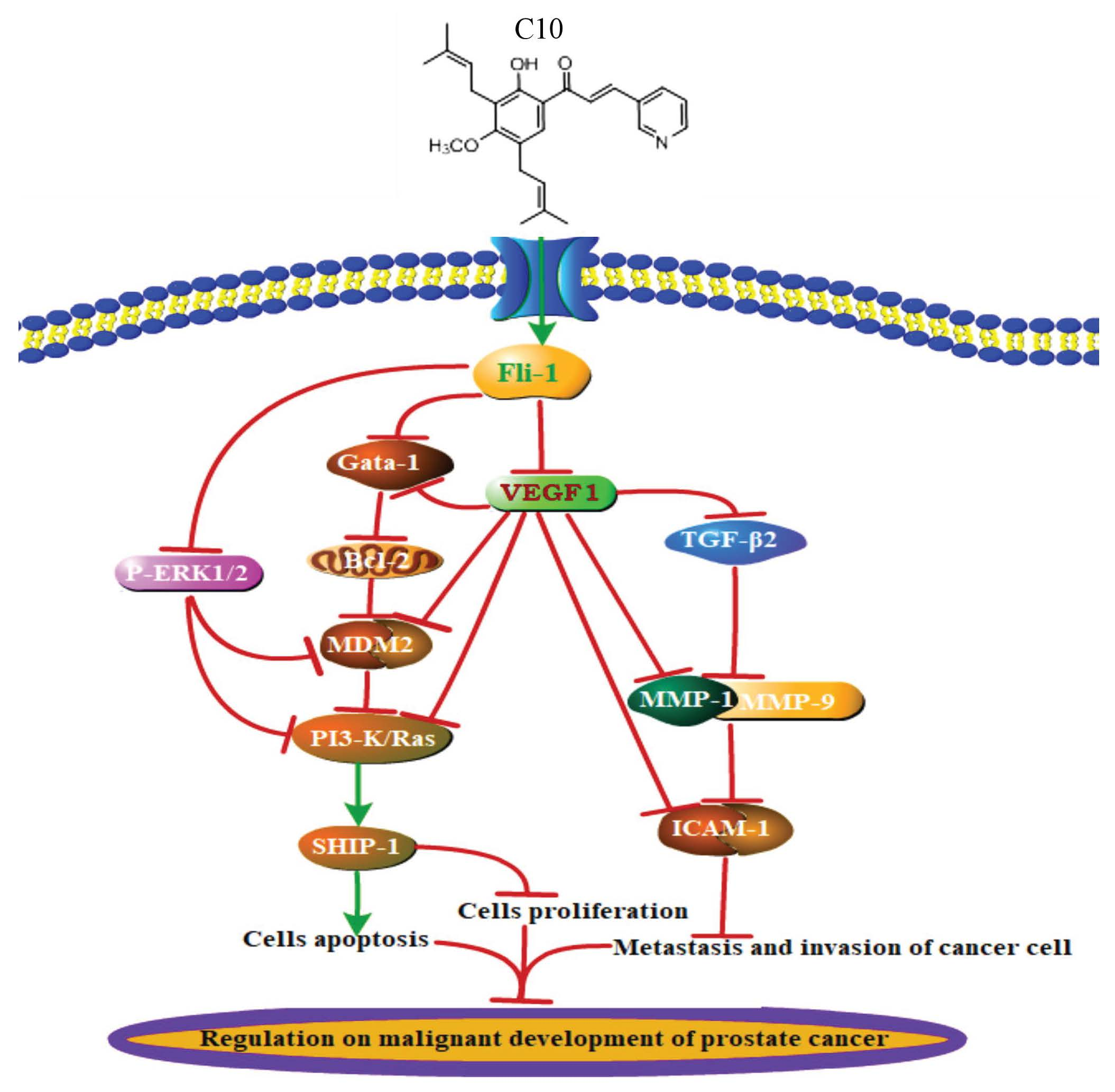
:
1. Introduction
2. Results
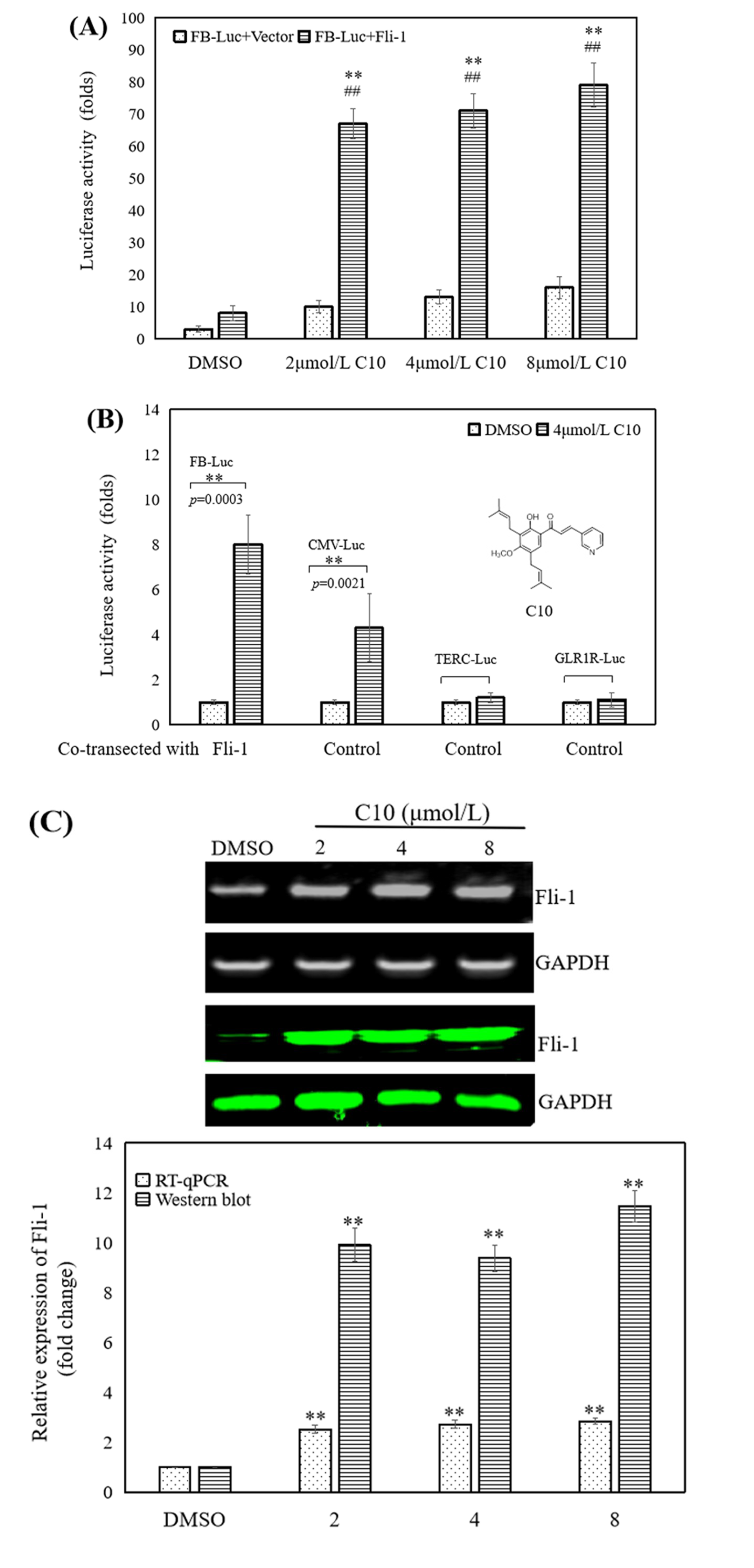
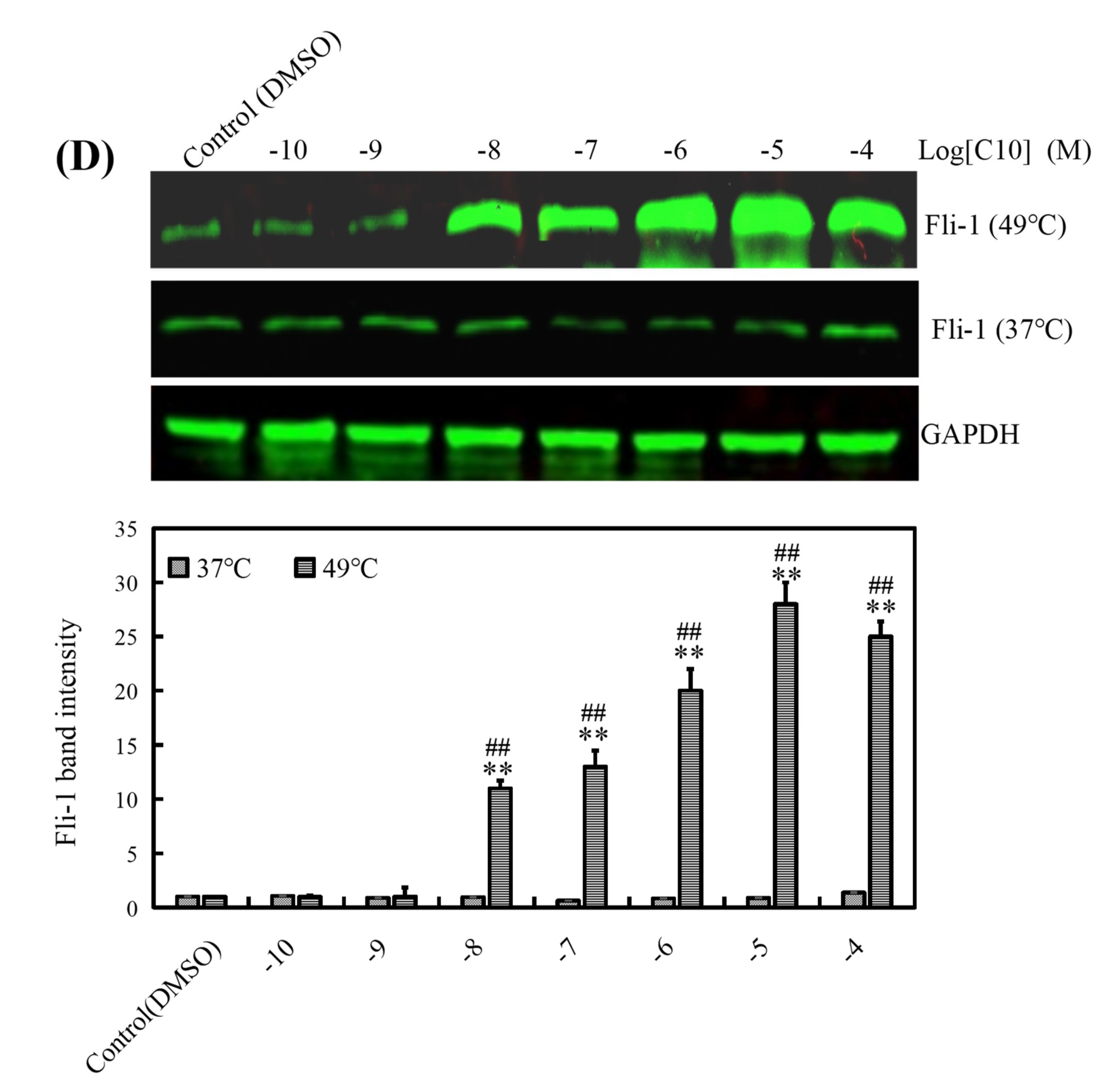
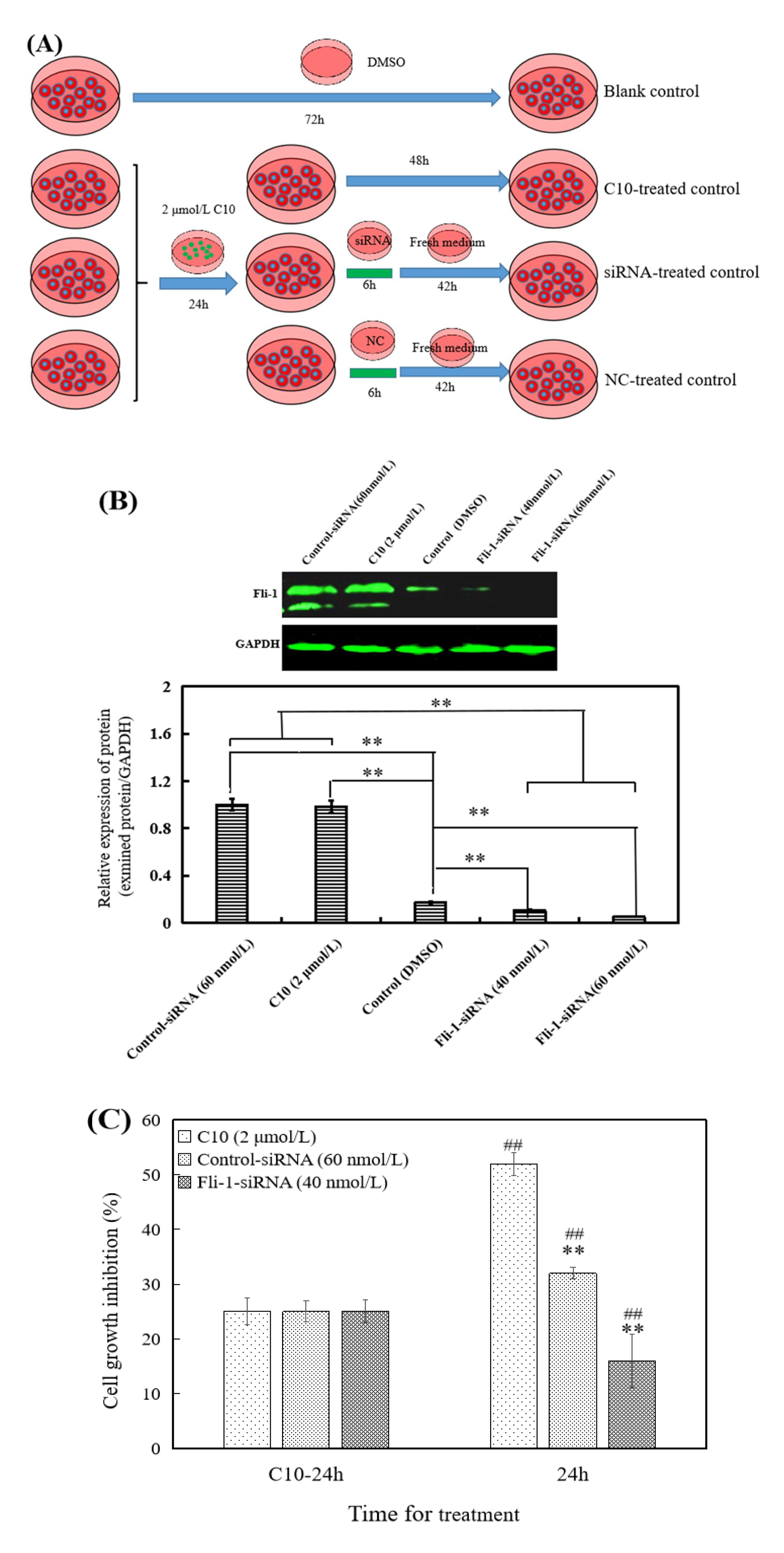
2.1. C10 Promoted the Fli-1 Expression in PC3 Cells
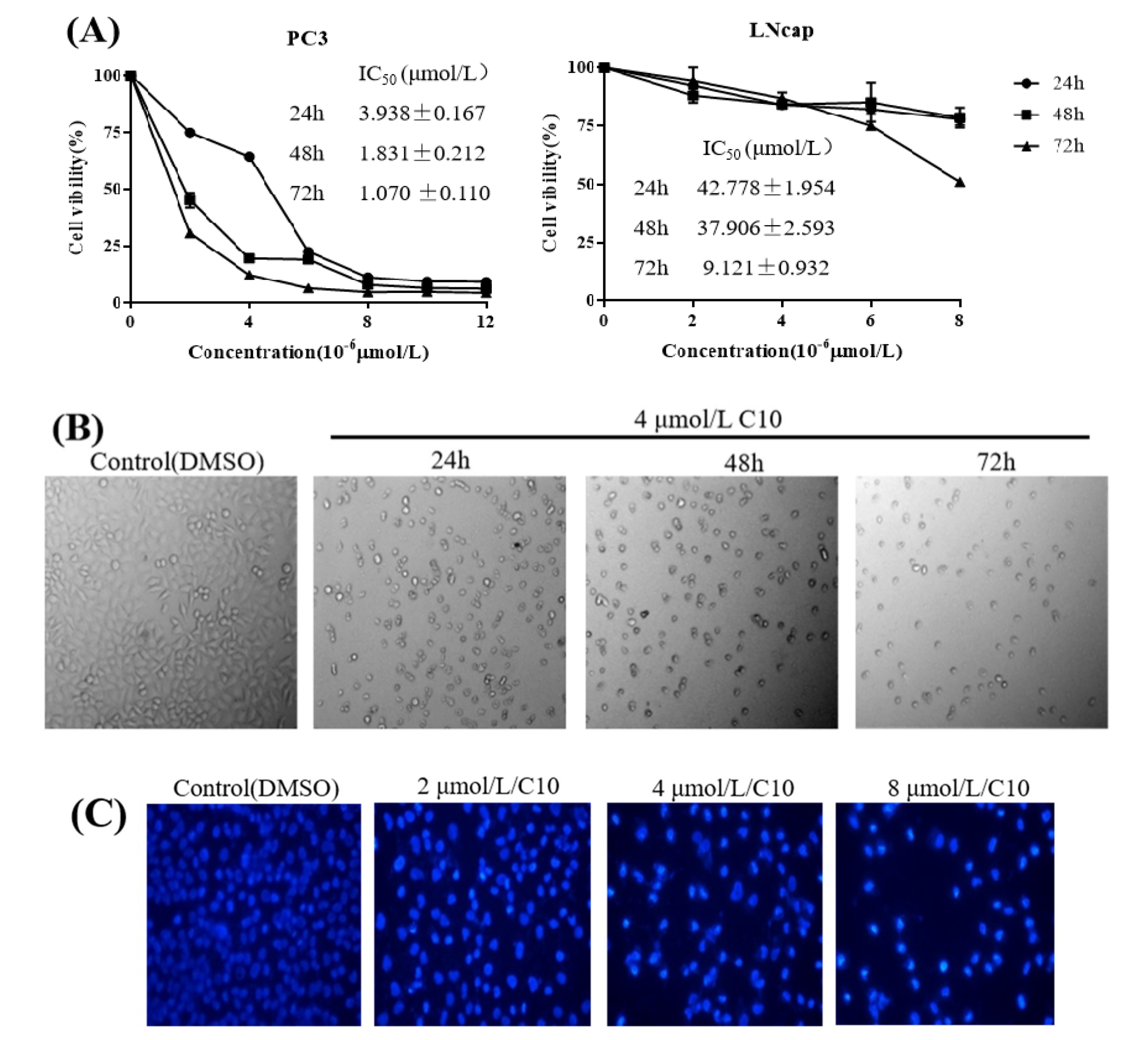
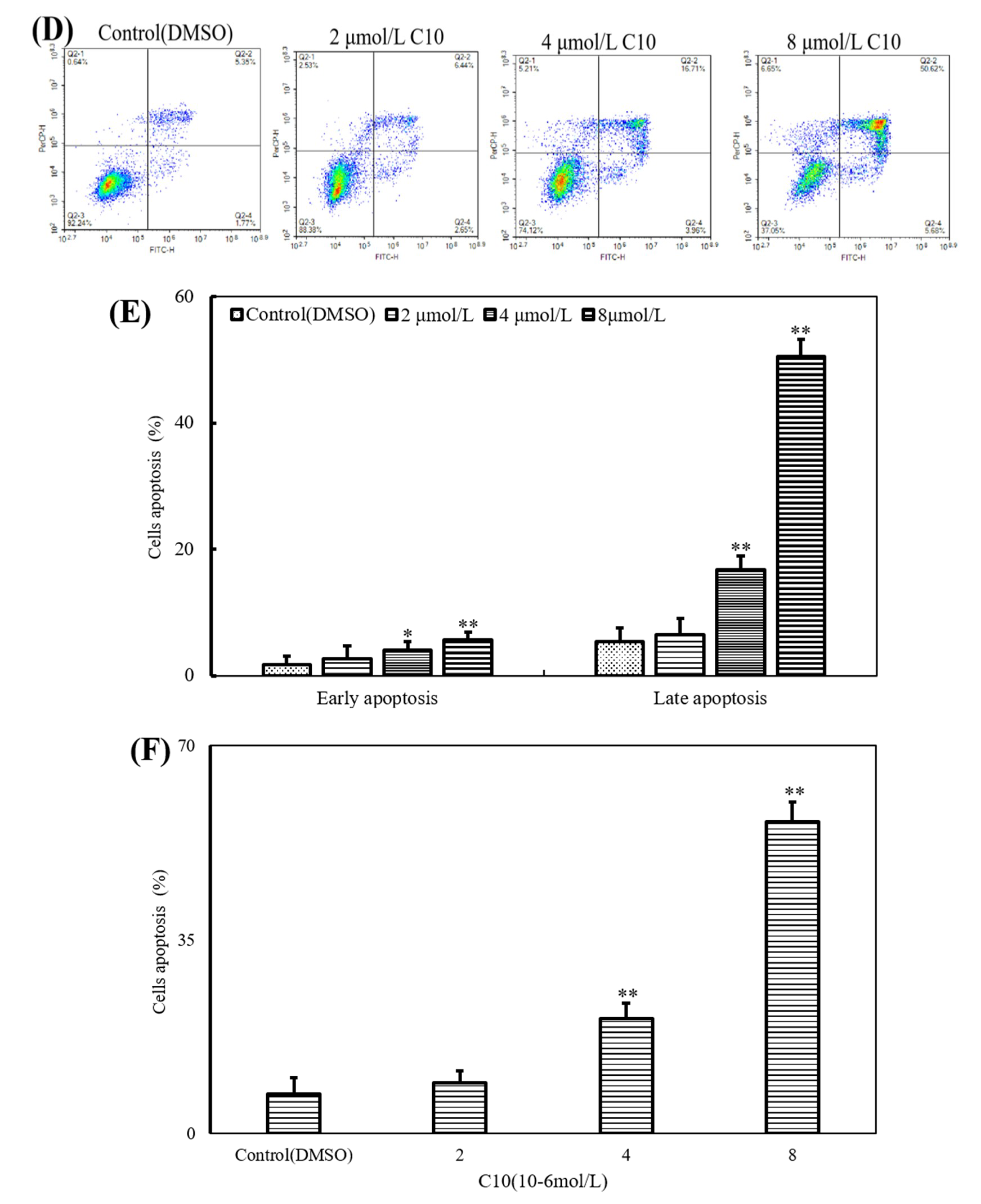
2.2. C10 Inhibited the Proliferation of PC3 Cells by Inducing Apoptosis
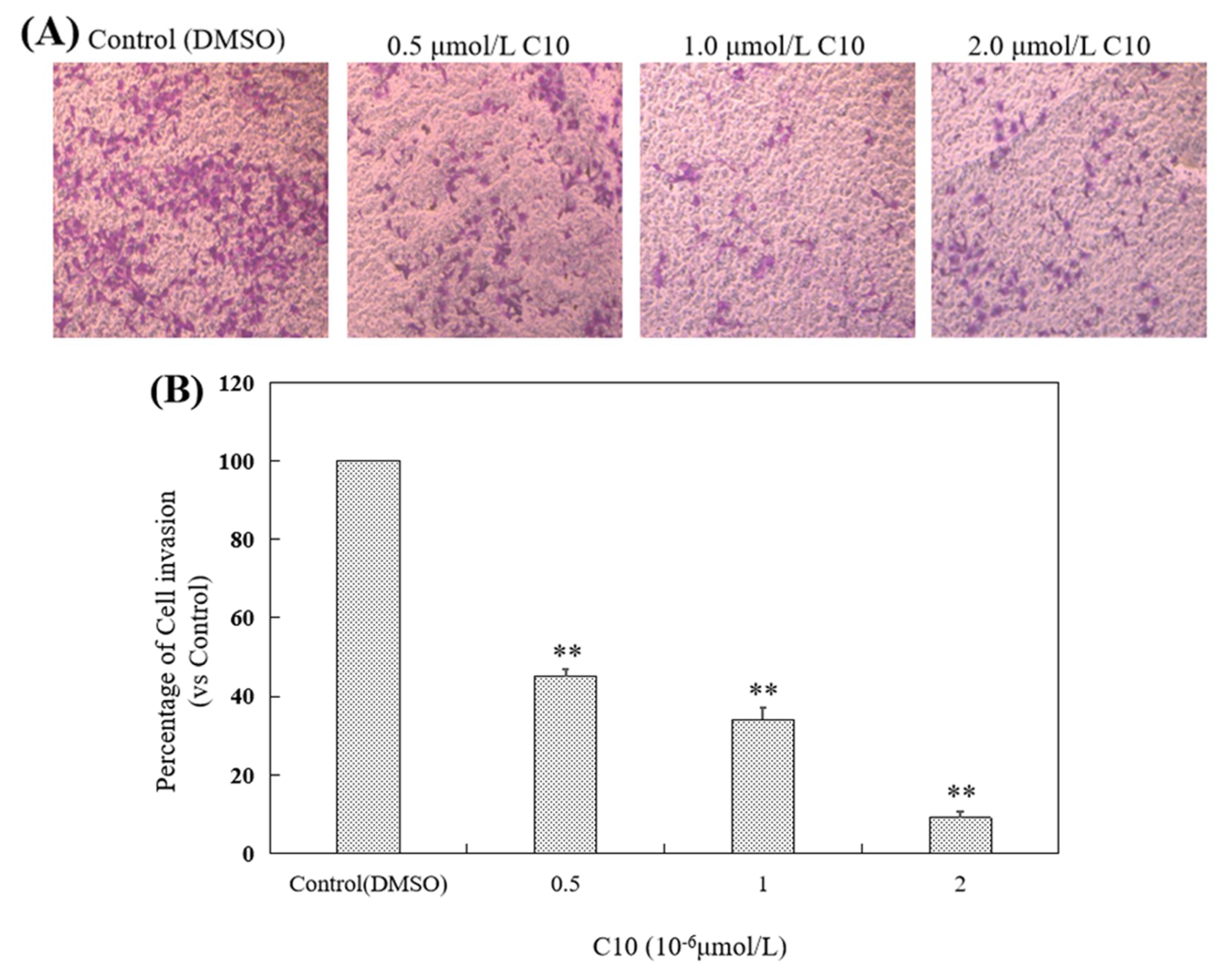
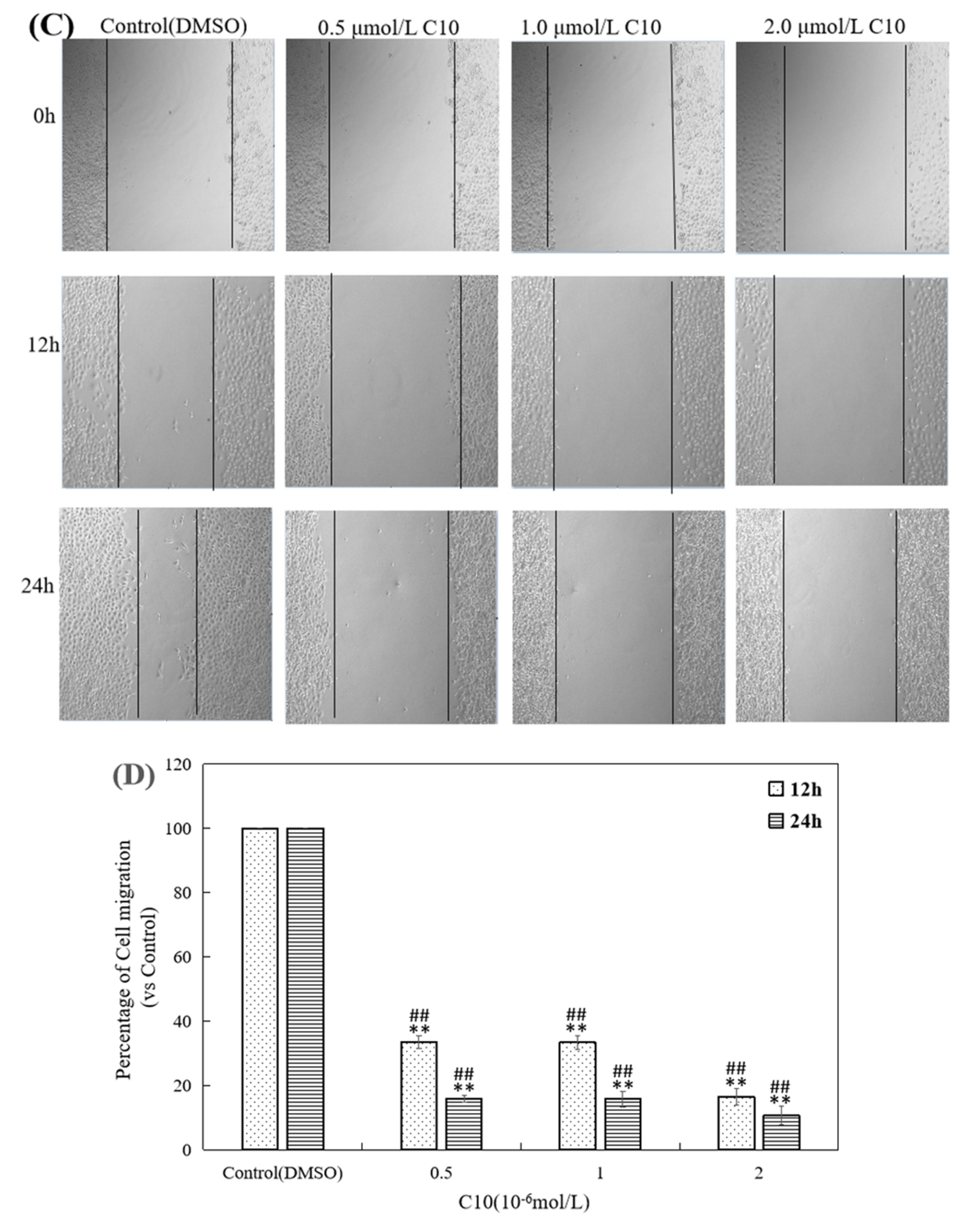
2.3. Inhibitory Effect of C10 on Migration and Invasion of PC3 Cells
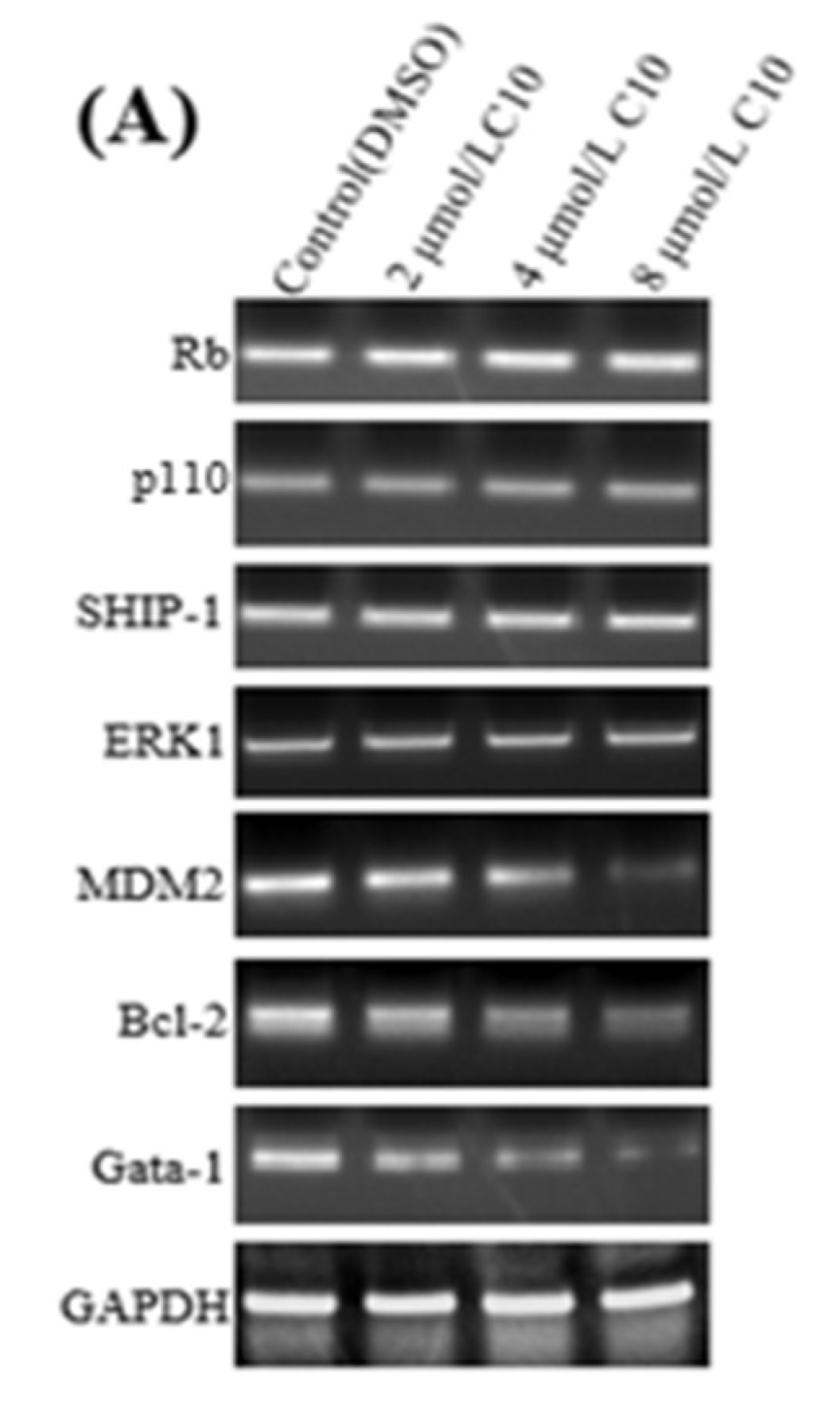
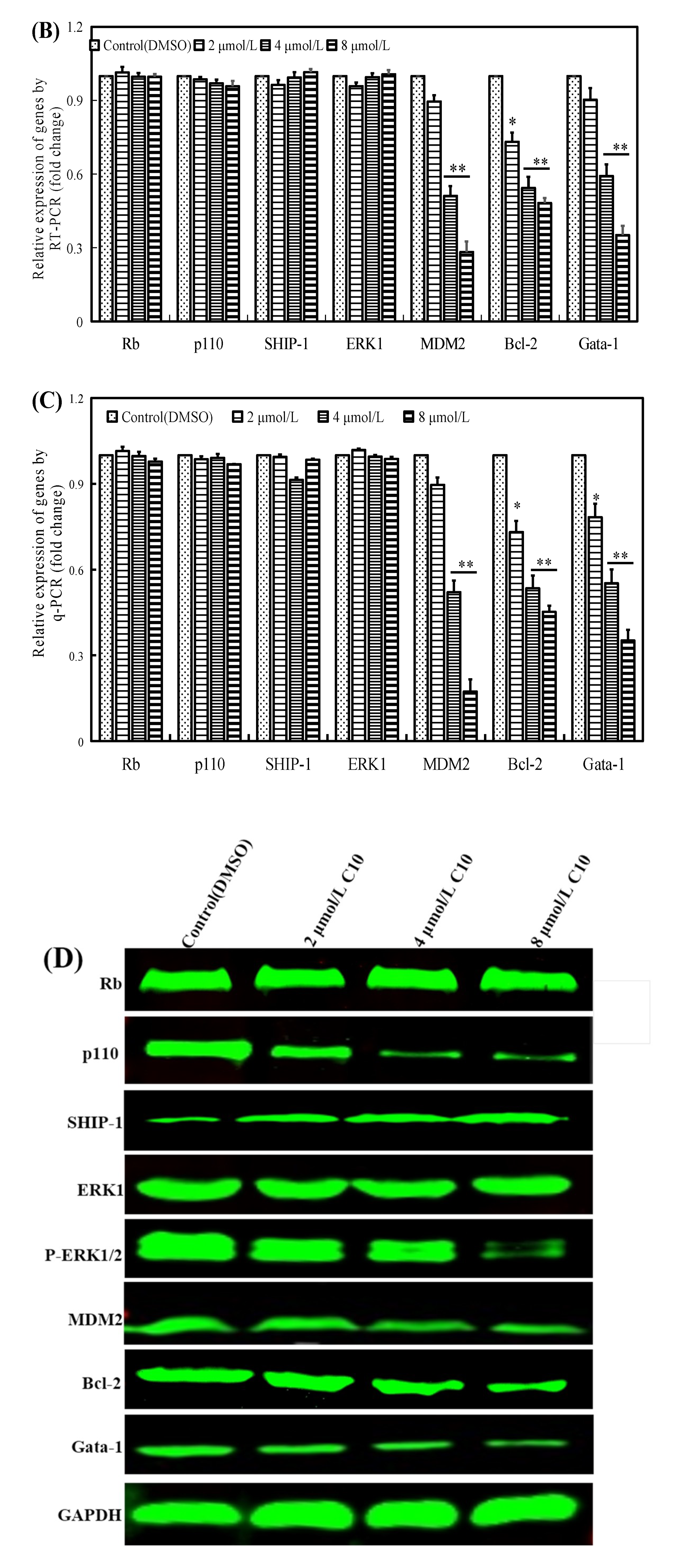
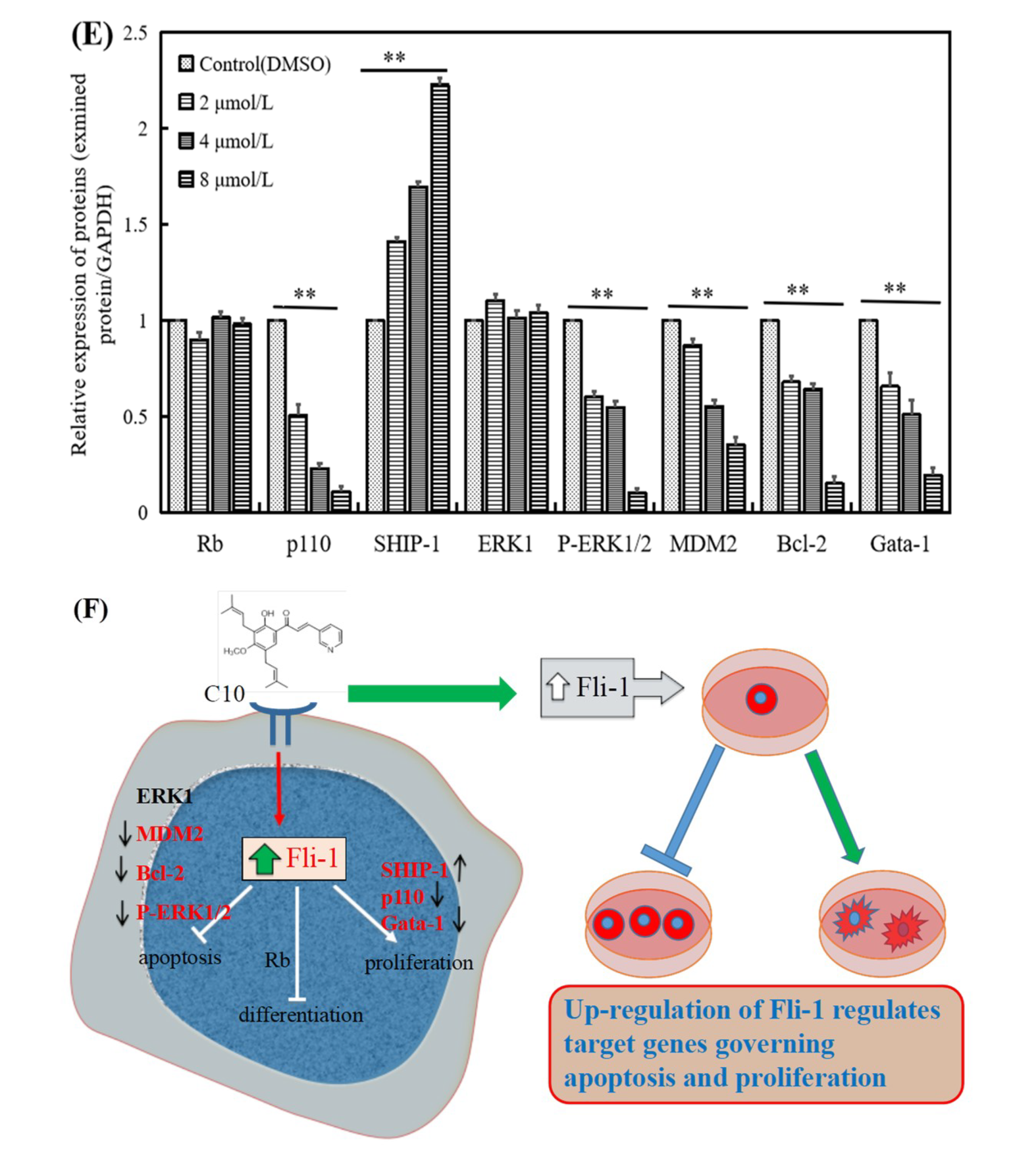
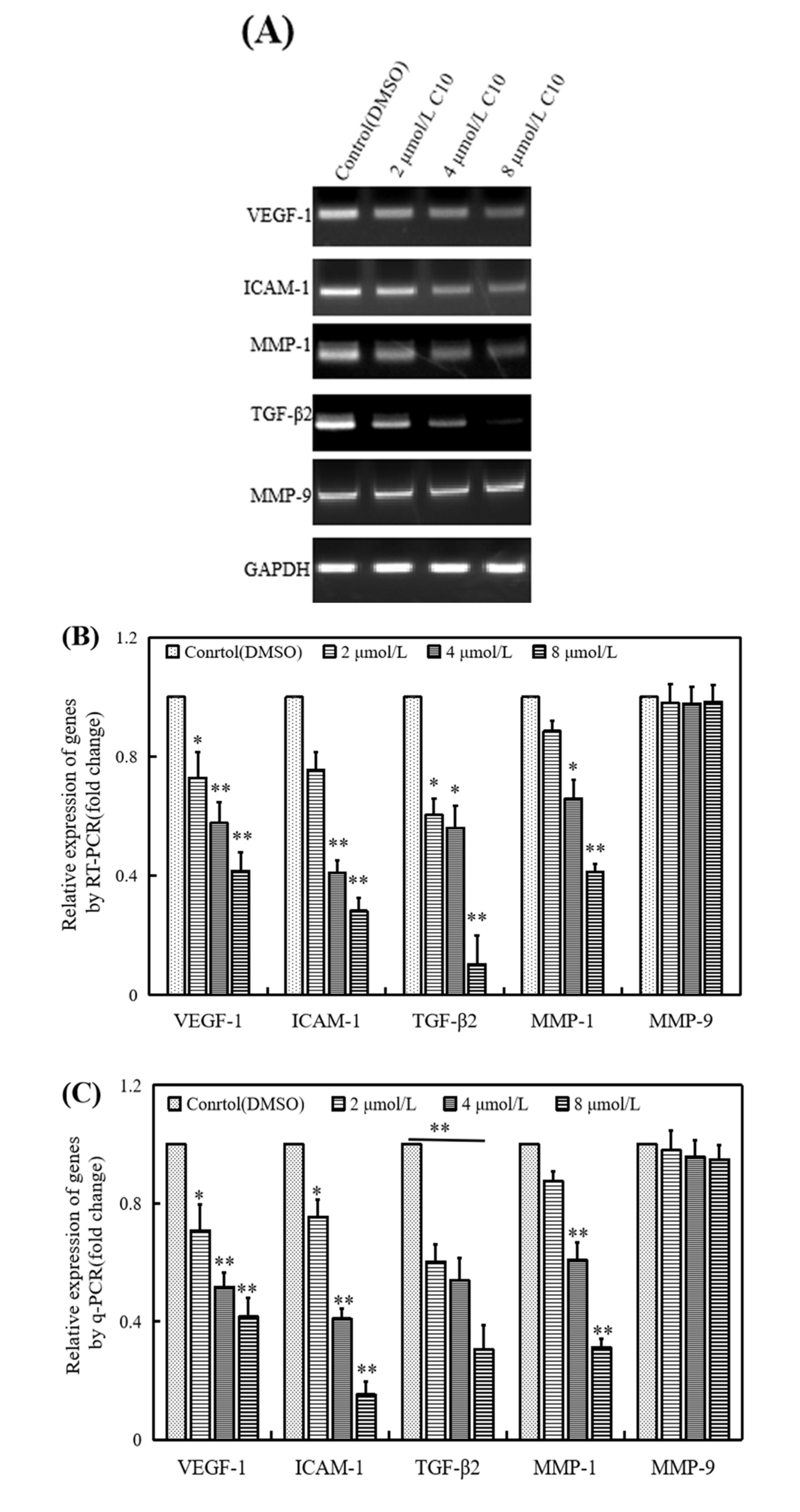
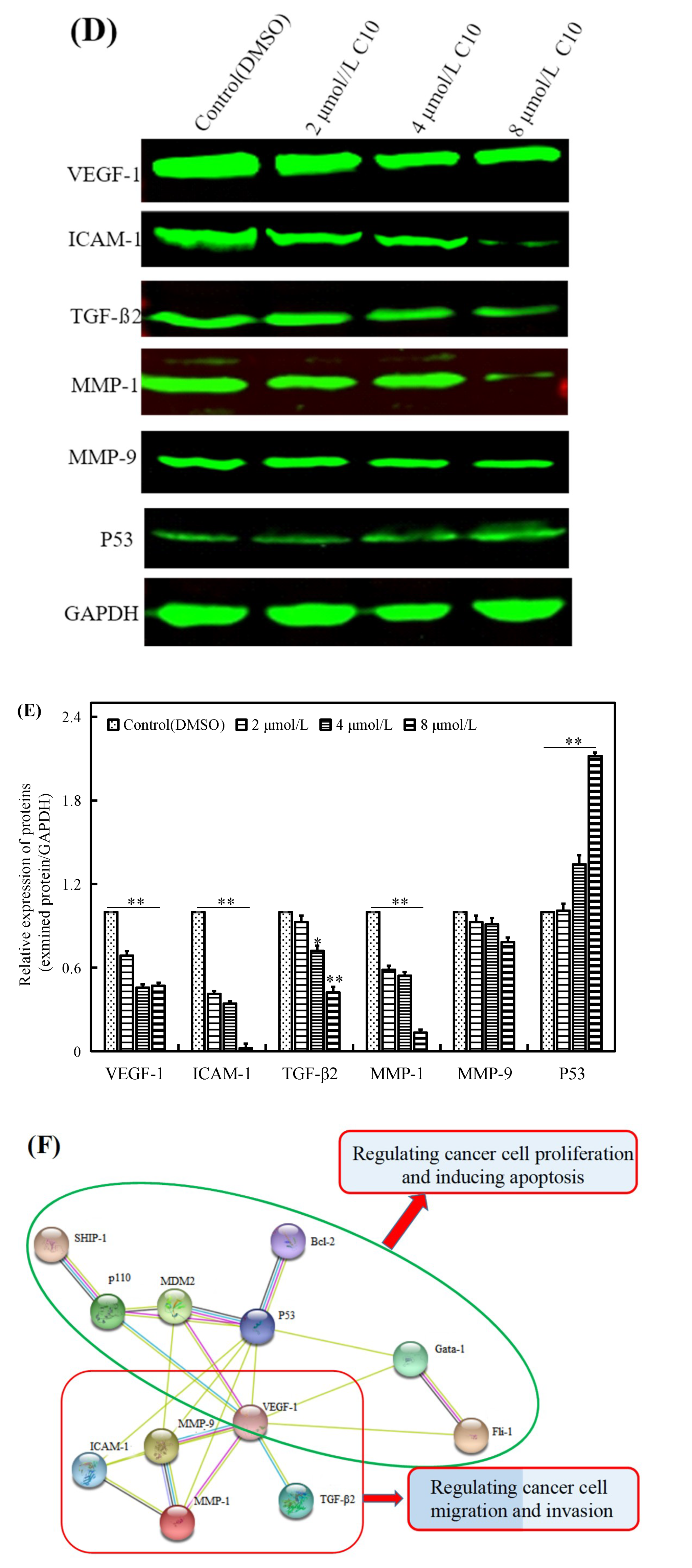
2.4. Regulatory Effect of C10 on the Expression of Fli-1 Target Genes and Metastasis-Associated Genes in PC3 Cells
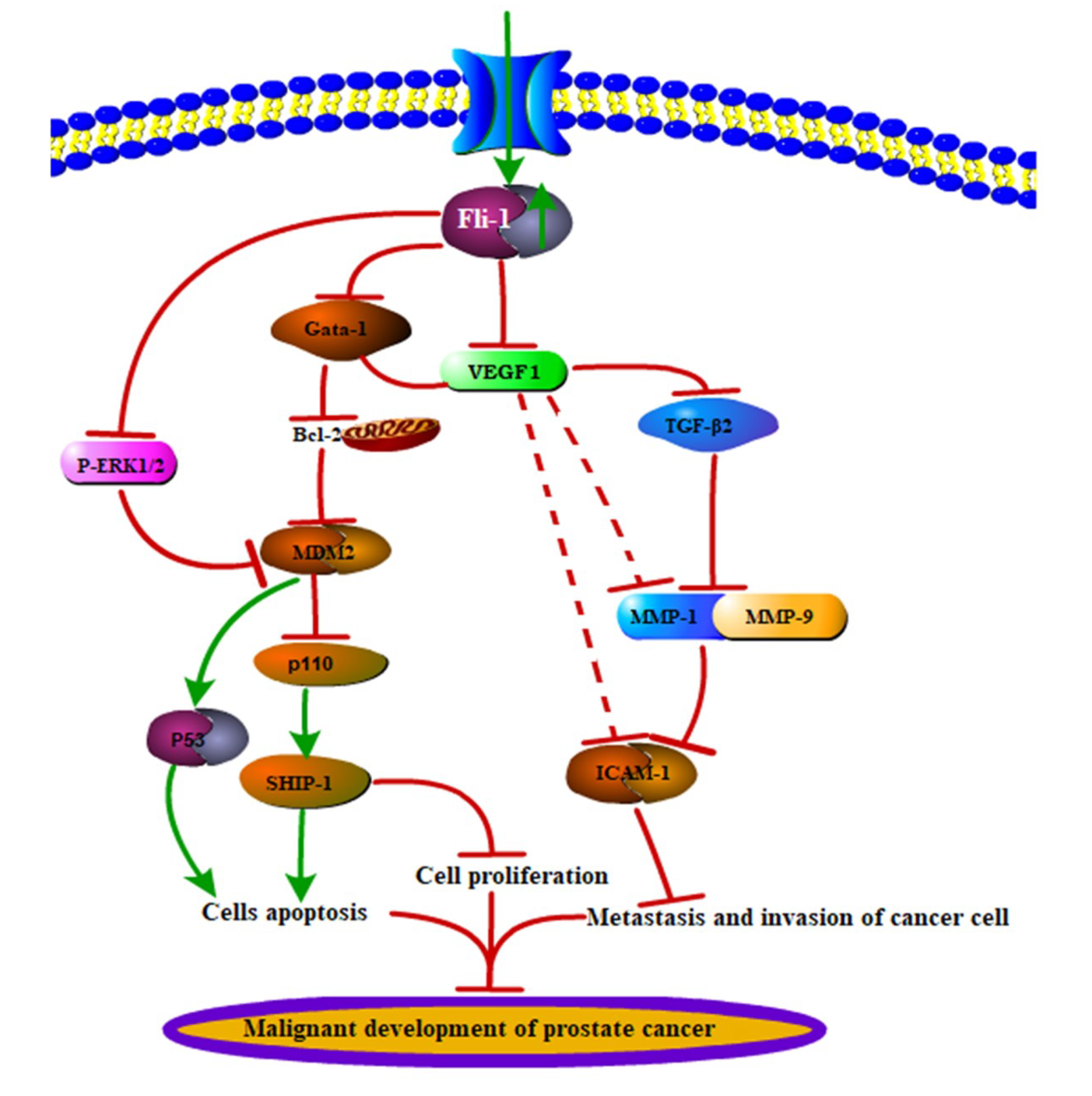
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Antibodies
4.3. Cell Proliferation Assay
4.4. Scratch Repair Assay
4.5. Transwell Chamber Invasion Assay
4.6. Cell Apoptosis Assay
4.7. Genes Expression Assay
4.8. Cellular Thermal Shift Assay
4.9. Promoter Activity Assay
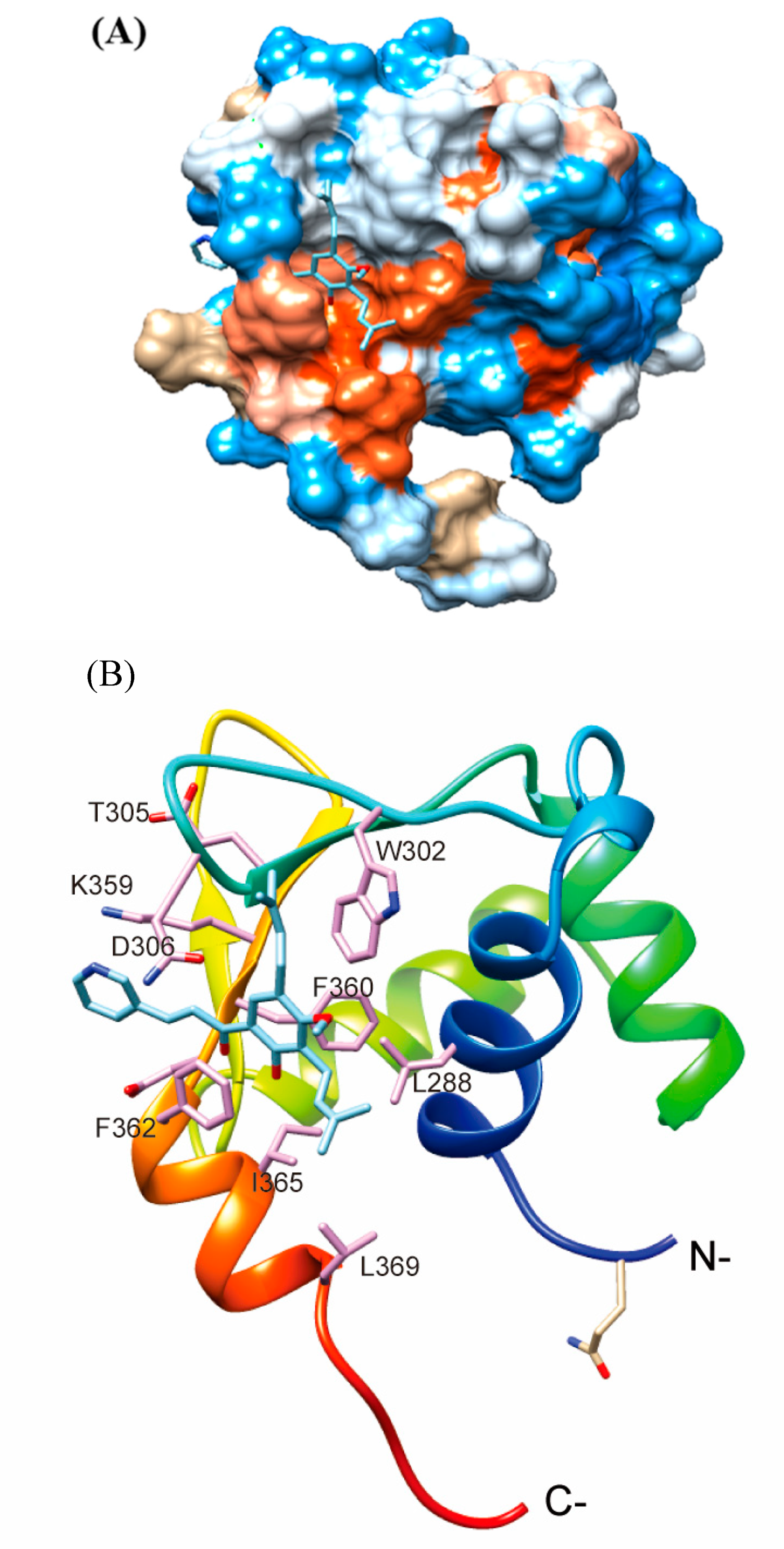
4.10. Molecular Docking Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Fli-1 | Friend leukemia integration 1 |
| PCa | Prostate cancer |
| DMEM | Dulbecco’s Modified Eagle Medium |
| MTT | 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide, |
| DMSO | Dimethyl sulfoxide |
| PBS | Phosphate buffer saline |
| RNA | Ribonucleic Aci |
| PI | Propidium Iodide |
| FITC | Fluoresceine isothiocyanate |
| ERK1 | Extracellular regulated protein kinases 1 |
| PCR | Polymerase Chain Reaction |
| Rb | Retinoblastoma |
| Bcl-2 | B-cell lymphoma-2 |
| ICAM-1 | intercellular cell adhesion molecule-1 |
| TGF-β2 | transforming growth factor-β |
| MMP-1 | matrix metalloproteinase 1 |
| MMP-9 | matrix metalloproteinase 9 |
| VEGF1 | Vascular endothelial growth factor |
| SHIP-1 | Src homology-2 domain-containing inositol 5-phosphatase 1 |
| PI3-K | Phosphatidylinositol 3-kinase |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| BCA | Bicinchoninic acid |
| PVDF | PolyVinylideneFluoride |
References
- Guo, Z.; Chen, X.; Du, T.; Zhu, D.; Lai, Y.; Dong, W.; Wu, W.; Lin, C.; Liu, L.; Huang, H. Elevated levels of epithelial cell transforming sequence 2 predicts poor prognosis for prostate cancer. Med. Oncol. 2017, 34, 13. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.; Deep, G.; Agarwal, C.; Agarwal, R. The strategies to control prostate cancer by chemoprevention approaches. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2014, 760, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.; Ramos, A.A.; Almeida, T.; Azqueta, A.; Rocha, E. Drug resistance in glioblastoma and cytotoxicity of seaweed compounds, alone and in combination with anticancer drugs: A mini review. Phytomedicine 2018, 48, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, B. Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis: A Review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef] [Green Version]
- Zou, D.M.; Brewer, M.; Garcia, F.; Feugang, J.M.; Jian, W.; Zang, R.; Liu, H.; Zou, C. Cactus pear: A natural product in cancer chemoprevention. Nutr. J. 2005, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Selvasundaram, R.; Manoharan, S.; Buddhan, R.; Neelakandan, M.; Naidu, R.M. Chemopreventive potential of esculetin in 7,12-dimethylbenz(a)anthracene-induced hamster buccal pouch carcinogenesis. Mol. Cell. Biochem. 2018, 448, 145–153. [Google Scholar] [CrossRef]
- Kingston, D.G.; Newman, D.J. Natural products as drug leads: An old process or the new hope for drug discovery? Idrugs Investig. Drugs J. 2005, 8, 990–992. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 3, 629–661. [Google Scholar] [CrossRef] [Green Version]
- John, M. Natural products in cancer chemotherapy: Past, present and future. Nat. Rev. Cancer 2002, 2, 143–148. [Google Scholar]
- Bishayee, A.; Sethi, G. Bioactive natural products in cancer prevention and therapy: Progress and promise. Semin. Cancer Biol. 2016, 40–41, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007, 9, 767. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Xu, D.; Gu, J.; Xue, C.; Yang, B.; Fu, L.; Song, S.; Liu, D.; Zhou, W.; Lv, J. Saikosaponin-d inhibits proliferation by up-regulating autophagy via the CaMKK β –AMPK–mTOR pathway in ADPKD cells. Mol. Cell. Biochem. 2018, 449, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, J.; Chen, R.; Deng, Y.; Yi, P. Prenatal Exposure to Lipopolysaccharide Alters Renal DNA Methyltransferase Expression in Rat Offspring. PLoS ONE 2017, 12, e0169206. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wonsey, D.R.; Lemieux, M.E.; Kung, A.L. Differential Disruption of EWS-FLI1 Binding by DNA-Binding Agents. PLoS ONE 2013, 8, e69714. [Google Scholar] [CrossRef]
- Eiji, S.; Sarah, W.; Shuzo, S.; Gary, G.; Watson, D.K.; Zhang, X.K. The transcription factor Fli-1 regulates monocyte, macrophage and dendritic cell development in mice. Immunology 2013, 139, 318–327. [Google Scholar]
- Orlikova, B.; Tasdemir, D.; Golais, F.; Dicato, M.; Diederich, M. Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr. 2011, 6, 125–147. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Zhang, Y.; Wang, X.; Zeng, X.; Hu, Z.; Liu, Y.; Xie, Y.; Liang, G.; Zhu, J.; Luo, H. Novel 3′,5′-diprenylated chalcones inhibited the proliferation of cancer cells in vitro by inducing cell apoptosis and arresting cell cycle phase. Eur. J. Med. Chem. 2017, 133, 227. [Google Scholar] [CrossRef]
- Peng, F.; Wang, G.; Li, X.; Cao, D.; Yang, Z.; Ma, L.; Ye, H.; Liang, X.; Ran, Y.; Chen, J. Rational design, synthesis, and pharmacological properties of pyranochalcone derivatives as potent anti-inflammatory agents. Eur. J. Med. Chem. 2012, 54, 272–280. [Google Scholar] [CrossRef]
- Belostotsky, R.; Ben-Shalom, E.; Rinat, C.; Becker-Cohen, R.; Feinstein, S.; Zeligson, S.; Segel, R.; Elpeleg, O.; Nassar, S.; Frishberg, Y. Mutations in the Mitochondrial Seryl-tRNA Synthetase Cause Hyperuricemia, Pulmonary Hypertension, Renal Failure in Infancy and Alkalosis, HUPRA Syndrome. Am. J. Hum. Genet. 2011, 88, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Q.; Wen, Z.H.; Wan, K.; Yuan, D.; Zeng, X.; Liang, G.; Luo, H. A novel synthesized 3′, 5′-diprenylated chalcone mediates the proliferation of human leukemia cells by regulating apoptosis and autophagy pathways. Biomed. Pharmacother. 2018, 106, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Nowling, T.K.; Fulton, J.D.; Chikeharris, K.; Gilkeson, G.S. Ets factors and a newly identified polymorphism regulate Fli1 promoter activity in lymphocytes. Mol. Immunol. 2008, 45, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Xia, L.; Yao, Y.; Yan, C.; Fan, Y.; Gajendran, B.; Yang, J.; Li, Y.-J.; Chen, J.; Filmus, J.; et al. Identification of diterpenoid compounds that interfere with Fli-1 DNA binding to suppress leukemogenesis. Cell Death Dis. 2019, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yao, Y.; Zhang, G.; Wang, Y.; Deng, B.; Song, J.; Li, X.; Han, F.; Xiao, X.; Yang, J. A screen for Fli-1 transcriptional modulators identifies PKC agonists that induce erythroid to megakaryocytic differentiation and suppress leukemogenesis. Oncotarget 2017, 8, 16728–16743. [Google Scholar] [CrossRef] [Green Version]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundbck, T.; Nordlund, P.R.; Molina, D.M. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100. [Google Scholar] [CrossRef]
- Martin, B.J.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting apoptosis pathways in cancer therapy. Curr. Cancer Drug Targets 2005, 55, 178–194. [Google Scholar] [CrossRef]
- Seth, A.; Watson, D.K. ETS transcription factors and their emerging roles in human cancer. Eur. J. Cancer 2005, 41, 2462–2478. [Google Scholar] [CrossRef]
- Paulette, M.F.; Herrmann, F.R.; Wiam, B.; Kunle, O.; Luigi, T.; Guido, S.; Cheney, R.T.; Jeff, G.; Remedios, P.; Paulette, M.F. Friend leukaemia integration-1 expression in malignant and benign tumours: A multiple tumour tissue microarray analysis using polyclonal antibody. J. Clin. Pathol. 2007, 60, 694–700. [Google Scholar]
- Truong, A.; Ben-David, Y. The role of Fli-1 in normal cell function and malignant transformation. Oncogene 2001, 19, 6482–6489. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Luo, H.; Liu, T.; Zacksenhaus, E.; Ben-David, Y. The ets transcription factor Fli-1 in development, cancer and disease. Oncogene 2015, 34, 2022–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Hu, L.; Li, W.; Wang, G.; Li, Y.; Yan, L.; Cui, J. Oncogenic Fli-1 is a potential prognostic marker for the progression of epithelial ovarian cancer. BMC Cancer 2014, 14, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Zhang, T.; Li, W.; Mu, R.; Zhang, L.; Li, Y.; Cui, J. Overexpression of Fli-1 is Associated with Adverse Prognosis of Endometrial Cancer. Cancer Investig. 2015, 33, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Scheiber, M.N.; Watson, P.M.; Rumboldt, T.; Stanley, C.; Wilson, R.C.; Findlay, V.J.; Anderson, P.E.; Watson, D.K. FLI1 Expression is Correlated with Breast Cancer Cellular Growth, Migration, and Invasion and Altered Gene Expression. Neoplasia 2014, 16, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Gautam, V.; Costantini, S.; Paladino, A.; Colonna, G. Interactomic and pharmacological insights on human sirt-1. Front. Pharmacol. 2012, 3, 40. [Google Scholar] [CrossRef] [Green Version]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 2015, 323, 643–646. [Google Scholar] [CrossRef]
- Lu, Y.; Qin, T.; Li, J.; Wang, L.; Zhang, Q.; Jiang, Z.; Mao, J. MicroRNA-140-5p inhibits invasion and angiogenesis through targeting VEGF-A in breast cancer. Cancer Gene Ther. 2017, 24, 386–392. [Google Scholar] [CrossRef] [Green Version]
- Benedicto, A.; Romayor, I.; Arteta, B. Role of liver ICAM-1 in metastasis. Oncol. Lett. 2017, 14, 3883–3892. [Google Scholar] [CrossRef] [Green Version]
- Brand, S.; Dambacher, J.; Beigel, F.; Olszak, T.; Diebold, J.; Otte, J.M.; Göke, B.; Eichhorst, S.T. CXCR4 and CXCL12 are inversely expressed in colorectal cancer cells and modulate cancer cell migration, invasion and MMP-9 activation. Exp. Cell Res. 2005, 310, 117–130. [Google Scholar] [CrossRef]
- Liu, H. The role of MMP-1 in breast cancer growth and metastasis to the brain in a xenograft model. BMC Cancer 2012, 12, 583. [Google Scholar] [CrossRef] [Green Version]
- Takatsugu, O.; Kikuo, N.; Eiji, H. Alternative antiandrogen therapy in patients with castration-resistant prostate cancer: A single-center experience. Int. J. Urol. 2010, 17, 950–955. [Google Scholar]
- Wang, K.; Ruan, H.; Xu, T.; Liu, L.; Liu, D.; Yang, H.; Zhang, X.; Chen, K. Recent advances on the progressive mechanism and therapy in castration-resistant prostate cancer. Oncotargets Ther. 2018, 11, 3167–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.L.; Sun, B.F.; Huang, L.R.; Yuan, H.B.; Zhang, S.; Chen, J.; Yu, Z.J.; Luo, H. Potent Inhibition of miR-34b on Migration and Invasion in Metastatic Prostate Cancer Cells by Regulating the TGF-β Pathway. Int. J. Mol. Sci. 2017, 18, 2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, C.; Zhang, G.; Han, F.; Fu, S.; Cao, Y.; Zhang, F.; Zhang, Q.; Meslamani, J.; Xu, Y.; Ji, D.; et al. Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. Proc. Natl. Acad. Sci. USA 2018, 31, 7949–7954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.J.; Liu, G.; Xia, L.; Xiao, X.; Liu, J.C.; Menezes, M.E.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B. Suppression of Her2/Neu mammary tumor development inmda-7/IL-24transgenic mice. Oncotarget 2015, 6, 36943–36954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Ren, C.; Ryou, C.; Li, J. Direct interaction of DNMT inhibitors to PrPC suppresses pathogenic process of prion. Acta Pharm. Sin. B 2019, 9, 952–959. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Primer Sequence (5′→3′) |
|---|---|---|
| Rb | Forward | CTCTCGTCAGGCTTGAGTTTG |
| Reverse | GACATCTCATCTAGGTCAACTGC | |
| MDM-2 | Forward | GAATCATCGGACTCAGGTACATC |
| Reverse | TCTGTCTCACTAATTGCTCTCCT | |
| Bcl-2 | Forward | GGTGGGGTCATGTGTGTGG |
| Reverse | CGGTTCAGGTACTCAGTCATCC | |
| Gata-1 | Forward | CTGTCCCCAATAGTGCTTATGG |
| Reverse | GAATAGGCTGCTGAATTGAGGG | |
| SHIP-1 | Forward | GCGTGCTGTATCGGAATTGC |
| Reverse | TGGTGAAGAACCTCATGGAGAC | |
| p110 | Forward | TATTTGGACTTTGCGACAAGACT |
| Reverse | TCGAACGTACGGTCTGGATAG | |
| ERK1 | Forward | TACACCAACCTCTCGTACATCG |
| Reverse | CATGTCTGAAGCGCAGTAAGATT | |
| VEGF-1 | Forward | AGGGCAGAATCATCACGAAGT |
| Reverse | AGGGTCTCGATTGGATGGCA | |
| ICAM-1 | Forward | ATGCCCAGACATCTGTGTCC |
| Reverse | GGGGTCTCTATGCCCAACAA | |
| TGF-β2 | Forward | CAGCACACTCGATATGGACCA |
| Reverse | CCTCGGGCTCAGGATAGTCT | |
| MMP-1 | Forward | AAAATTACACGCCAGATTTGCC |
| Reverse | GGTGTGACATTACTCCAGAGTTG | |
| MMP-9 | Forward | TGTACCGCTATGGTTACACTCG |
| Reverse | GGCAGGGACAGTTGCTTCT | |
| Fli-1 | Forward | CAGCCCCACAAGATCAACCC |
| Reverse | CACCGGAGACTCCCTGGAT | |
| GAPDH | Forward | GGAGCGAGATCCCTCCAAAAT |
| Reverse | GGCTGTTGTCATACTTCTCATGG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Xu, B.; Yu, J.; Huang, L.; Zeng, X.; Shen, X.; Ren, C.; Ben-David, Y.; Luo, H. Fli-1 Activation through Targeted Promoter Activity Regulation Using a Novel 3’, 5’-diprenylated Chalcone Inhibits Growth and Metastasis of Prostate Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2216. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062216
Ma Y, Xu B, Yu J, Huang L, Zeng X, Shen X, Ren C, Ben-David Y, Luo H. Fli-1 Activation through Targeted Promoter Activity Regulation Using a Novel 3’, 5’-diprenylated Chalcone Inhibits Growth and Metastasis of Prostate Cancer Cells. International Journal of Molecular Sciences. 2020; 21(6):2216. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062216
Chicago/Turabian StyleMa, Youfen, Bixue Xu, Jia Yu, Lirong Huang, Xiaoping Zeng, Xiangchun Shen, Chunyan Ren, Yaacov Ben-David, and Heng Luo. 2020. "Fli-1 Activation through Targeted Promoter Activity Regulation Using a Novel 3’, 5’-diprenylated Chalcone Inhibits Growth and Metastasis of Prostate Cancer Cells" International Journal of Molecular Sciences 21, no. 6: 2216. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062216