Neuroprotective Strategies for Retinal Ganglion Cell Degeneration: Current Status and Challenges Ahead

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Obstacles to RGC Survival and Regeneration upon Injury: Insights from Development to Disease Models
3. Potential Therapeutic Targets Aiming RGC Neuroprotection
3.1. Neuroprotective Therapies
3.1.1. Neurotrophic Factors
Nerve Growth Factor (NGF)
Brain-Derived Neurotrophic Factor (BDNF)
Glial Cell Line-Derived Neurotrophic Factor (GDNF)
Ciliary Neurotrophic Factor (CNTF)
Other Trophic Factors
3.1.2. Glutamate Receptors Antagonists
3.1.3. Alpha-2 Adrenergic Receptors Agonists
3.1.4. Calcium Channel Blockers
3.1.5. Antioxidants
3.1.6. Nitric Oxide Synthase Inhibitors
3.1.7. Adenosinergic System
3.2. Cell-Based Therapies
3.3. Glia-Mediated Neuroprotection
4. Clinical Trials Targeting RGCs Neuroprotection
5. Different Types of RGCs and their Susceptibility after Retinal Damage
6. Potential Pitfalls in Translating Preclinical Studies into the Clinics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A1 | Adenosine A1 receptor |
| A2A | Adenosine A2A receptor |
| A2B | Adenosine A2B receptor |
| A3 | Adenosine A3 receptor |
| ARTN | Artemin |
| ATP | Adenosine triphosphate |
| BCDVA | Best corrected distance visual acuity |
| BDNF | Brain-derived neurotrophic factor |
| bFGF | Basic fibroblast growth factor |
| cAMP | Cyclic adenosine monophosphate |
| CART | Cocaine and amphetamine-regulated transcript |
| CAT | Catalase |
| CHA | N(6)-cyclohexyl-adenosine |
| CNS | Central nervous system |
| CNTF | Ciliary neurotrophic factor |
| CNTFR | CNTF receptors |
| Cop-1 | Copolymer-1 |
| Cx3cr1 | CX3C chemokine receptor 1 |
| DS-RGCs | Directionally selective ganglion cells |
| ED | Embryonic day |
| EHP | Elevated hydrostatic pressure |
| EPO | Erythropoietin |
| ERG | Electroretinography |
| GCL | Ganglion cell layer |
| GDNF | Glial cell-line derived neurotrophic factor |
| GLAST | Glutamate/aspartate transporter |
| GPx | Glutathione peroxidase |
| GSH | Glutathione |
| IGF-1 | Insulin-like growth factor-1 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| INL | Inner nuclear layer |
| iNOS | Inducible nitric oxide synthase |
| IOP | Intraocular pressure |
| ipRGCs | Intrinsically photosensitive melanopsin-containing RGCs |
| J-RGCs | JamB expressing RGCs |
| KLF | Krüppel-like family |
| MAG | Myelin-associated glycoprotein |
| MHC-II | Major histocompatibility complex class II |
| mTOR | Mammalian target of rapamycin |
| ND4 | NADH dehydrogenase subunit 4 |
| NFL | Nerve fiber layer |
| NGF | Nerve growth factor |
| NgR | Nogo receptor |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NMDA | N-methyl-D-aspartate |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NRTN | Neurturin |
| NT-3 | Neurotrophin-3 |
| NT-4/5 | Neurotrophin-4/5 |
| OCT | Optical coherence tomography |
| OHT | Ocular hypertension |
| ONC | Optic nerve crush |
| ONH | Optic nerve head |
| ONL | Outer nuclear layer |
| PEDF | Pigment epithelium derived factor |
| PI3K | Phosphoinositide 3-kinases |
| PND | Postnatal day |
| PNS | Peripheral nervous system |
| PSPN | Persephin |
| PTEN | Phosphatase and tensin homologue |
| RAGE | Receptors for advanced glycation end-products |
| RGCs | Retinal ganglion cells |
| rhNGF | Recombinant human nerve growth factor |
| ROS | Reactive oxygen species |
| Sema3A | Semaphorin-3A |
| Sema5A | Semaphorin-5A |
| SOD | Superoxide dismutase |
| TLRs | Toll-like receptors |
| TNF | Tumour necrosis factor |
| TrK | Tyrosine kinase |
| TSPO | Translocator protein |
| VEGF-A | Vascular endothelial growth factor A |
| Zn2+ | Zinc |
| αRGCs | alpha retinal ganglion cells |
| βRGCs | beta retinal ganglion cells |
References
- Kolb, H.; Fernandez, E.; Nelson, R. Webvision: The Organization of the Retina and Visual System. In Webvision: The Organization of the Retina and Visual System; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. [Google Scholar]
- Carelli, V.; La Morgia, C.; Ross-Cisneros, F.N.; Sadun, A.A. Optic neuropathies: The tip of the neurodegeneration iceberg. Hum. Mol. Genet. 2017, 26, R139–R150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, J.L.; Espinosa, J.S.; Xu, Y.; Davidson, N.; Kovacs, G.T.; Barres, B.A. Retinal ganglion cells do not extend axons by default: Promotion by neurotrophic signaling and electrical activity. Neuron 2002, 33, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Dratviman-Storobinsky, O.; Hasanreisoglu, M.; Offen, D.; Barhum, Y.; Weinberger, D.; Goldenberg-Cohen, N. Progressive damage along the optic nerve following induction of crush injury or rodent anterior ischemic optic neuropathy in transgenic mice. Mol. Vis. 2008, 14, 2171–2179. [Google Scholar] [PubMed]
- Kutsarova, E.; Munz, M.; Ruthazer, E.S. Rules for shaping neural connections in the developing brain. Front. Neural Circuits 2016, 10, 111. [Google Scholar] [CrossRef] [Green Version]
- Guerin, M.B.; McKernan, D.P.; O’Brien, C.J.; Cotter, T.G. Retinal ganglion cells: Dying to survive. Int. J. Dev. Biol. 2006, 50, 665–674. [Google Scholar] [CrossRef]
- Goldberg, J.L.; Klassen, M.P.; Hua, Y.; Barres, B.A. Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science 2002, 296, 1860–1864. [Google Scholar] [CrossRef]
- Martins, J.; Elvas, F.; Brudzewsky, D.; Martins, T.; Kolomiets, B.; Tralhao, P.; Gotzsche, C.R.; Cavadas, C.; Castelo-Branco, M.; Woldbye, D.P.; et al. Activation of neuropeptide y receptors modulates retinal ganglion cell physiology and exerts neuroprotective actions in vitro. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Barres, B.A.; Silverstein, B.E.; Corey, D.P.; Chun, L.L. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron 1988, 1, 791–803. [Google Scholar] [CrossRef]
- Ming, G.L.; Song, H.J.; Berninger, B.; Holt, C.E.; Tessier-Lavigne, M.; Poo, M.M. cAMP-dependent growth cone guidance by netrin-1. Neuron 1997, 19, 1225–1235. [Google Scholar] [CrossRef] [Green Version]
- Rodger, J.; Goto, H.; Cui, Q.; Chen, P.B.; Harvey, A.R. cAMP regulates axon outgrowth and guidance during optic nerve regeneration in goldfish. Mol. Cell. Neurosci. 2005, 30, 452–464. [Google Scholar] [CrossRef]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.R.; Chen, H.Y.; Hu, Z.Z.; Xie, P.; Liu, Q.H. PTEN knockdown with the Y444F mutant AAV2 vector promotes axonal regeneration in the adult optic nerve. Neural Regen. Res. 2018, 13, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.L.; Blackmore, M.G.; Hu, Y.; Kaestner, K.H.; Bixby, J.L.; Lemmon, V.P.; Goldberg, J.L. KLF family members regulate intrinsic axon regeneration ability. Science 2009, 326, 298–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steketee, M.B.; Oboudiyat, C.; Daneman, R.; Trakhtenberg, E.; Lamoureux, P.; Weinstein, J.E.; Heidemann, S.; Barres, B.A.; Goldberg, J.L. Regulation of intrinsic axon growth ability at retinal ganglion cell growth cones. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4369–4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Andereggen, L.; Yuki, K.; Omura, K.; Yin, Y.; Gilbert, H.Y.; Erdogan, B.; Asdourian, M.S.; Shrock, C.; de Lima, S.; et al. Mobile zinc increases rapidly in the retina after optic nerve injury and regulates ganglion cell survival and optic nerve regeneration. Proc. Natl. Acad. Sci. USA 2017, 114, E209–E218. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.L.; Goldberg, J.L. Multiple transcription factor families regulate axon growth and regeneration. Dev. Neurobiol. 2011, 71, 1186–1211. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Schlamp, C.L.; Poulsen, G.L.; Jackson, M.W.; Griep, A.E.; Nickells, R.W. p53 regulates apoptotic retinal ganglion cell death induced by N-methyl-D-aspartate. Mol. Vis. 2002, 8, 341–350. [Google Scholar]
- Di Giovanni, S.; Knights, C.D.; Rao, M.; Yakovlev, A.; Beers, J.; Catania, J.; Avantaggiati, M.L.; Faden, A.I. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006, 25, 4084–4096. [Google Scholar] [CrossRef] [Green Version]
- Maes, M.E.; Schlamp, C.L.; Nickells, R.W. BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog. Retin. Eye Res. 2017, 57, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Isenmann, S.; Wahl, C.; Krajewski, S.; Reed, J.C.; Bahr, M. Up-regulation of Bax protein in degenerating retinal ganglion cells precedes apoptotic cell death after optic nerve lesion in the rat. Eur. J. Neurosci. 1997, 9, 1763–1772. [Google Scholar] [CrossRef]
- Kaneda, K.; Kashii, S.; Kurosawa, T.; Kaneko, S.; Akaike, A.; Honda, Y.; Minami, M.; Satoh, M. Apoptotic DNA fragmentation and upregulation of Bax induced by transient ischemia of the rat retina. Brain Res. 1999, 815, 11–20. [Google Scholar] [CrossRef]
- Libby, R.T.; Li, Y.; Savinova, O.V.; Barter, J.; Smith, R.S.; Nickells, R.W.; John, S.W. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005, 1, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.F.; Schneider, G.E.; Martinou, J.C.; Tonegawa, S. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature 1997, 385, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.M.; McGuinness, U.M.; Aguayo, A.J. Axons from CNS neurones regenerate into PNS grafts. Nature 1980, 284, 264–265. [Google Scholar] [CrossRef]
- Vidal-Sanz, M.; Bray, G.M.; Villegas-Perez, M.P.; Thanos, S.; Aguayo, A.J. Axonal regeneration and synapse formation in the superior colliculus by retinal ganglion cells in the adult rat. J. Neurosci. Off. J. Soc. Neurosci. 1987, 7, 2894–2909. [Google Scholar] [CrossRef]
- Yiu, G.; He, Z. Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 2006, 7, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Pasterkamp, R.J.; Giger, R.J.; Ruitenberg, M.J.; Holtmaat, A.J.; De Wit, J.; De Winter, F.; Verhaagen, J. Expression of the gene encoding the chemorepellent semaphorin III is induced in the fibroblast component of neural scar tissue formed following injuries of adult but not neonatal CNS. Mol. Cell. Neurosci. 1999, 13, 143–166. [Google Scholar] [CrossRef]
- Pasterkamp, R.J.; Anderson, P.N.; Verhaagen, J. Peripheral nerve injury fails to induce growth of lesioned ascending dorsal column axons into spinal cord scar tissue expressing the axon repellent Semaphorin3A. Eur. J. Neurosci. 2001, 13, 457–471. [Google Scholar] [CrossRef]
- Van Horck, F.P.G.; Weinl, C.; Holt, C.E. Retinal axon guidance: Novel mechanisms for steering. Curr. Opin. Neurobiol. 2004, 14, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Tillo, M.; Ruhrberg, C.; Mackenzie, F. Emerging roles for semaphorins and VEGFs in synaptogenesis and synaptic plasticity. Cell Adhes. Migr. 2012, 6, 541–546. [Google Scholar] [CrossRef]
- Chan-Juan, H.; Sen, L.; Li-Qianyu, A.; Jian, Y.; Rong-Di, Y. MicroRNA-30b regulates the polarity of retinal ganglion cells by inhibiting semaphorin-3A. Mol. Vis. 2019, 25, 722–730. [Google Scholar] [PubMed]
- Dallimore, E.J.; Cui, Q.; Beazley, L.D.; Harvey, A.R. Postnatal innervation of the rat superior colliculus by axons of late-born retinal ganglion cells. Eur. J. Neurosci. 2002, 16, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- De Winter, F.; Cui, Q.; Symons, N.; Verhaagen, J.; Harvey, A.R. Expression of class-3 semaphorins and their receptors in the neonatal and adult rat retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4554–4562. [Google Scholar] [CrossRef] [Green Version]
- Zylbersztejn, K.; Petkovic, M.; Burgo, A.; Deck, M.; Garel, S.; Marcos, S.; Bloch-Gallego, E.; Nothias, F.; Serini, G.; Bagnard, D.; et al. The vesicular SNARE Synaptobrevin is required for Semaphorin 3A axonal repulsion. J. Cell Biol. 2012, 196, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Shirvan, A.; Kimron, M.; Holdengreber, V.; Ziv, I.; Ben-Shaul, Y.; Melamed, S.; Melamed, E.; Barzilai, A.; Solomon, A.S. Anti-semaphorin 3A antibodies rescue retinal ganglion cells from cell death following optic nerve axotomy. J. Biol. Chem. 2002, 277, 49799–49807. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, J.L.; Vargas, M.E.; Wang, J.T.; Mandemakers, W.; Oster, S.F.; Sretavan, D.W.; Barres, B.A. An oligodendrocyte lineage-specific semaphorin, Sema5A, inhibits axon growth by retinal ganglion cells. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 4989–4999. [Google Scholar] [CrossRef]
- Fisher, J.; Levkovitch-Verbin, H.; Schori, H.; Yoles, E.; Butovsky, O.; Kaye, J.F.; Ben-Nun, A.; Schwartz, M. Vaccination for neuroprotection in the mouse optic nerve: Implications for optic neuropathies. J. Neurosci. 2001, 21, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Kipnis, J.; Yoles, E.; Porat, Z.; Cohen, A.; Mor, F.; Sela, M.; Cohen, I.R.; Schwartz, M. T cell immunity to copolymer 1 confers neuroprotection on the damaged optic nerve: Possible therapy for optic neuropathies. Proc. Natl. Acad. Sci. USA 2000, 97, 7446–7451. [Google Scholar] [CrossRef] [Green Version]
- Schori, H.; Kipnis, J.; Yoles, E.; WoldeMussie, E.; Ruiz, G.; Wheeler, L.A.; Schwartz, M. Vaccination for protection of retinal ganglion cells against death from glutamate cytotoxicity and ocular hypertension: Implications for glaucoma. Proc. Natl. Acad. Sci. USA 2001, 98, 3398–3403. [Google Scholar] [CrossRef] [Green Version]
- Bakalash, S.; Ben-Shlomo, G.; Aloni, E.; Shaked, I.; Wheeler, L.; Ofri, R.; Schwartz, M. T-cell-based vaccination for morphological and functional neuroprotection in a rat model of chronically elevated intraocular pressure. J. Mol. Med. 2005, 83, 904–916. [Google Scholar] [CrossRef]
- Pernet, V.; Joly, S.; Christ, F.; Dimou, L.; Schwab, M.E. Nogo-A and myelin-associated glycoprotein differently regulate oligodendrocyte maturation and myelin formation. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 7435–7444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernet, V. Nogo-A in the visual system development and in ocular diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Solomon, A.M.; Westbrook, T.; Field, G.D.; McGee, A.W. Nogo receptor 1 is expressed by nearly all retinal ganglion cells. PLoS ONE 2018, 13, e0196565. [Google Scholar] [CrossRef] [PubMed]
- Pernet, V.; Joly, S.; Dalkara, D.; Schwarz, O.; Christ, F.; Schaffer, D.; Flannery, J.G.; Schwab, M.E. Neuronal Nogo-A upregulation does not contribute to ER stress-associated apoptosis but participates in the regenerative response in the axotomized adult retina. Cell Death Differ. 2012, 19, 1096–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vajda, F.; Jordi, N.; Dalkara, D.; Joly, S.; Christ, F.; Tews, B.; Schwab, M.E.; Pernet, V. Cell type-specific Nogo-A gene ablation promotes axonal regeneration in the injured adult optic nerve. Cell Death Differ. 2015, 22, 323–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mdzomba, J.B.; Jordi, N.; Rodriguez, L.; Joly, S.; Bretzner, F.; Pernet, V. Nogo-A inactivation improves visual plasticity and recovery after retinal injury. Cell Death Dis. 2018, 9, 727. [Google Scholar] [CrossRef]
- Wong, E.V.; David, S.; Jacob, M.H.; Jay, D.G. Inactivation of myelin-associated glycoprotein enhances optic nerve regeneration. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 3112–3117. [Google Scholar] [CrossRef]
- Liao, X.-X.; Chen, D.; Shi, J.; Sun, Y.-Q.; Sun, S.-J.; So, K.-F.; Fu, Q.-L. The expression patterns of Nogo-A, Myelin Associated Glycoprotein and Oligodendrocyte Myelin Glycoprotein in the Retina After Ocular hypertension. Neurochem. Res. 2011, 36, 1955–1961. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.-L.; Liao, X.-X.; Li, X.; Chen, D.; Shi, J.; Wen, W.; Lee, D.H.S.; So, K.-F. Soluble Nogo-66 receptor prevents synaptic dysfunction and rescues retinal ganglion cell loss in chronic glaucoma. Investig. Opthalmol. Vis. Sci. 2011, 52, 8374. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Wang, F.; Teng, Y.; Zhao, S.G.; Cui, H.; Pan, S.H. Axonal regeneration of optic nerve after crush in Nogo66 receptor knockout mice. Neurosci. Lett. 2009, 460, 223–226. [Google Scholar] [CrossRef]
- Vecino, E.; Ugarte, M.; Nash, M.S.; Osborne, N.N. NMDA induces BDNF expression in the albino rat retina in vivo. Neuroreport 1999, 10, 1103–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecino, E.; Caminos, E.; Ugarte, M.; Martin-Zanca, D.; Osborne, N.N. Immunohistochemical distribution of neurotrophins and their receptors in the rat retina and the effects of ischemia and reperfusion. Gen. Pharmacol. 1998, 30, 305–314. [Google Scholar] [CrossRef]
- Hofer, M.; Pagliusi, S.R.; Hohn, A.; Leibrock, J.; Barde, Y.A. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 1990, 9, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, C.; Ernfors, P.; Persson, H.; Olson, L. Localization of brain-derived neurotrophic factor mRNA to neurons in the brain by in situ hybridization. Exp. Neurol. 1990, 109, 141–152. [Google Scholar] [CrossRef]
- Herzog, K.H.; von Bartheld, C.S. Contributions of the optic tectum and the retina as sources of brain-derived neurotrophic factor for retinal ganglion cells in the chick embryo. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 2891–2906. [Google Scholar] [CrossRef]
- Ma, Y.T.; Hsieh, T.; Forbes, M.E.; Johnson, J.E.; Frost, D.O. BDNF injected into the superior colliculus reduces developmental retinal ganglion cell death. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 2097–2107. [Google Scholar] [CrossRef]
- Di Polo, A.; Aigner, L.J.; Dunn, R.J.; Bray, G.M.; Aguayo, A.J. Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Muller cells temporarily rescues injured retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3978–3983. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Weber, A.J. BDNF enhances retinal ganglion cell survival in cats with optic nerve damage. Investig. Ophthalmol. Vis. Sci. 2001, 42, 966–974. [Google Scholar]
- Mey, J.; Thanos, S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993, 602, 304–317. [Google Scholar] [CrossRef]
- Mansour-Robaey, S.; Clarke, D.B.; Wang, Y.C.; Bray, G.M.; Aguayo, A.J. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc. Natl. Acad. Sci. USA 1994, 91, 1632–1636. [Google Scholar] [CrossRef] [Green Version]
- Peinado-Ramon, P.; Salvador, M.; Villegas-Perez, M.P.; Vidal-Sanz, M. Effects of axotomy and intraocular administration of NT-4, NT-3, and brain-derived neurotrophic factor on the survival of adult rat retinal ganglion cells. A quantitative in vivo study. Investig. Ophthalmol. Vis. Sci. 1996, 37, 489–500. [Google Scholar]
- Domenici, L.; Origlia, N.; Falsini, B.; Cerri, E.; Barloscio, D.; Fabiani, C.; Sanso, M.; Giovannini, L. Rescue of retinal function by BDNF in a mouse model of glaucoma. PLoS ONE 2014, 9, e115579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galindo-Romero, C.; Valiente-Soriano, F.J.; Jimenez-Lopez, M.; Garcia-Ayuso, D.; Villegas-Perez, M.P.; Vidal-Sanz, M.; Agudo-Barriuso, M. Effect of brain-derived neurotrophic factor on mouse axotomized retinal ganglion cells and phagocytic microglia. Investig. Ophthalmol. Vis. Sci. 2013, 54, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-W.; Lu, Q.; You, S.-W.; Zhi, Y.; Yip, H.K.; Wu, W.; So, K.-F.; Cui, Q. CNTF and BDNF have similar effects on retinal ganglion cell survival but differential effects on nitric oxide synthase expression soon after optic nerve injury. Investig. Opthalmol. Vis. Sci. 2005, 46, 1497. [Google Scholar] [CrossRef]
- Meyer-Franke, A.; Kaplan, M.R.; Pfrieger, F.W.; Barres, B.A. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron 1995, 15, 805–819. [Google Scholar] [CrossRef] [Green Version]
- Leaver, S.G.; Cui, Q.; Plant, G.W.; Arulpragasam, A.; Hisheh, S.; Verhaagen, J.; Harvey, A.R. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006, 13, 1328–1341. [Google Scholar] [CrossRef] [Green Version]
- Muller, A.; Hauk, T.G.; Leibinger, M.; Marienfeld, R.; Fischer, D. Exogenous CNTF stimulates axon regeneration of retinal ganglion cells partially via endogenous CNTF. Mol. Cell. Neurosci. 2009, 41, 233–246. [Google Scholar] [CrossRef]
- Maffei, L.; Carmignoto, G.; Perry, V.H.; Candeo, P.; Ferrari, G. Schwann cells promote the survival of rat retinal ganglion cells after optic nerve section. Proc. Natl. Acad. Sci. USA 1990, 87, 1855–1859. [Google Scholar] [CrossRef] [Green Version]
- Carmignoto, G.; Maffei, L.; Candeo, P.; Canella, R.; Comelli, C. Effect of NGF on the survival of rat retinal ganglion cells following optic nerve section. J. Neurosci. Off. J. Soc. Neurosci. 1989, 9, 1263–1272. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.E. Retina ganglion cell degeneration in glaucoma: An opportunity missed? A review. Clin. Exp. Ophthalmol. 2012, 40, 364–368. [Google Scholar] [CrossRef]
- Aires, I.D.; Ambrosio, A.F.; Santiago, A.R. Modeling human glaucoma: Lessons from the in vitro models. Ophthalmic Res. 2017, 57, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mak, H.K.; Chan, Y.K.; Lin, C.; Kong, C.; Leung, C.K.S.; Shum, H.C. An in vitro pressure model towards studying the response of primary retinal ganglion cells to elevated hydrostatic pressures. Sci. Rep. 2019, 9, 9057. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Guy, J.; Anderson, D.R. Blockade of rapid axonal transport. Effect of intraocular pressure elevation in primate optic nerve. Arch. Ophthalmol. 1979, 97, 525–531. [Google Scholar] [CrossRef]
- Quigley, H.A.; McKinnon, S.J.; Zack, D.J.; Pease, M.E.; Kerrigan-Baumrind, L.A.; Kerrigan, D.F.; Mitchell, R.S. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3460–3466. [Google Scholar]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 764–774. [Google Scholar]
- Park, H.L.; Kim, S.W.; Kim, J.H.; Park, C.K. Increased levels of synaptic proteins involved in synaptic plasticity after chronic intraocular pressure elevation and modulation by brain-derived neurotrophic factor in a glaucoma animal model. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Aires, I.D.; Boia, R.; Rodrigues-Neves, A.C.; Madeira, M.H.; Marques, C.; Ambrosio, A.F.; Santiago, A.R. Blockade of microglial adenosine A2A receptor suppresses elevated pressure-induced inflammation, oxidative stress, and cell death in retinal cells. Glia 2019, 67, 896–914. [Google Scholar] [CrossRef] [Green Version]
- Madeira, M.H.; Elvas, F.; Boia, R.; Goncalves, F.Q.; Cunha, R.A.; Ambrosio, A.F.; Santiago, A.R. Adenosine A2AR blockade prevents neuroinflammation-induced death of retinal ganglion cells caused by elevated pressure. J. Neuroinflamm. 2015, 12, 115. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Harada, C.; Parada, L.F. Molecular regulation of visual system development: More than meets the eye. Genes Dev. 2007, 21, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Namekata, K.; Guo, X.; Harada, C.; Harada, T. Neuroprotection, growth factors and BDNF-TrkB signalling in retinal degeneration. Int. J. Mol. Sci. 2016, 17, 1584. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.V.; Bull, N.D.; Martin, K.R. Neurotrophic factor delivery as a protective treatment for glaucoma. Exp. Eye Res. 2011, 93, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.J.; Tian, X.S.; Ruan, Z.; Chen, Y.T.; Wu, L.; Gong, Q.; Wang, W.; Zhang, H.Y. Dysregulation of neurotrophic and inflammatory systems accompanied by decreased CREB signaling in ischemic rat retina. Exp. Eye Res. 2014, 125, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.; Deppmeier, L.M.; Wentzien, S.K.; Hsu, I.; Morrison, J.C. Chronology of optic nerve head and retinal responses to elevated intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2000, 41, 431–442. [Google Scholar]
- Pietrucha-Dutczak, M.; Amadio, M.; Govoni, S.; Lewin-Kowalik, J.; Smedowski, A. The Role of endogenous neuroprotective mechanisms in the prevention of retinal ganglion cells degeneration. Front. Neurosci. 2018, 12, 834. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.; Forster, V.; Hicks, D.; Vecino, E. In vivo expression of neurotrophins and neurotrophin receptors is conserved in adult porcine retina in vitro. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4532–4541. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ederra, J.; Hitchcock, P.F.; Vecino, E. Two classes of astrocytes in the adult human and pig retina in terms of their expression of high affinity NGF receptor (TrkA). Neurosci. Lett. 2003, 337, 127–130. [Google Scholar] [CrossRef]
- Vecino, E.; Caminos, E.; Becker, E.; Martín-Zanca, D.; Osborne, N.N. Expression of neurotrophins and their receptors within the glial cells of retina and optic nerve. In Understanding glial cells; Springer: Boston, MA, USA, 1998; pp. 149–166. [Google Scholar] [CrossRef]
- Garcia, M.; Forster, V.; Hicks, D.; Vecino, E. Effects of muller glia on cell survival and neuritogenesis in adult porcine retina in vitro. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3735–3743. [Google Scholar]
- Freeman, R.S.; Burch, R.L.; Crowder, R.J.; Lomb, D.J.; Schoell, M.C.; Straub, J.A.; Xie, L. NGF deprivation-induced gene expression: After ten years, where do we stand? Prog. Brain Res. 2004, 146, 111–126. [Google Scholar] [CrossRef]
- Lomb, D.J.; Desouza, L.A.; Franklin, J.L.; Freeman, R.S. Prolyl hydroxylase inhibitors depend on extracellular glucose and hypoxia-inducible factor (HIF)-2alpha to inhibit cell death caused by nerve growth factor (NGF) deprivation: Evidence that HIF-2alpha has a role in NGF-promoted survival of sympathetic neurons. Mol. Pharmacol. 2009, 75, 1198–1209. [Google Scholar] [CrossRef] [Green Version]
- Roberti, G.; Mantelli, F.; Macchi, I.; Massaro-Giordano, M.; Centofanti, M. Nerve growth factor modulation of retinal ganglion cell physiology. J. Cell. Physiol. 2014, 229, 1130–1133. [Google Scholar] [CrossRef]
- Colafrancesco, V.; Parisi, V.; Sposato, V.; Rossi, S.; Russo, M.A.; Coassin, M.; Lambiase, A.; Aloe, L. Ocular application of nerve growth factor protects degenerating retinal ganglion cells in a rat model of glaucoma. J. Glaucoma 2011, 20, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloe, L.; Rocco, M.L.; Balzamino, B.O.; Micera, A. Nerve growth factor: A focus on neuroscience and therapy. Curr. Neuropharmacol. 2015, 13, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Wang, H.; Liao, S.; Gao, Y.; Liao, R.; Little, P.J.; Xu, J.; Feng, Z.P.; Zheng, Y.; Zheng, W. Nerve growth factor protects retinal ganglion cells against injury induced by retinal ischemia-reperfusion in rats. Growth Factors 2015, 33, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Lambiase, A.; Aloe, L.; Centofanti, M.; Parisi, V.; Bao, S.N.; Mantelli, F.; Colafrancesco, V.; Manni, G.L.; Bucci, M.G.; Bonini, S.; et al. Experimental and clinical evidence of neuroprotection by nerve growth factor eye drops: Implications for glaucoma. Proc. Natl. Acad. Sci. USA 2009, 106, 13469–13474. [Google Scholar] [CrossRef] [Green Version]
- Vecino, E.; Garcia-Crespo, D.; Garcia, M.; Martinez-Millan, L.; Sharma, S.C.; Carrascal, E. Rat retinal ganglion cells co-express brain derived neurotrophic factor (BDNF) and its receptor TrkB. Vis. Res. 2002, 42, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Pernet, V.; Di Polo, A. Synergistic action of brain-derived neurotrophic factor and lens injury promotes retinal ganglion cell survival, but leads to optic nerve dystrophy in vivo. Brain A J. Neurol. 2006, 129, 1014–1026. [Google Scholar] [CrossRef] [Green Version]
- Pietrucha-Dutczak, M.; Smedowski, A.; Liu, X.; Matuszek, I.; Varjosalo, M.; Lewin-Kowalik, J. Candidate proteins from predegenerated nerve exert time-specific protection of retinal ganglion cells in glaucoma. Sci. Rep. 2017, 7, 14540. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Xu, J.; Brahimi, F.; Zhuo, Y.; Sarunic, M.V.; Saragovi, H.U. An agonistic TrkB mAb causes sustained TrkB activation, delays RGC death, and protects the retinal structure in optic nerve axotomy and in glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4722–4731. [Google Scholar] [CrossRef]
- Ghaffariyeh, A.; Honarpisheh, N.; Shakiba, Y.; Puyan, S.; Chamacham, T.; Zahedi, F.; Zarrineghbal, M. Brain-derived neurotrophic factor in patients with normal-tension glaucoma. Optometry 2009, 80, 635–638. [Google Scholar] [CrossRef]
- Oddone, F.; Roberti, G.; Micera, A.; Busanello, A.; Bonini, S.; Quaranta, L.; Agnifili, L.; Manni, G. Exploring serum levels of brain derived neurotrophic factor and nerve growth factor across glaucoma stages. PLoS ONE 2017, 12, e0168565. [Google Scholar] [CrossRef] [Green Version]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Koeberle, P.D.; Ball, A.K. Effects of GDNF on retinal ganglion cell survival following axotomy. Vis. Res. 1998, 38, 1505–1515. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.; Wang, J.; Matheson, C.R.; Urich, J.L. Glial cell line-derived neurotrophic factor (GDNF) promotes the survival of axotomized retinal ganglion cells in adult rats: Comparison to and combination with brain-derived neurotrophic factor (BDNF). J. Neurobiol. 1999, 38, 382–390. [Google Scholar] [CrossRef]
- Kyhn, M.V.; Klassen, H.; Johansson, U.E.; Warfvinge, K.; Lavik, E.; Kiilgaard, J.F.; Prause, J.U.; Scherfig, E.; Young, M.; la Cour, M. Delayed administration of glial cell line-derived neurotrophic factor (GDNF) protects retinal ganglion cells in a pig model of acute retinal ischemia. Exp. Eye Res. 2009, 89, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Checa-Casalengua, P.; Jiang, C.; Bravo-Osuna, I.; Tucker, B.A.; Molina-Martinez, I.T.; Young, M.J.; Herrero-Vanrell, R. Retinal ganglion cells survival in a glaucoma model by GDNF/Vit E PLGA microspheres prepared according to a novel microencapsulation procedure. J. Control. Release Off. J. Control. Release Soc. 2011, 156, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.S.; Khoobehi, A.; Lavik, E.B.; Langer, R.; Young, M.J. Neuroprotection of retinal ganglion cells in DBA/2J mice with GDNF-loaded biodegradable microspheres. J. Pharm. Sci. 2007, 96, 558–568. [Google Scholar] [CrossRef]
- Koeberle, P.D.; Bahr, M. The upregulation of GLAST-1 is an indirect antiapoptotic mechanism of GDNF and neurturin in the adult CNS. Cell Death Differ. 2008, 15, 471–483. [Google Scholar] [CrossRef]
- Del Rio, P.; Irmler, M.; Arango-Gonzalez, B.; Favor, J.; Bobe, C.; Bartsch, U.; Vecino, E.; Beckers, J.; Hauck, S.M.; Ueffing, M. GDNF-induced osteopontin from Muller glial cells promotes photoreceptor survival in the Pde6brd1 mouse model of retinal degeneration. Glia 2011, 59, 821–832. [Google Scholar] [CrossRef]
- Ernst, M.; Jenkins, B.J. Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. TIG 2004, 20, 23–32. [Google Scholar] [CrossRef]
- Kirsch, M.; Lee, M.Y.; Meyer, V.; Wiese, A.; Hofmann, H.D. Evidence for multiple, local functions of ciliary neurotrophic factor (CNTF) in retinal development: Expression of CNTF and its receptors and in vitro effects on target cells. J. Neurochem. 1997, 68, 979–990. [Google Scholar] [CrossRef]
- Wen, R.; Song, Y.; Liu, Y.; Li, Y.; Zhao, L.; Laties, A.M. CNTF negatively regulates the phototransduction machinery in rod photoreceptors: Implication for light-induced photostasis plasticity. Adv. Exp. Med. Biol. 2008, 613, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Mathews, M.K.; Guo, Y.; Langenberg, P.; Bernstein, S.L. Ciliary neurotrophic factor (CNTF)-mediated ganglion cell survival in a rodent model of non-arteritic anterior ischaemic optic neuropathy (NAION). Br. J. Ophthalmol. 2015, 99, 133–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pease, M.E.; Zack, D.J.; Berlinicke, C.; Bloom, K.; Cone, F.; Wang, Y.; Klein, R.L.; Hauswirth, W.W.; Quigley, H.A. Effect of CNTF on retinal ganglion cell survival in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, K.; Rau, C.R.; Storch, M.K.; Sattler, M.B.; Demmer, I.; Weissert, R.; Taheri, N.; Kuhnert, A.V.; Bahr, M.; Diem, R. Ciliary neurotrophic factor protects retinal ganglion cells from secondary cell death during acute autoimmune optic neuritis in rats. Brain Pathol. 2004, 14, 378–387. [Google Scholar] [CrossRef]
- Fischer, D.; Leibinger, M. Promoting optic nerve regeneration. Prog. Retin. Eye Res. 2012, 31, 688–701. [Google Scholar] [CrossRef]
- Muller, A.; Hauk, T.G.; Fischer, D. Astrocyte-derived CNTF switches mature RGCs to a regenerative state following inflammatory stimulation. Brain A J. Neurol. 2007, 130, 3308–3320. [Google Scholar] [CrossRef] [Green Version]
- Shpak, A.A.; Guekht, A.B.; Druzhkova, T.A.; Kozlova, K.I.; Gulyaeva, N.V. Ciliary neurotrophic factor in patients with primary open-angle glaucoma and age-related cataract. Mol. Vis. 2017, 23, 799–809. [Google Scholar]
- Zhou, X.; Li, F.; Kong, L.; Chodosh, J.; Cao, W. Anti-inflammatory effect of pigment epithelium-derived factor in DBA/2J mice. Mol. Vis. 2009, 15, 438–450. [Google Scholar]
- Yang, X.; Wei, A.; Liu, Y.; He, G.; Zhou, Z.; Yu, Z. IGF-1 protects retinal ganglion cells from hypoxia-induced apoptosis by activating the Erk-1/2 and Akt pathways. Mol. Vis. 2013, 19, 1901–1912. [Google Scholar]
- Kermer, P.; Klocker, N.; Labes, M.; Bahr, M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Foxton, R.H.; Finkelstein, A.; Vijay, S.; Dahlmann-Noor, A.; Khaw, P.T.; Morgan, J.E.; Shima, D.T.; Ng, Y.S. VEGF-A is necessary and sufficient for retinal neuroprotection in models of experimental glaucoma. Am. J. Pathol. 2013, 182, 1379–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, B.; Wang, R.; Gao, X.; Dong, X.; Ji, X. Effect of vascular endothelial growth factor on retinal ganglion cells of rats with chronic intraocular hypertension. Int. J. Clin. Exp. Pathol. 2014, 7, 5717–5724. [Google Scholar] [PubMed]
- Brar, V.S.; Sharma, R.K.; Murthy, R.K.; Chalam, K.V. Bevacizumab neutralizes the protective effect of vascular endothelial growth factor on retinal ganglion cells. Mol. Vis. 2010, 16, 1848–1853. [Google Scholar] [PubMed]
- Lee, W.J.; Kim, Y.K.; Kim, Y.W.; Jeoung, J.W.; Kim, S.H.; Heo, J.W.; Yu, H.G.; Park, K.H. Rate of macular ganglion cell-inner plexiform layer thinning in glaucomatous eyes with vascular endothelial growth factor inhibition. J. Glaucoma 2017, 26, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Sisk, D.R.; Kuwabara, T. Histologic changes in the inner retina of albino rats following intravitreal injection of monosodium L-glutamate. Graefes Arch. Clin. Exp. Ophthalmol. 1985, 223, 250–258. [Google Scholar] [CrossRef]
- Sucher, N.J.; Lipton, S.A.; Dreyer, E.B. Molecular basis of glutamate toxicity in retinal ganglion cells. Vis. Res. 1997, 37, 3483–3493. [Google Scholar] [CrossRef] [Green Version]
- Vorwerk, C.K.; Lipton, S.A.; Zurakowski, D.; Hyman, B.T.; Sabel, B.A.; Dreyer, E.B. Chronic low-dose glutamate is toxic to retinal ganglion cells. Toxicity blocked by memantine. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1618–1624. [Google Scholar]
- Dreyer, E.B.; Zurakowski, D.; Schumer, R.A.; Podos, S.M.; Lipton, S.A. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch. Ophthalmol. 1996, 114, 299–305. [Google Scholar] [CrossRef]
- Brooks, D.E.; Garcia, G.A.; Dreyer, E.B.; Zurakowski, D.; Franco-Bourland, R.E. Vitreous body glutamate concentration in dogs with glaucoma. Am. J. Vet. Res. 1997, 58, 864–867. [Google Scholar]
- Chaudhary, P.; Ahmed, F.; Sharma, S.C. MK801-a neuroprotectant in rat hypertensive eyes. Brain Res. 1998, 792, 154–158. [Google Scholar] [CrossRef]
- Guo, L.; Salt, T.E.; Maass, A.; Luong, V.; Moss, S.E.; Fitzke, F.W.; Cordeiro, M.F. Assessment of neuroprotective effects of glutamate modulation on glaucoma-related retinal ganglion cell apoptosis in vivo. Investig. Ophthalmol. Vis. Sci. 2006, 47, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Prospects for clinically tolerated NMDA antagonists: Open-channel blockers and alternative redox states of nitric oxide. Trends Neurosci. 1993, 16, 527–532. [Google Scholar] [CrossRef]
- Lagreze, W.A.; Knorle, R.; Bach, M.; Feuerstein, T.J. Memantine is neuroprotective in a rat model of pressure-induced retinal ischemia. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1063–1066. [Google Scholar]
- Hare, W.A.; WoldeMussie, E.; Lai, R.K.; Ton, H.; Ruiz, G.; Chun, T.; Wheeler, L. Efficacy and safety of memantine treatment for reduction of changes associated with experimental glaucoma in monkey, I: Functional measures. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2625–2639. [Google Scholar] [CrossRef] [PubMed]
- Danesh-Meyer, H.V.; Levin, L.A. Neuroprotection: Extrapolating from neurologic diseases to the eye. Am. J. Ophthalmol. 2009, 148, 186–191.e2. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.A.; Gil, D.W.; WoldeMussie, E. Role of alpha-2 adrenergic receptors in neuroprotection and glaucoma. Surv. Ophthalmol. 2001, 45 (Suppl. 3), S290–S294, discussion S295–S296. [Google Scholar] [CrossRef]
- Hernandez, M.; Urcola, J.H.; Vecino, E. Retinal ganglion cell neuroprotection in a rat model of glaucoma following brimonidine, latanoprost or combined treatments. Exp. Eye Res. 2008, 86, 798–806. [Google Scholar] [CrossRef]
- Pinar-Sueiro, S.; Urcola, H.; Rivas, M.A.; Vecino, E. Prevention of retinal ganglion cell swelling by systemic brimonidine in a rat experimental glaucoma model. Clin. Exp. Ophthalmol. 2011, 39, 799–807. [Google Scholar] [CrossRef]
- Ahmed, F.A.; Hegazy, K.; Chaudhary, P.; Sharma, S.C. Neuroprotective effect of alpha(2) agonist (brimonidine) on adult rat retinal ganglion cells after increased intraocular pressure. Brain Res. 2001, 913, 133–139. [Google Scholar] [CrossRef]
- Yoles, E.; Wheeler, L.A.; Schwartz, M. Alpha2-adrenoreceptor agonists are neuroprotective in a rat model of optic nerve degeneration. Investig. Ophthalmol. Vis. Sci. 1999, 40, 65–73. [Google Scholar]
- Donello, J.E.; Padillo, E.U.; Webster, M.L.; Wheeler, L.A.; Gil, D.W. alpha(2)-Adrenoceptor agonists inhibit vitreal glutamate and aspartate accumulation and preserve retinal function after transient ischemia. J. Pharmacol. Exp. Ther. 2001, 296, 216–223. [Google Scholar] [PubMed]
- Kalapesi, F.B.; Coroneo, M.T.; Hill, M.A. Human ganglion cells express the alpha-2 adrenergic receptor: Relevance to neuroprotection. Br. J. Ophthalmol. 2005, 89, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung, T.; Oen, F.T.; Wong, H.T.; Chan, Y.H.; Khoo, B.K.; Liu, Y.P.; Ho, C.L.; See, J.; Thean, L.H.; Viswanathan, A.C.; et al. Randomised controlled trial comparing the effect of brimonidine and timolol on visual field loss after acute primary angle closure. Br. J. Ophthalmol. 2004, 88, 88–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, W.S.; Ruiz, L.; Crish, S.D.; Wheeler, L.A.; Calkins, D.J. Brimonidine prevents axonal and somatic degeneration of retinal ganglion cell neurons. Mol. Neurodegener. 2011, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.C.; Chang, H.W. Comparison of the effects of brimonidine 0.2% and timolol 0.5% on retinal nerve fiber layer thickness in ocular hypertensive patients: A prospective, unmasked study. J. Ocul. Pharmacol. Ther. 2005, 21, 475–482. [Google Scholar] [CrossRef]
- Doozandeh, A.; Yazdani, S. Neuroprotection in glaucoma. J. Ophthalmic Vis. Res. 2016, 11, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Crish, S.D.; Calkins, D.J. Neurodegeneration in glaucoma: Progression and calcium-dependent intracellular mechanisms. Neuroscience 2011, 176, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [CrossRef]
- Osborne, N.N.; Wood, J.P.; Cupido, A.; Melena, J.; Chidlow, G. Topical flunarizine reduces IOP and protects the retina against ischemia-excitotoxicity. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1456–1464. [Google Scholar]
- Yamada, H.; Chen, Y.N.; Aihara, M.; Araie, M. Neuroprotective effect of calcium channel blocker against retinal ganglion cell damage under hypoxia. Brain Res. 2006, 1071, 75–80. [Google Scholar] [CrossRef]
- Koseki, N.; Araie, M.; Tomidokoro, A.; Nagahara, M.; Hasegawa, T.; Tamaki, Y.; Yamamoto, S. A placebo-controlled 3-year study of a calcium blocker on visual field and ocular circulation in glaucoma with low-normal pressure. Ophthalmology 2008, 115, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Mayama, C. Calcium channels and their blockers in intraocular pressure and glaucoma. Eur. J. Pharmacol. 2014, 739, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Bagnis, A.; Sacca, S.C. The role of oxidative stress in glaucoma. Mutat. Res. 2006, 612, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Geiger, L.K.; Kortuem, K.R.; Alexejun, C.; Levin, L.A. Reduced redox state allows prolonged survival of axotomized neonatal retinal ganglion cells. Neuroscience 2002, 109, 635–642. [Google Scholar] [CrossRef]
- Caprioli, J.; Munemasa, Y.; Kwong, J.M.; Piri, N. Overexpression of thioredoxins 1 and 2 increases retinal ganglion cell survival after pharmacologically induced oxidative stress, optic nerve transection, and in experimental glaucoma. Trans. Am. Ophthalmol. Soc. 2009, 107, 161–165. [Google Scholar]
- Swanson, K.I.; Schlieve, C.R.; Lieven, C.J.; Levin, L.A. Neuroprotective effect of sulfhydryl reduction in a rat optic nerve crush model. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3737–3741. [Google Scholar] [CrossRef]
- Nucci, C.; Tartaglione, R.; Cerulli, A.; Mancino, R.; Spano, A.; Cavaliere, F.; Rombola, L.; Bagetta, G.; Corasaniti, M.T.; Morrone, L.A. Retinal damage caused by high intraocular pressure-induced transient ischemia is prevented by coenzyme Q10 in rat. Int. Rev. Neurobiol. 2007, 82, 397–406. [Google Scholar] [CrossRef]
- Russo, R.; Cavaliere, F.; Rombola, L.; Gliozzi, M.; Cerulli, A.; Nucci, C.; Fazzi, E.; Bagetta, G.; Corasaniti, M.T.; Morrone, L.A. Rational basis for the development of coenzyme Q10 as a neurotherapeutic agent for retinal protection. Prog. Brain Res. 2008, 173, 575–582. [Google Scholar] [CrossRef]
- Nakajima, Y.; Inokuchi, Y.; Nishi, M.; Shimazawa, M.; Otsubo, K.; Hara, H. Coenzyme Q10 protects retinal cells against oxidative stress in vitro and in vivo. Brain Res. 2008, 1226, 226–233. [Google Scholar] [CrossRef]
- Pinar-Sueiro, S.; Martinez-Fernandez, R.; Lage-Medina, S.; Aldamiz-Echevarria, L.; Vecino, E. Optic neuropathy in methylmalonic acidemia: The role of neuroprotection. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S199–S203. [Google Scholar] [CrossRef]
- Gherghel, D.; Griffiths, H.R.; Hilton, E.J.; Cunliffe, I.A.; Hosking, S.L. Systemic reduction in glutathione levels occurs in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Aydemir, O.; Naziroglu, M.; Celebi, S.; Yilmaz, T.; Kukner, A.S. Antioxidant effects of alpha-, gamma- and succinate-tocopherols in guinea pig retina during ischemia-reperfusion injury. Pathophysiol. Off. J. Int. Soc. Pathophysiol. 2004, 11, 167–171. [Google Scholar] [CrossRef]
- Dilsiz, N.; Sahaboglu, A.; Yildiz, M.Z.; Reichenbach, A. Protective effects of various antioxidants during ischemia-reperfusion in the rat retina. Graefes Arch. Clin. Exp. Ophthalmol. 2006, 244, 627–633. [Google Scholar] [CrossRef]
- Ko, M.L.; Peng, P.H.; Hsu, S.Y.; Chen, C.F. Dietary deficiency of vitamin E aggravates retinal ganglion cell death in experimental glaucoma of rats. Curr. Eye Res. 2010, 35, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, Q.; Wang, R.; Chen, Z.; Wu, J.; Xia, F.; Fan, X.Q. Methane attenuates retinal ischemia/reperfusion injury via anti-oxidative and anti-apoptotic pathways. Brain Res. 2016, 1646, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Schultz, R.; Witte, O.W.; Schmeer, C. Increased frataxin levels protect retinal ganglion cells after acute ischemia/reperfusion in the mouse retina in vivo. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4115–4124. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qi, Y.; Yang, X. Neuroprotective effects of crocin against oxidative stress induced by ischemia/reperfusion injury in rat retina. Ophthalmic Res. 2015, 54, 157–168. [Google Scholar] [CrossRef]
- Nebbioso, M.; Scarsella, G.; Tafani, M.; Pescosolido, N. Mechanisms of ocular neuroprotection by antioxidant molecules in animal models. J. Biol. Regul. Homeost. Agents 2013, 27, 197–209. [Google Scholar]
- Jiang, W.; Tang, L.; Zeng, J.; Chen, B. Adeno-associated virus mediated SOD gene therapy protects the retinal ganglion cells from chronic intraocular pressure elevation induced injury via attenuating oxidative stress and improving mitochondrial dysfunction in a rat model. Am. J. Transl. Res. 2016, 8, 799–810. [Google Scholar]
- Park, S.H.; Kim, J.H.; Kim, Y.H.; Park, C.K. Expression of neuronal nitric oxide synthase in the retina of a rat model of chronic glaucoma. Vis. Res. 2007, 47, 2732–2740. [Google Scholar] [CrossRef] [Green Version]
- Aslan, M.; Cort, A.; Yucel, I. Oxidative and nitrative stress markers in glaucoma. Free Radic. Biol. Med. 2008, 45, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Siu, A.W.; Leung, M.C.; To, C.H.; Siu, F.K.; Ji, J.Z.; So, K.F. Total retinal nitric oxide production is increased in intraocular pressure-elevated rats. Exp. Eye Res. 2002, 75, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Shareef, S.; Sawada, A.; Neufeld, A.H. Isoforms of nitric oxide synthase in the optic nerves of rat eyes with chronic moderately elevated intraocular pressure. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2884–2891. [Google Scholar]
- Hangai, M.; Yoshimura, N.; Hiroi, K.; Mandai, M.; Honda, Y. Inducible nitric oxide synthase in retinal ischemia-reperfusion injury. Exp. Eye Res. 1996, 63, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, A.H.; Kawai, S.; Das, S.; Vora, S.; Gachie, E.; Connor, J.R.; Manning, P.T. Loss of retinal ganglion cells following retinal ischemia: The role of inducible nitric oxide synthase. Exp. Eye Res. 2002, 75, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Neufeld, A.H. Expression of nitric oxide synthase-2 (NOS-2) in reactive astrocytes of the human glaucomatous optic nerve head. Glia 2000, 30, 178–186. [Google Scholar] [CrossRef]
- Neufeld, A.H.; Hernandez, M.R.; Gonzalez, M. Nitric oxide synthase in the human glaucomatous optic nerve head. Arch. Ophthalmol. 1997, 115, 497–503. [Google Scholar] [CrossRef]
- Neufeld, A.H.; Sawada, A.; Becker, B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proc. Natl. Acad. Sci. USA 1999, 96, 9944–9948. [Google Scholar] [CrossRef] [Green Version]
- Geyer, O.; Almog, J.; Lupu-Meiri, M.; Lazar, M.; Oron, Y. Nitric oxide synthase inhibitors protect rat retina against ischemic injury. FEBS Lett. 1995, 374, 399–402. [Google Scholar] [CrossRef] [Green Version]
- Libby, R.T.; Howell, G.R.; Pang, I.H.; Savinova, O.V.; Mehalow, A.K.; Barter, J.W.; Smith, R.S.; Clark, A.F.; John, S.W. Inducible nitric oxide synthase, Nos2, does not mediate optic neuropathy and retinopathy in the DBA/2J glaucoma model. BMC Neurosci. 2007, 8, 108. [Google Scholar] [CrossRef] [Green Version]
- Pang, I.H.; Johnson, E.C.; Jia, L.; Cepurna, W.O.; Shepard, A.R.; Hellberg, M.R.; Clark, A.F.; Morrison, J.C. Evaluation of inducible nitric oxide synthase in glaucomatous optic neuropathy and pressure-induced optic nerve damage. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1313–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oku, H.; Goto, W.; Kobayashi, T.; Okuno, T.; Hirao, M.; Sugiyama, T.; Yoneda, S.; Hara, H.; Ikeda, T. Adenosine protects cultured retinal neurons against NMDA-induced cell death through A1 receptors. Curr. Eye Res. 2004, 29, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Barnes, S.; Baldridge, W.H. Adenosine inhibits calcium channel currents via A1 receptors on salamander retinal ganglion cells in a mini-slice preparation. J. Neurochem. 2002, 81, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.K.; Osborne, N.N. Involvement of adenosine in retinal ischemia. Studies on the rat. Investig. Ophthalmol. Vis. Sci. 1996, 37, 2603–2611. [Google Scholar]
- Perigolo-Vicente, R.; Ritt, K.; Pereira, M.R.; Torres, P.M.; Paes-de-Carvalho, R.; Giestal-de-Araujo, E. IL-6 treatment increases the survival of retinal ganglion cells in vitro: The role of adenosine A1 receptor. Biochem. Biophys. Res. Commun. 2013, 430, 512–518. [Google Scholar] [CrossRef] [Green Version]
- Mendonca Torres, P.M.; de Araujo, E.G. Interleukin-6 increases the survival of retinal ganglion cells in vitro. J. Neuroimmunol. 2001, 117, 43–50. [Google Scholar] [CrossRef]
- Sappington, R.M.; Chan, M.; Calkins, D.J. Interleukin-6 protects retinal ganglion cells from pressure-induced death. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2932–2942. [Google Scholar] [CrossRef] [Green Version]
- Murphy, P.G.; Borthwick, L.A.; Altares, M.; Gauldie, J.; Kaplan, D.; Richardson, P.M. Reciprocal actions of interleukin-6 and brain-derived neurotrophic factor on rat and mouse primary sensory neurons. Eur. J. Neurosci. 2000, 12, 1891–1899. [Google Scholar] [CrossRef]
- Mailavaram, R.P.; Al-Attraqchi, O.H.A.; Kar, S.; Ghosh, S. Current status in the design and development of agonists and antagonists of adenosine A3 receptor as potential therapeutic agents. Curr. Pharm. Des. 2019, 25, 2772–2787. [Google Scholar] [CrossRef]
- Zhang, M.; Budak, M.T.; Lu, W.; Khurana, T.S.; Zhang, X.; Laties, A.M.; Mitchell, C.H. Identification of the A3 adenosine receptor in rat retinal ganglion cells. Mol. Vis. 2006, 12, 937–948. [Google Scholar]
- Hu, H.; Lu, W.; Zhang, M.; Zhang, X.; Argall, A.J.; Patel, S.; Lee, G.E.; Kim, Y.C.; Jacobson, K.A.; Laties, A.M.; et al. Stimulation of the P2X7 receptor kills rat retinal ganglion cells in vivo. Exp. Eye Res. 2010, 91, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, M.; Laties, A.M.; Mitchell, C.H. Balance of purines may determine life or death of retinal ganglion cells as A3 adenosine receptors prevent loss following P2X7 receptor stimulation. J. Neurochem. 2006, 98, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, H.; Zhang, X.; Lu, W.; Lim, J.; Eysteinsson, T.; Jacobson, K.A.; Laties, A.M.; Mitchell, C.H. The A3 adenosine receptor attenuates the calcium rise triggered by NMDA receptors in retinal ganglion cells. Neurochem. Int. 2010, 56, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, K.I.; Iwao, K.; Inoue, T.; Haga, A.; Tsutsumi, T.; Mochita, M.I.; Fujimoto, T.; Tanihara, H. Stimulation of the adenosine A3 receptor, not the A1 or A2 receptors, promote neurite outgrowth of retinal ganglion cells. Exp. Eye Res. 2018, 170, 160–168. [Google Scholar] [CrossRef]
- Galvao, J.; Elvas, F.; Martins, T.; Cordeiro, M.F.; Ambrosio, A.F.; Santiago, A.R. Adenosine A3 receptor activation is neuroprotective against retinal neurodegeneration. Exp. Eye Res. 2015, 140, 65–74. [Google Scholar] [CrossRef]
- Cen, L.P.; Ng, T.K. Stem cell therapy for retinal ganglion cell degeneration. Neural Regen. Res. 2018, 13, 1352–1353. [Google Scholar] [CrossRef]
- Bennicelli, J.L.; Bennett, J. Stem cells set their sights on retinitis pigmentosa. eLife 2013, 2, e01291. [Google Scholar] [CrossRef]
- Siqueira, R.C. Stem cell therapy for retinal diseases: Update. Stem Cell Res. Ther. 2011, 2, 50. [Google Scholar] [CrossRef] [Green Version]
- Zarbin, M. Cell-based therapy for degenerative retinal disease. Trends Mol. Med. 2016, 22, 115–134. [Google Scholar] [CrossRef]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiraku, M.; Sasai, Y. Mouse embryonic stem cell culture for generation of three-dimensional retinal and cortical tissues. Nat. Protoc. 2011, 7, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.; Berry, M.; Logan, A.; Scott, R.A.; Leadbeater, W.; Scheven, B.A. Stem cell treatment of degenerative eye disease. Stem Cell Res. 2015, 14, 243–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankin, M.H.; Lund, R.D. Directed early axonal outgrowth from retinal transplants into host rat brains. J. Neurobiol. 1990, 21, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Lund, R.D.; Hankin, M.H. Pathfinding by retinal ganglion cell axons: Transplantation studies in genetically and surgically blind mice. J. Comp. Neurol. 1995, 356, 481–489. [Google Scholar] [CrossRef]
- Hertz, J.; Qu, B.; Hu, Y.; Patel, R.D.; Valenzuela, D.A.; Goldberg, J.L. Survival and integration of developing and progenitor-derived retinal ganglion cells following transplantation. Cell Transplant. 2014, 23, 855–872. [Google Scholar] [CrossRef]
- Venugopalan, P.; Wang, Y.; Nguyen, T.; Huang, A.; Muller, K.J.; Goldberg, J.L. Transplanted neurons integrate into adult retinas and respond to light. Nat. Commun. 2016, 7, 10472. [Google Scholar] [CrossRef]
- Tang, R.; Jing, L.; Willard, V.P.; Wu, C.L.; Guilak, F.; Chen, J.; Setton, L.A. Differentiation of human induced pluripotent stem cells into nucleus pulposus-like cells. Stem Cell Res. Ther. 2018, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Razavi, S.; Razavi, M.R.; Zarkesh Esfahani, H.; Kazemi, M.; Mostafavi, F.S. Comparing brain-derived neurotrophic factor and ciliary neurotrophic factor secretion of induced neurotrophic factor secreting cells from human adipose and bone marrow-derived stem cells. Dev. Growth Differ. 2013, 55, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Tan, H.B.; Wang, X.M.; Rong, H.; Cui, H.P.; Cui, H. Bone marrow mesenchymal stem cells protect against retinal ganglion cell loss in aged rats with glaucoma. Clin. Interv. Aging 2013, 8, 1467–1470. [Google Scholar] [CrossRef] [Green Version]
- Osborne, A.; Sanderson, J.; Martin, K.R. Neuroprotective Effects of Human Mesenchymal Stem Cells and Platelet-Derived Growth Factor on Human Retinal Ganglion Cells. Stem Cells 2018, 36, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Mead, B.; Amaral, J.; Tomarev, S. Mesenchymal stem cell-derived small extracellular vesicles promote neuroprotection in rodent models of glaucoma. Investig. Ophthalmol. Vis. Sci. 2018, 59, 702–714. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory response in the CNS: Friend or foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [Green Version]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Sriram, K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin. Drug Saf. 2005, 4, 433–442. [Google Scholar] [CrossRef]
- Silverman, S.M.; Wong, W.T. Microglia in the retina: Roles in development, maturity, and disease. Ann. Rev. Vis. Sci. 2018, 4, 45–77. [Google Scholar] [CrossRef]
- Karlstetter, M.; Ebert, S.; Langmann, T. Microglia in the healthy and degenerating retina: Insights from novel mouse models. Immunobiology 2010, 215, 685–691. [Google Scholar] [CrossRef]
- Karlstetter, M.; Langmann, T. Microglia in the aging retina. Adv. Exp. Med. Biol. 2014, 801, 207–212. [Google Scholar] [CrossRef]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in retinal degeneration. Front. Immunol. 2019, 10, 1975. [Google Scholar] [CrossRef] [Green Version]
- Takeda, A.; Shinozaki, Y.; Kashiwagi, K.; Ohno, N.; Eto, K.; Wake, H.; Nabekura, J.; Koizumi, S. Microglia mediate non-cell-autonomous cell death of retinal ganglion cells. Glia 2018, 66, 2366–2384. [Google Scholar] [CrossRef]
- Ullian, E.M.; Barkis, W.B.; Chen, S.; Diamond, J.S.; Barres, B.A. Invulnerability of retinal ganglion cells to NMDA excitotoxicity. Mol. Cell. Neurosci. 2004, 26, 544–557. [Google Scholar] [CrossRef]
- Neufeld, A.H. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch. Ophthalmol. 1999, 117, 1050–1056. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Neufeld, A.H. Activated microglia in the human glaucomatous optic nerve head. J. Neurosci. Res. 2001, 64, 523–532. [Google Scholar] [CrossRef]
- Bosco, A.; Steele, M.R.; Vetter, M.L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011, 519, 599–620. [Google Scholar] [CrossRef] [Green Version]
- Bosco, A.; Romero, C.O.; Breen, K.T.; Chagovetz, A.A.; Steele, M.R.; Ambati, B.K.; Vetter, M.L. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Dis. Models Mech. 2015, 8, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Madeira, M.H.; Boia, R.; Elvas, F.; Martins, T.; Cunha, R.A.; Ambrosio, A.F.; Santiago, A.R. Selective A2A receptor antagonist prevents microglia-mediated neuroinflammation and protects retinal ganglion cells from high intraocular pressure-induced transient ischemic injury. Transl. Res. J. Lab. Clin. Med. 2016, 169, 112–128. [Google Scholar] [CrossRef]
- Cho, K.J.; Kim, J.H.; Park, H.Y.; Park, C.K. Glial cell response and iNOS expression in the optic nerve head and retina of the rat following acute high IOP ischemia-reperfusion. Brain Res. 2011, 1403, 67–77. [Google Scholar] [CrossRef]
- Madeira, M.H.; Ortin-Martinez, A.; Nadal-Nicolas, F.; Ambrosio, A.F.; Vidal-Sanz, M.; Agudo-Barriuso, M.; Santiago, A.R. Caffeine administration prevents retinal neuroinflammation and loss of retinal ganglion cells in an animal model of glaucoma. Sci. Rep. 2016, 6, 27532. [Google Scholar] [CrossRef]
- Rodrigues-Neves, A.C.; Aires, I.D.; Vindeirinho, J.; Boia, R.; Madeira, M.H.; Goncalves, F.Q.; Cunha, R.A.; Santos, P.F.; Ambrosio, A.F.; Santiago, A.R. Elevated Pressure Changes the Purinergic System of Microglial Cells. Front. Pharmacol. 2018, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Santiago, A.R.; Baptista, F.I.; Santos, P.F.; Cristovao, G.; Ambrosio, A.F.; Cunha, R.A.; Gomes, C.A. Role of microglia adenosine A(2A) receptors in retinal and brain neurodegenerative diseases. Mediat. Inflamm. 2014, 2014, 465694. [Google Scholar] [CrossRef]
- Boia, R.; Elvas, F.; Madeira, M.H.; Aires, I.D.; Rodrigues-Neves, A.C.; Tralhao, P.; Szabo, E.C.; Baqi, Y.; Muller, C.E.; Tome, A.R.; et al. Treatment with A2A receptor antagonist KW6002 and caffeine intake regulate microglia reactivity and protect retina against transient ischemic damage. Cell Death Dis. 2017, 8, e3065. [Google Scholar] [CrossRef] [Green Version]
- Boia, R.; Ambrosio, A.F.; Santiago, A.R. Therapeutic opportunities for caffeine and A2A receptor antagonists in retinal diseases. Ophthalmic Res. 2016, 55, 212–218. [Google Scholar] [CrossRef]
- Tezel, G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Neufeld, A.H. Tumor necrosis factor-alpha: A potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia 2000, 32, 42–50. [Google Scholar] [CrossRef]
- Roh, M.; Zhang, Y.; Murakami, Y.; Thanos, A.; Lee, S.C.; Vavvas, D.G.; Benowitz, L.I.; Miller, J.W. Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS ONE 2012, 7, e40065. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Sun, Q.; Xue, S.; Guan, H.; Ji, M. Activation of P2X7R- NLRP3 pathway in Retinal microglia contribute to Retinal Ganglion Cells death in chronic ocular hypertension (COH). Exp. Eye Res. 2019, 188, 107771. [Google Scholar] [CrossRef]
- Fernandez-Albarral, J.A.; Ramirez, A.I.; de Hoz, R.; Lopez-Villarin, N.; Salobrar-Garcia, E.; Lopez-Cuenca, I.; Licastro, E.; Inarejos-Garcia, A.M.; Almodovar, P.; Pinazo-Duran, M.D.; et al. Neuroprotective and Anti-Inflammatory Effects of a Hydrophilic Saffron Extract in a Model of Glaucoma. Int. J. Mol. Sci. 2019, 20, 4110. [Google Scholar] [CrossRef] [Green Version]
- Howell, G.R.; Macalinao, D.G.; Sousa, G.L.; Walden, M.; Soto, I.; Kneeland, S.C.; Barbay, J.M.; King, B.L.; Marchant, J.K.; Hibbs, M.; et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Investig. 2011, 121, 1429–1444. [Google Scholar] [CrossRef]
- Mirzaei, M.; Gupta, V.B.; Chick, J.M.; Greco, T.M.; Wu, Y.; Chitranshi, N.; Wall, R.V.; Hone, E.; Deng, L.; Dheer, Y.; et al. Age-related neurodegenerative disease associated pathways identified in retinal and vitreous proteome from human glaucoma eyes. Sci. Rep. 2017, 7, 12685. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Yang, X.; Luo, C.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Kaplan, H.J. Oxidative stress and the regulation of complement activation in human glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5071–5082. [Google Scholar] [CrossRef]
- Tyler, C.M.; Boulanger, L.M. Complement-mediated microglial clearance of developing retinal ganglion cell axons. Neuron 2012, 74, 597–599. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Rosen, A.M.; Stevens, B. The role of the classical complement cascade in synapse loss during development and glaucoma. Adv. Exp. Med. Biol. 2010, 703, 75–93. [Google Scholar] [CrossRef]
- Wang, K.; Peng, B.; Lin, B. Fractalkine receptor regulates microglial neurotoxicity in an experimental mouse glaucoma model. Glia 2014, 62, 1943–1954. [Google Scholar] [CrossRef]
- Breen, K.T.; Anderson, S.R.; Steele, M.R.; Calkins, D.J.; Bosco, A.; Vetter, M.L. Loss of fractalkine signaling exacerbates axon transport dysfunction in a chronic model of glaucoma. Front. Neurosci. 2016, 10, 526. [Google Scholar] [CrossRef] [Green Version]
- Bosco, A.; Inman, D.M.; Steele, M.R.; Wu, G.; Soto, I.; Marsh-Armstrong, N.; Hubbard, W.C.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1437–1446. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Ruzafa, N.; Vecino, E. Effect of Muller cells on the survival and neuritogenesis in retinal ganglion cells. Arch. Soc. Esp. Oftalmol. 2015, 90, 522–526. [Google Scholar] [CrossRef]
- Ruzafa, N.; Pereiro, X.; Lepper, M.F.; Hauck, S.M.; Vecino, E. A proteomics approach to identify candidate proteins secreted by muller glia that protect ganglion cells in the retina. Proteomics 2018, 18, e1700321. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Biedermann, B.; Francke, M.; Iandiev, I.; Grosche, J.; Wiedemann, P.; Albrecht, J.; Reichenbach, A. Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem. Int. 2009, 54, 143–160. [Google Scholar] [CrossRef]
- Garcia, M.; Vecino, E. Role of Muller glia in neuroprotection and regeneration in the retina. Histol. Histopathol. 2003, 18, 1205–1218. [Google Scholar] [CrossRef]
- Heidinger, V.; Hicks, D.; Sahel, J.; Dreyfus, H. Ability of retinal Muller glial cells to protect neurons against excitotoxicity in vitro depends upon maturation and neuron-glial interactions. Glia 1999, 25, 229–239. [Google Scholar] [CrossRef]
- Kawasaki, A.; Otori, Y.; Barnstable, C.J. Muller cell protection of rat retinal ganglion cells from glutamate and nitric oxide neurotoxicity. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3444–3450. [Google Scholar]
- Kitano, S.; Morgan, J.; Caprioli, J. Hypoxic and excitotoxic damage to cultured rat retinal ganglion cells. Exp. Eye Res. 1996, 63, 105–112. [Google Scholar] [CrossRef]
- Izumi, Y.; Kirby, C.O.; Benz, A.M.; Olney, J.W.; Zorumski, C.F. Muller cell swelling, glutamate uptake, and excitotoxic neurodegeneration in the isolated rat retina. Glia 1999, 25, 379–389. [Google Scholar] [CrossRef]
- Carter-Dawson, L.; Crawford, M.L.; Harwerth, R.S.; Smith, E.L., 3rd; Feldman, R.; Shen, F.F.; Mitchell, C.K.; Whitetree, A. Vitreal glutamate concentration in monkeys with experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2633–2637. [Google Scholar]
- Biedermann, B.; Bringmann, A.; Franze, K.; Faude, F.; Wiedemann, P.; Reichenbach, A. GABA(A) receptors in Muller glial cells of the human retina. Glia 2004, 46, 302–310. [Google Scholar] [CrossRef]
- Hurley, J.B.; Lindsay, K.J.; Du, J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef] [Green Version]
- Matteucci, A.; Gaddini, L.; Villa, M.; Varano, M.; Parravano, M.; Monteleone, V.; Cavallo, F.; Leo, L.; Mallozzi, C.; Malchiodi-Albedi, F.; et al. Neuroprotection by rat Muller glia against high glucose-induced neurodegeneration through a mechanism involving ERK1/2 activation. Exp. Eye Res. 2014, 125, 20–29. [Google Scholar] [CrossRef]
- Vohra, R.; Kolko, M. Neuroprotection of the inner retina: Muller cells and lactate. Neural Regen. Res. 2018, 13, 1741–1742. [Google Scholar] [CrossRef]
- Eastlake, K.; Luis, J.; Limb, G.A. Potential of muller glia for retina neuroprotection. Curr. Eye Res. 2020, 45, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Reichelt, W.; Stabel-Burow, J.; Pannicke, T.; Weichert, H.; Heinemann, U. The glutathione level of retinal Muller glial cells is dependent on the high-affinity sodium-dependent uptake of glutamate. Neuroscience 1997, 77, 1213–1224. [Google Scholar] [CrossRef]
- Harada, T.; Harada, C.; Nakamura, K.; Quah, H.M.; Okumura, A.; Namekata, K.; Saeki, T.; Aihara, M.; Yoshida, H.; Mitani, A.; et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J. Clin. Investig. 2007, 117, 1763–1770. [Google Scholar] [CrossRef] [Green Version]
- Eichler, W.; Savkovic-Cvijic, H.; Burger, S.; Beck, M.; Schmidt, M.; Wiedemann, P.; Reichenbach, A.; Unterlauft, J.D. Muller cell-derived PEDF mediates neuroprotection via STAT3 activation. Cell. Physiol. Biochem. 2017, 44, 1411–1424. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Srinivasan, B.; Wordinger, R.J.; Roque, R.S. Glutamate stimulates neurotrophin expression in cultured Muller cells. Brain Res. Mol. Brain Res. 2003, 111, 189–197. [Google Scholar] [CrossRef]
- Harada, T.; Harada, C.; Nakayama, N.; Okuyama, S.; Yoshida, K.; Kohsaka, S.; Matsuda, H.; Wada, K. Modification of glial-neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron 2000, 26, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Eastlake, K.; Banerjee, P.J.; Angbohang, A.; Charteris, D.G.; Khaw, P.T.; Limb, G.A. Muller glia as an important source of cytokines and inflammatory factors present in the gliotic retina during proliferative vitreoretinopathy. Glia 2016, 64, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Sotozono, C.; Ikeda, T.; Kinoshita, S. Interleukin-6 (IL-6) production by cytokine-stimulated human Muller cells. Curr. Eye Res. 2001, 22, 341–347. [Google Scholar] [CrossRef]
- Busik, J.V.; Mohr, S.; Grant, M.B. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes 2008, 57, 1952–1965. [Google Scholar] [CrossRef] [Green Version]
- Mohr, S.; Xi, X.; Tang, J.; Kern, T.S. Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes 2002, 51, 1172–1179. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Wax, M.B. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 8693–8700. [Google Scholar] [CrossRef]
- Pereiro, X.; Ruzafa, N.; Acera, A.; Fonollosa, A.; Rodriguez, F.D.; Vecino, E. Dexamethasone protects retinal ganglion cells but not Muller glia against hyperglycemia in vitro. PLoS ONE 2018, 13, e0207913. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, R.K.; Miller, L.J.; Singh, P.K.; Kanwar, M. Muller glia in retinal innate immunity: A perspective on their roles in endophthalmitis. Crit. Rev. Immunol. 2013, 33, 119–135. [Google Scholar] [CrossRef]
- Zong, H.; Ward, M.; Madden, A.; Yong, P.H.; Limb, G.A.; Curtis, T.M.; Stitt, A.W. Hyperglycaemia-induced pro-inflammatory responses by retinal Muller glia are regulated by the receptor for advanced glycation end-products (RAGE). Diabetologia 2010, 53, 2656–2666. [Google Scholar] [CrossRef] [Green Version]
- Bringmann, A.; Wiedemann, P. Muller glial cells in retinal disease. Ophthalmologica 2012, 227, 1–19. [Google Scholar] [CrossRef]
- Greenberg, M.E.; Xu, B.; Lu, B.; Hempstead, B.L. New insights in the biology of BDNF synthesis and release: Implications in CNS function. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 12764–12767. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Shruthi, S.; Sumitha, R.; Varghese, A.M.; Ashok, S.; Chandrasekhar Sagar, B.K.; Sathyaprabha, T.N.; Nalini, A.; Kramer, B.W.; Raju, T.R.; Vijayalakshmi, K.; et al. Brain-Derived Neurotrophic Factor Facilitates Functional Recovery from ALS-Cerebral Spinal Fluid-Induced Neurodegenerative Changes in the NSC-34 Motor Neuron Cell Line. Neuro Degener. Dis. 2017, 17, 44–58. [Google Scholar] [CrossRef]
- Ferrari, M.P.; Mantelli, F.; Sacchetti, M.; Antonangeli, M.I.; Cattani, F.; D’Anniballe, G.; Sinigaglia, F.; Ruffini, P.A.; Lambiase, A. Safety and pharmacokinetics of escalating doses of human recombinant nerve growth factor eye drops in a double-masked, randomized clinical trial. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2014, 28, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Cantor, L.B. Brimonidine in the treatment of glaucoma and ocular hypertension. Ther. Clin. Risk Manag. 2006, 2, 337–346. [Google Scholar] [CrossRef] [Green Version]
- WoldeMussie, E.; Ruiz, G.; Wijono, M.; Wheeler, L.A. Neuroprotection of retinal ganglion cells by brimonidine in rats with laser-induced chronic ocular hypertension. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2849–2855. [Google Scholar]
- Grieb, P. Neuroprotective properties of citicoline: Facts, doubts and unresolved issues. CNS Drugs 2014, 28, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Parisi, V.; Manni, G.; Colacino, G.; Bucci, M.G. Cytidine-5′-diphosphocholine (citicoline) improves retinal and cortical responses in patients with glaucoma. Ophthalmology 1999, 106, 1126–1134. [Google Scholar] [CrossRef]
- Parisi, V.; Oddone, F.; Ziccardi, L.; Roberti, G.; Coppola, G.; Manni, G. Citicoline and retinal ganglion cells: Effects on morphology and function. Curr. Neuropharmacol. 2018, 16, 919–932. [Google Scholar] [CrossRef]
- Parisi, V. Electrophysiological assessment of glaucomatous visual dysfunction during treatment with cytidine-5′-diphosphocholine (citicoline): A study of 8 years of follow-up. Doc. Ophthalmol. Adv. Ophthalmol. 2005, 110, 91–102. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Liebmann, J.M.; Cioffi, G.A.; Goldberg, I.; Brandt, J.D.; Johnson, C.A.; Zangwill, L.M.; Schneider, S.; Badger, H.; Bejanian, M. Oral memantine for the treatment of glaucoma. Ophthalmology 2018, 125, 1874–1885. [Google Scholar] [CrossRef] [Green Version]
- Steigerwalt, R.D., Jr.; Cesarone, M.R.; Pascarella, A.; De Angelis, M.; Nebbioso, M.; Belcaro, G.; Feragalli, B. Ocular and optic nerve ischemia: Recognition and treatment with intravenous prostaglandin E1. Panminerva Med. 2011, 53, 119–124. [Google Scholar]
- Rosenthal, R.; Fromm, M. Endothelin antagonism as an active principle for glaucoma therapy. Br. J. Pharmacol. 2011, 162, 806–816. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, S.; Zhang, X.; Huang, W.; Jonas, J.B. Intravitreal triamcinolone acetonide, retinal microglia and retinal ganglion cell apoptosis in the optic nerve crush model. Acta Ophthalmol. 2016, 94, e305–e311. [Google Scholar] [CrossRef]
- Sheng, Y.; Zhu, Y.; Wu, L. Effect of high dosage of methylprednisolone on rat retinal ganglion cell apoptosis after optic nerve crush. Yan Ke Xue Bao Eye Sci. 2004, 20, 181–186. [Google Scholar]
- Rath, E.Z.; Hazan, Z.; Adamsky, K.; Solomon, A.; Segal, Z.I.; Levin, L.A. Randomized controlled phase 2a study of RPh201 in previous nonarteritic anterior ischemic optic neuropathy. J. Neuroophthalmol. 2019, 39, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Shaheer, M.; Amjad, A.; Saleem, Z. Retinal ganglion cell complex changes after intravitreal bevacizumab for diabetic macular edema. J. Coll. Phys. Surg. Pak. JCPSP 2019, 29, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Entezari, M.; Esmaeili, M.; Yaseri, M. A pilot study of the effect of intravenous erythropoetin on improvement of visual function in patients with recent indirect traumatic optic neuropathy. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 1309–1313. [Google Scholar] [CrossRef]
- Kashkouli, M.B.; Pakdel, F.; Sanjari, M.S.; Haghighi, A.; Nojomi, M.; Homaee, M.H.; Heirati, A. Erythropoietin: A novel treatment for traumatic optic neuropathy—A pilot study. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 731–736. [Google Scholar] [CrossRef]
- Kilic, U.; Kilic, E.; Soliz, J.; Bassetti, C.I.; Gassmann, M.; Hermann, D.M. Erythropoietin protects from axotomy-induced degeneration of retinal ganglion cells by activating ERK-1/-2. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 249–251. [Google Scholar] [CrossRef]
- Meyerson, C.; Van Stavern, G.; McClelland, C. Leber hereditary optic neuropathy: Current perspectives. Clin. Ophthalmol. 2015, 9, 1165–1176. [Google Scholar] [CrossRef] [Green Version]
- Lyseng-Williamson, K.A. Idebenone: A Review in leber’s hereditary optic neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef]
- Kim, S.Y.; Shim, M.S.; Kim, K.Y.; Weinreb, R.N.; Wheeler, L.A.; Ju, W.K. Inhibition of cyclophilin D by cyclosporin A promotes retinal ganglion cell survival by preventing mitochondrial alteration in ischemic injury. Cell Death Dis. 2014, 5, e1105. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, Z.A. An attempt at a rational classification of theories of ageing. Biol. Rev. Camb. Philos. Soc. 1990, 65, 375–398. [Google Scholar] [CrossRef]
- Zeng, H.; Sanes, J.R. Neuronal cell-type classification: Challenges, opportunities and the path forward. Nat. Rev. Neurosci. 2017, 18, 530–546. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Sun, W.; Deng, Q.; He, S. Development of the mouse retina: Emerging morphological diversity of the ganglion cells. J. Neurobiol. 2004, 61, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y. A three-group classification of rat retinal ganglion cells: Histological and physiological studies. Brain Res. 1977, 119, 327–334. [Google Scholar] [CrossRef]
- Sanes, J.R.; Masland, R.H. The types of retinal ganglion cells: Current status and implications for neuronal classification. Ann. Rev. Neurosci. 2015, 38, 221–246. [Google Scholar] [CrossRef] [PubMed]
- Langer, K.B.; Ohlemacher, S.K.; Phillips, M.J.; Fligor, C.M.; Jiang, P.; Gamm, D.M.; Meyer, J.S. Retinal ganglion cell diversity and subtype specification from human pluripotent stem cells. Stem Cell Rep. 2018, 10, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.N.; De la Huerta, I.; Kim, I.J.; Zhang, Y.; Yamagata, M.; Chu, M.W.; Meister, M.; Sanes, J.R. Retinal ganglion cells with distinct directional preferences differ in molecular identity, structure, and central projections. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 7753–7762. [Google Scholar] [CrossRef] [Green Version]
- Yonehara, K.; Shintani, T.; Suzuki, R.; Sakuta, H.; Takeuchi, Y.; Nakamura-Yonehara, K.; Noda, M. Expression of SPIG1 reveals development of a retinal ganglion cell subtype projecting to the medial terminal nucleus in the mouse. PLoS ONE 2008, 3, e1533. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Qiao, M.; Bei, F.; Kim, I.J.; He, Z.; Sanes, J.R. Subtype-specific regeneration of retinal ganglion cells following axotomy: Effects of osteopontin and mTOR signaling. Neuron 2015, 85, 1244–1256. [Google Scholar] [CrossRef] [Green Version]
- Hattar, S.; Liao, H.W.; Takao, M.; Berson, D.M.; Yau, K.W. Melanopsin-containing retinal ganglion cells: Architecture, projections, and intrinsic photosensitivity. Science 2002, 295, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.A.; Li, H.; Zhang, Z.; Kiyama, T.; Panda, S.; Hattar, S.; Ribelayga, C.P.; Mills, S.L.; Wang, S.W. T-box transcription regulator Tbr2 is essential for the formation and maintenance of Opn4/melanopsin-expressing intrinsically photosensitive retinal ganglion cells. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 13083–13095. [Google Scholar] [CrossRef]
- Volgyi, B.; Chheda, S.; Bloomfield, S.A. Tracer coupling patterns of the ganglion cell subtypes in the mouse retina. J. Comp. Neurol. 2009, 512, 664–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.J.; Zhang, Y.; Meister, M.; Sanes, J.R. Laminar restriction of retinal ganglion cell dendrites and axons: Subtype-specific developmental patterns revealed with transgenic markers. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 1452–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huberman, A.D.; Manu, M.; Koch, S.M.; Susman, M.W.; Lutz, A.B.; Ullian, E.M.; Baccus, S.A.; Barres, B.A. Architecture and activity-mediated refinement of axonal projections from a mosaic of genetically identified retinal ganglion cells. Neuron 2008, 59, 425–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triplett, J.W.; Wei, W.; Gonzalez, C.; Sweeney, N.T.; Huberman, A.D.; Feller, M.B.; Feldheim, D.A. Dendritic and axonal targeting patterns of a genetically-specified class of retinal ganglion cells that participate in image-forming circuits. Neural Dev. 2014, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Rheaume, B.A.; Jereen, A.; Bolisetty, M.; Sajid, M.S.; Yang, Y.; Renna, K.; Sun, L.; Robson, P.; Trakhtenberg, E.F. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat. Commun. 2018, 9, 2759. [Google Scholar] [CrossRef]
- Ou, Y.; Jo, R.E.; Ullian, E.M.; Wong, R.O.; Della Santina, L. Selective vulnerability of specific retinal ganglion cell types and synapses after transient ocular hypertension. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 9240–9252. [Google Scholar] [CrossRef] [PubMed]
- Daniel, S.; Clark, A.F.; McDowell, C.M. Subtype-specific response of retinal ganglion cells to optic nerve crush. Cell Death Discov. 2018, 4, 7. [Google Scholar] [CrossRef]
- Cui, Q.; Ren, C.; Sollars, P.J.; Pickard, G.E.; So, K.F. The injury resistant ability of melanopsin-expressing intrinsically photosensitive retinal ganglion cells. Neuroscience 2015, 284, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ederra, J.; Garcia, M.; Hernandez, M.; Urcola, H.; Hernandez-Barbachano, E.; Araiz, J.; Vecino, E. The pig eye as a novel model of glaucoma. Exp. Eye Res. 2005, 81, 561–569. [Google Scholar] [CrossRef]
- Christensen, I.; Lu, B.; Yang, N.; Huang, K.; Wang, P.; Tian, N. The susceptibility of retinal ganglion cells to glutamatergic excitotoxicity is type-specific. Front. Neurosci. 2019, 13, 219. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.; Bruehl, C.; Salt, E.L.; Diem, R.; Draguhn, A.; Fairless, R. Selective vulnerability of alphaOFF retinal ganglion cells during onset of autoimmune optic neuritis. Neuroscience 2018, 393, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Puyang, Z.; Gong, H.Q.; He, S.G.; Troy, J.B.; Liu, X.; Liang, P.J. Different functional susceptibilities of mouse retinal ganglion cell subtypes to optic nerve crush injury. Exp. Eye Res. 2017, 162, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhao, Y.; Yoshida, M.; Chen, H.; Yang, J.F.; Kim, T.S.; Cang, J.; Troy, J.B.; Liu, X. Sustained ocular hypertension induces dendritic degeneration of mouse retinal ganglion cells that depends on cell type and location. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Sanz, M.; Galindo-Romero, C.; Valiente-Soriano, F.J.; Nadal-Nicolas, F.M.; Ortin-Martinez, A.; Rovere, G.; Salinas-Navarro, M.; Lucas-Ruiz, F.; Sanchez-Migallon, M.C.; Sobrado-Calvo, P.; et al. Shared and Differential Retinal Responses against Optic Nerve Injury and Ocular Hypertension. Front. Neurosci. 2017, 11, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Morgia, C.; Carelli, V.; Carbonelli, M. Melanopsin retinal ganglion cells and pupil: Clinical implications for neuro-ophthalmology. Front. Neurol. 2018, 9, 1047. [Google Scholar] [CrossRef] [Green Version]
- VanderWall, K.B.; Lu, B.; Wang, S.; Meyer, J.S. Differential susceptibility of rat retinal ganglion cells following optic nerve crush. bioRxiv 2018, 429282. [Google Scholar] [CrossRef]
- Siegert, S.; Scherf, B.G.; Del Punta, K.; Didkovsky, N.; Heintz, N.; Roska, B. Genetic address book for retinal cell types. Nat. Neurosci. 2009, 12, 1197–1204. [Google Scholar] [CrossRef]
- Norsworthy, M.W.; Bei, F.; Kawaguchi, R.; Wang, Q.; Tran, N.M.; Li, Y.; Brommer, B.; Zhang, Y.; Wang, C.; Sanes, J.R.; et al. Sox11 Expression Promotes Regeneration of Some Retinal Ganglion Cell Types but Kills Others. Neuron 2017, 94, 1112–1120.e4. [Google Scholar] [CrossRef] [Green Version]
- Harada, C.; Kimura, A.; Guo, X.; Namekata, K.; Harada, T. Recent advances in genetically modified animal models of glaucoma and their roles in drug repositioning. Br. J. Ophthalmol. 2019, 103, 161–166. [Google Scholar] [CrossRef] [Green Version]
- DeBusk, A.; Moster, M.L. Gene therapy in optic nerve disease. Curr. Opin. Ophthalmol. 2018, 29, 234–238. [Google Scholar] [CrossRef]
- Yang, S.; Ma, S.Q.; Wan, X.; He, H.; Pei, H.; Zhao, M.J.; Chen, C.; Wang, D.W.; Dong, X.Y.; Yuan, J.J.; et al. Long-term outcomes of gene therapy for the treatment of Leber’s hereditary optic neuropathy. EBioMedicine 2016, 10, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuer, W.J.; Schiffman, J.C.; Davis, J.L.; Porciatti, V.; Gonzalez, P.; Koilkonda, R.D.; Yuan, H.; Lalwani, A.; Lam, B.L.; Guy, J. Gene therapy for leber hereditary optic neuropathy: Initial results. Ophthalmology 2016, 123, 558–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Pei, H.; Zhao, M.J.; Yang, S.; Hu, W.K.; He, H.; Ma, S.Q.; Zhang, G.; Dong, X.Y.; Chen, C.; et al. Efficacy and safety of rAAV2-ND4 treatment for leber’s hereditary optic neuropathy. Sci. Rep. 2016, 6, 21587. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Zode, G.; Kasetti, R.B.; Ran, F.A.; Yan, W.; Sharma, T.P.; Bugge, K.; Searby, C.C.; Fingert, J.H.; Zhang, F.; et al. CRISPR-Cas9-based treatment of myocilin-associated glaucoma. Proc. Natl. Acad. Sci. USA 2017, 114, 11199–11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]






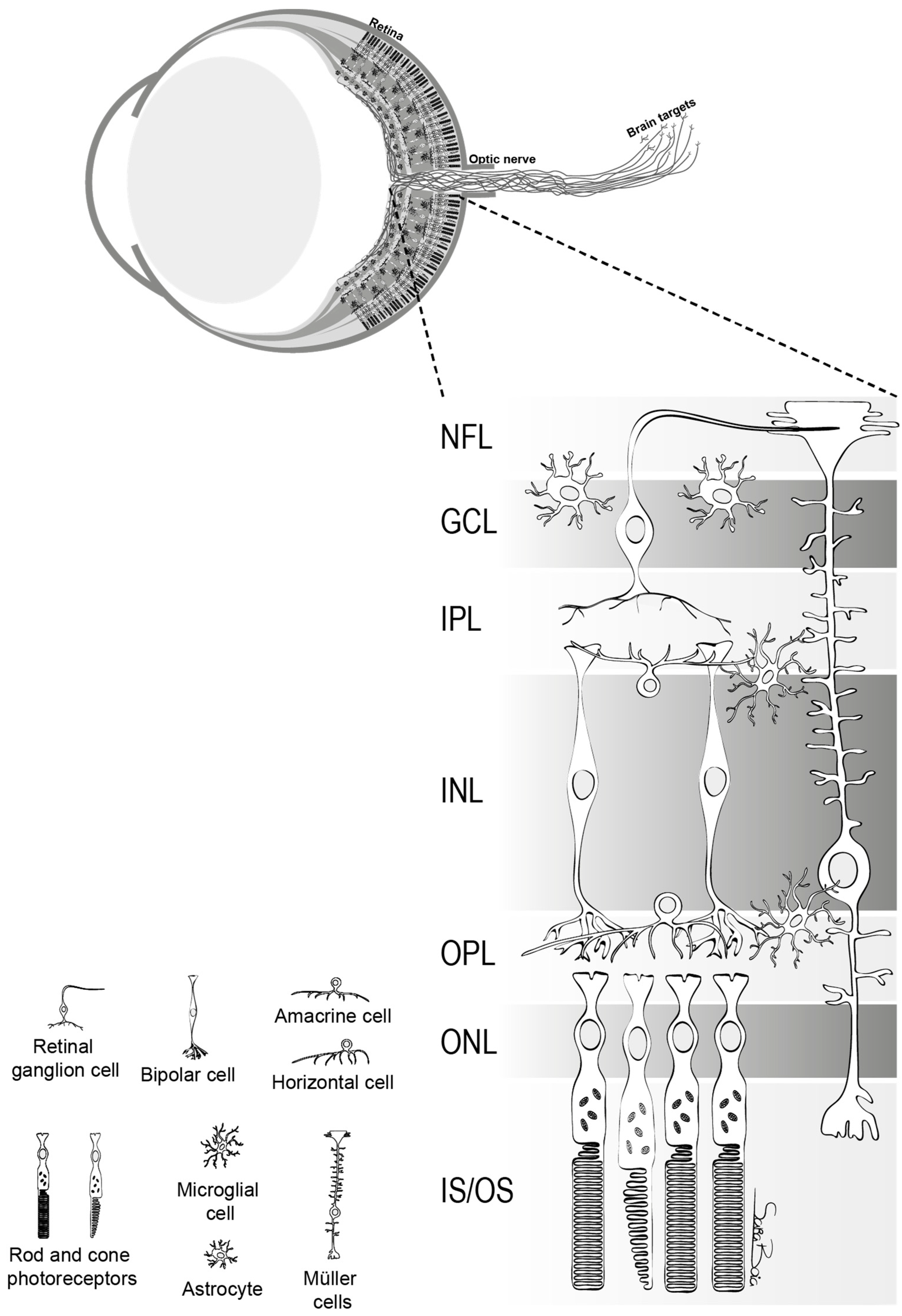{kind=link}
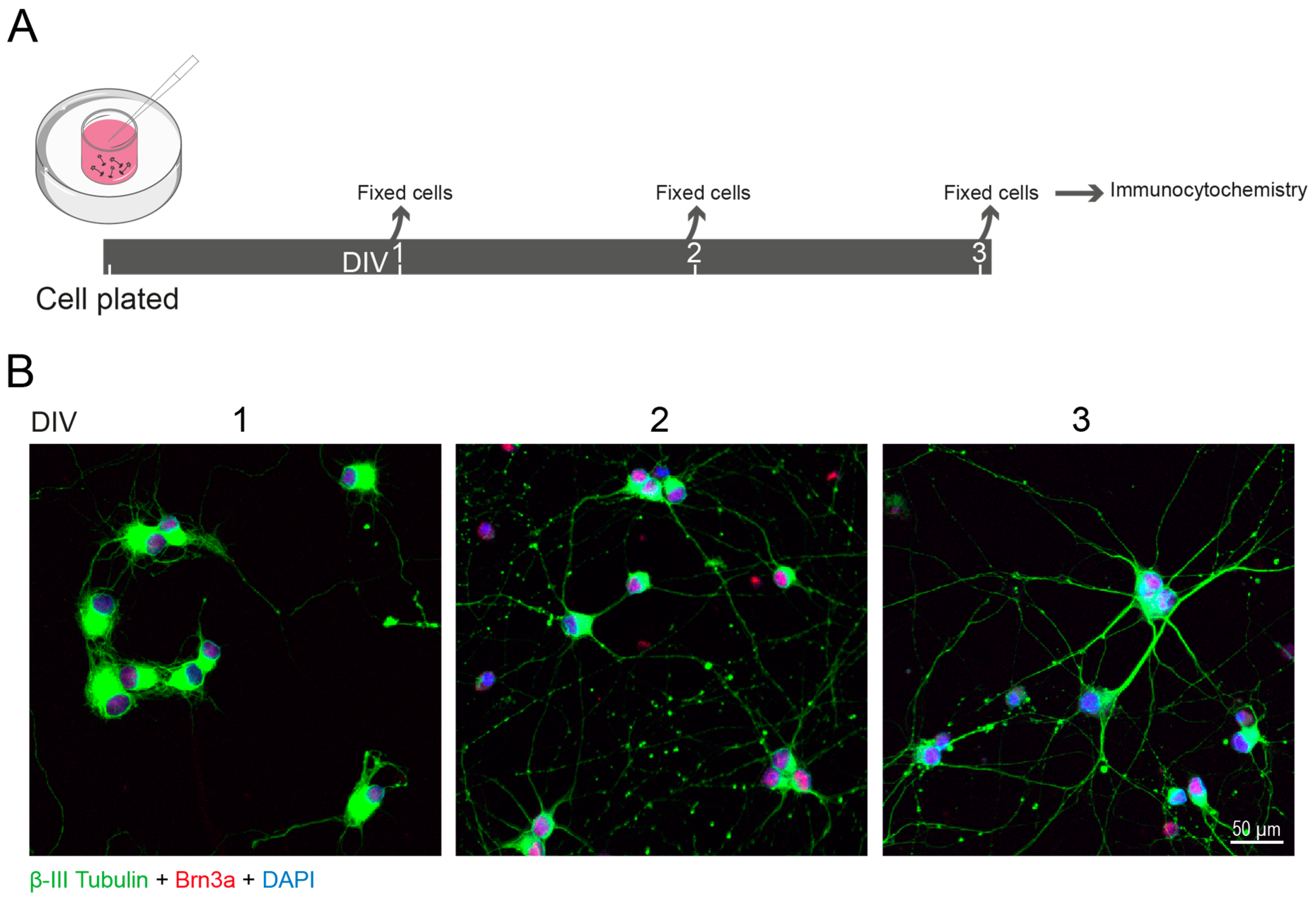{kind=link}
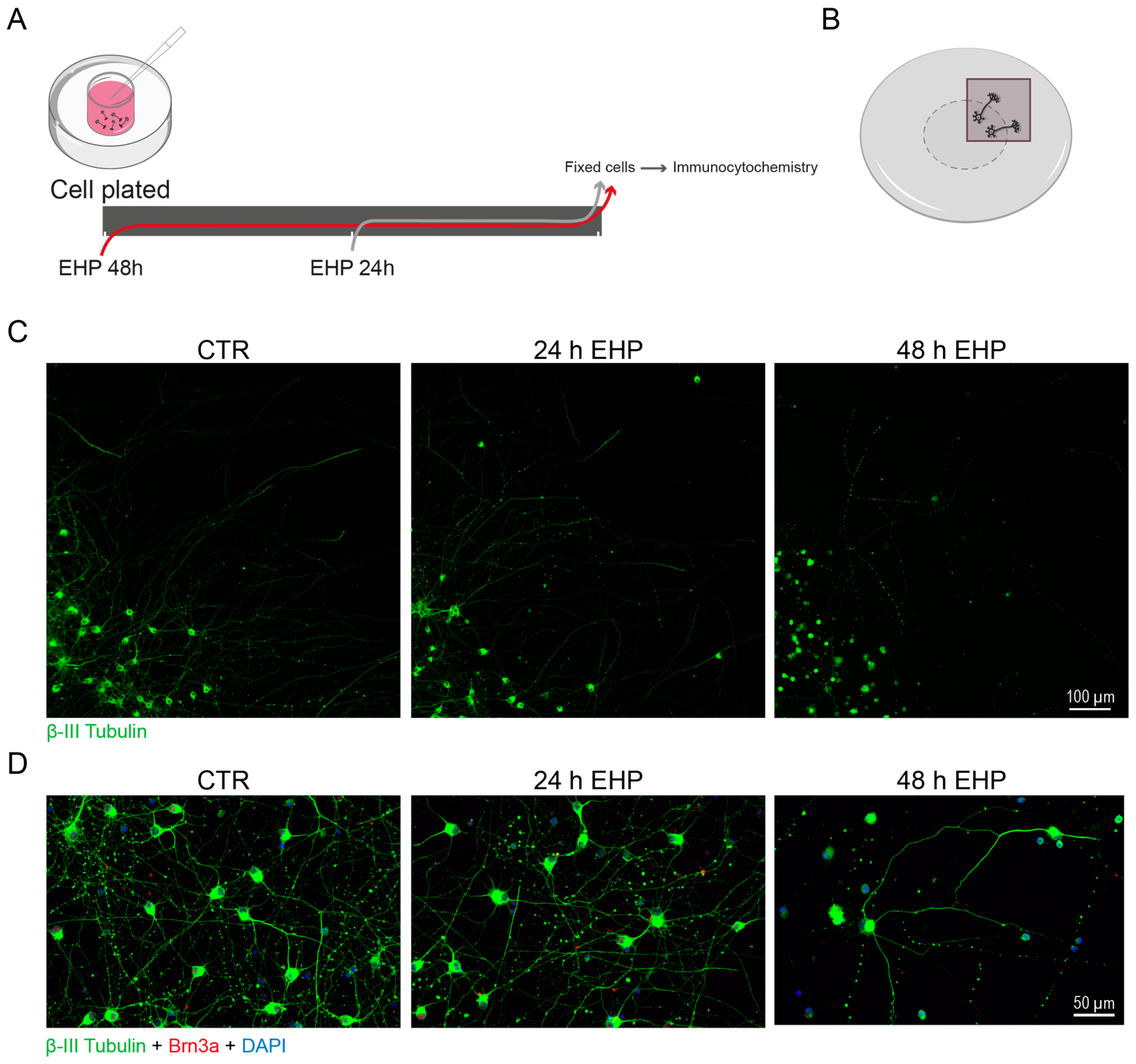{kind=link}
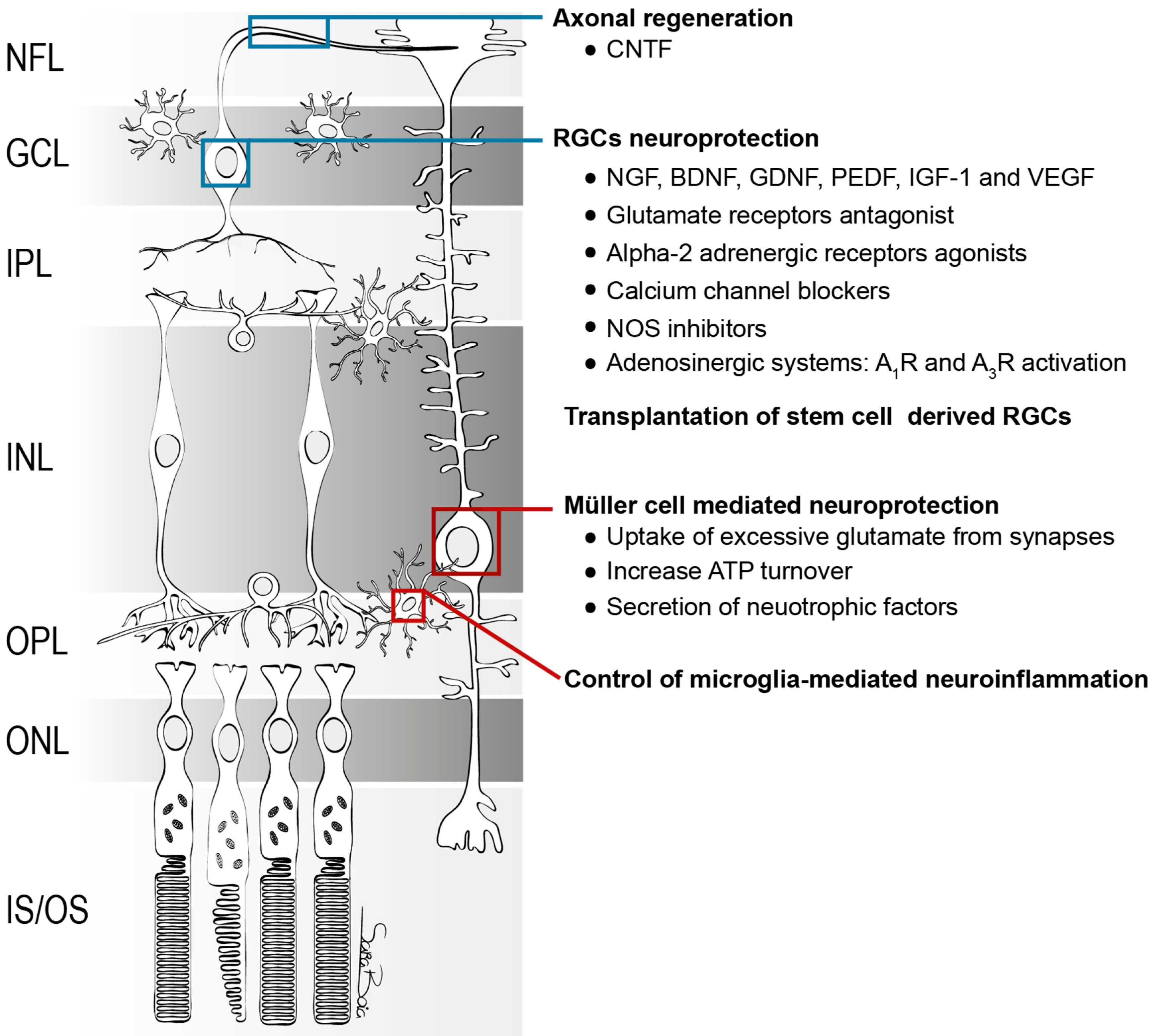{kind=link}
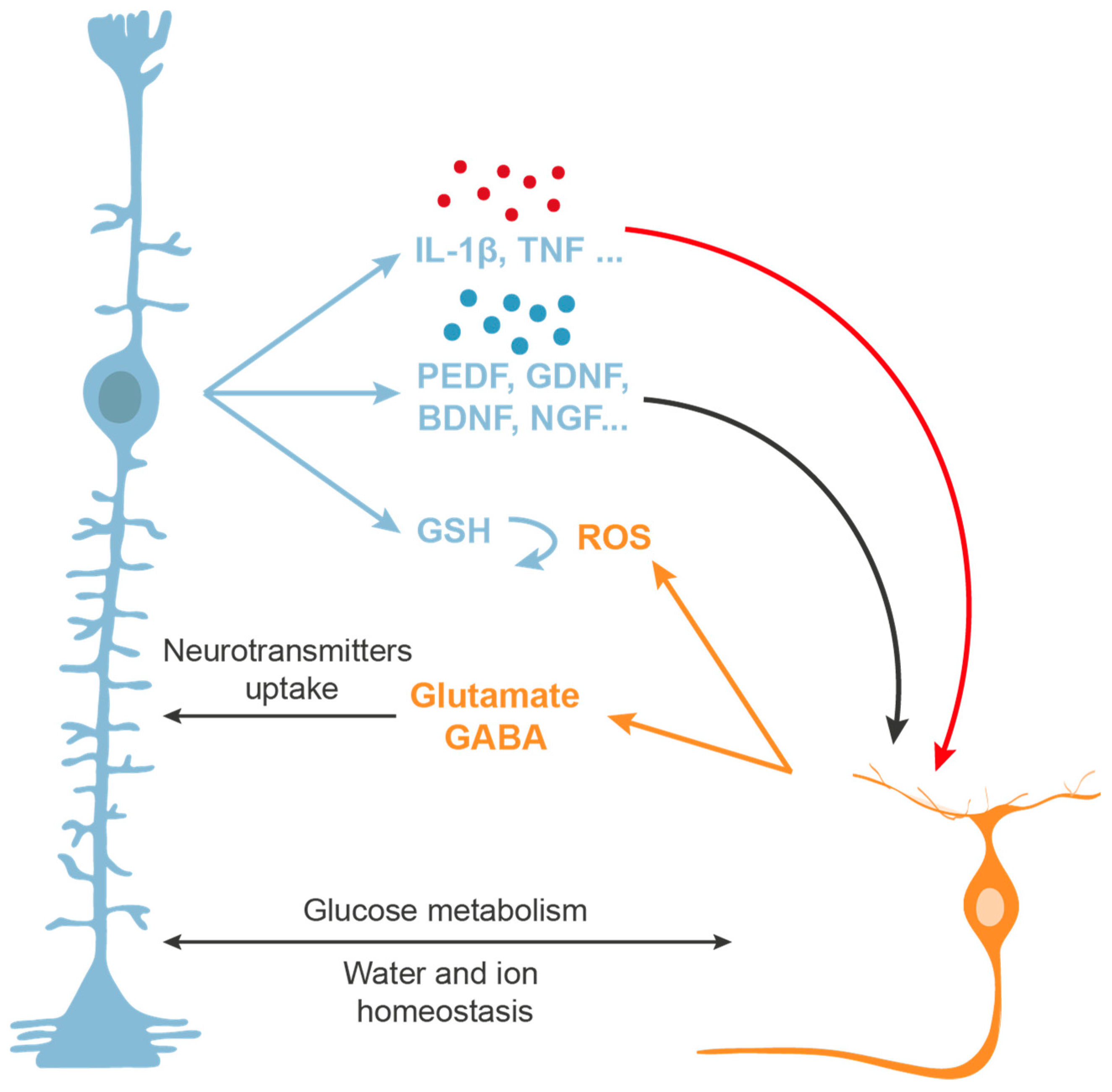{kind=link}
| Condition or Disease | Intervention | ClinicalTrials.gov Identifier | Phase | Starting Date |
|---|---|---|---|---|
| Glaucoma | NT-501 ECT implant | NCT02862938 | 2 | 2016 |
| Glaucoma | rhNGF | NCT02855450 | 1 | 2016 |
| Glaucoma, Primary Open Angle | NT-501 CNTF Implant | NCT01408472 | 1 | 2011 |
| Glaucoma, Open-Angle | Brimonidine Implant | NCT00693485 | 2 | 2008 |
| Glaucoma and Ischemic optic neuropathy | Citicoline | NCT00404729 | 4 | 2006 |
| Open-Angle Glaucoma | Memantine | NCT00141882 | 3 | 2005 |
| Open-Angle Glaucoma | Memantine | NCT00168350 | 3 | 2005 |
| Ischemic Optic Neuropathy | Alprostadil (prostaglandin E1) | NCT03851562 | 2 | 2019 |
| Ischemic Optic Neuropathy | Bosentan | NCT02377271 | 3 | 2015 |
| Ischemic Optic Neuropathy | Triamcinolone Acetonide | NCT02329288 | 3 | 2014 |
| Ischemic Optic Neuropathy | NT-501 CNTF Implant | NCT01411657 | 1 | 2011 |
| Non-arteritic Anterior Ischemic Optic Neuropathy | Prednisolone and Erythropoietin | NCT03715881 | 2 | 2018 |
| Non-arteritic Ischemic Optic Neuropathy | RPh201 | NCT03547206 | 3 | 2018 |
| Non-arteritic Anterior Ischemic Optic Neuropathy | Citicoline | NCT03046693 | 4 | 2017 |
| Non-arteritic Anterior Ischemic Optic Neuropathy | Methylprednisolone | NCT02439866 | 3 | 2015 |
| Non-arteritic Ischemic Optic Neuropathy | RPh201 | NCT02045212 | 2 | 2014 |
| Non-arteritic Ischemic Optic Neuropathy | Dalfampridine | NCT01975324 | 4 | 2013 |
| Non-arteritic Anterior Ischemic Optic Neuropathy | Avastin and Triamcinolone | NCT01330524 | 1 and 2 | 2011 |
| Non-arteritic Anterior Ischemic Optic Neuropathy | Bevacizumab | NCT00813059 | 2 | 2008 |
| Non-arteritic Anterior Ischemic Optic Neuropathy | Ranibizumab | NCT00561834 | 1 | 2007 |
| Non-arteritic Anterior Ischemic Optic Neuropathy | Levodopa-carbidopa | NCT00432393 | 4 | 2007 |
| Traumatic Optic Neuropathy | Recombinant human erythropoietin | NCT03308448 | 3 | 2017 |
| Traumatic Optic Neuropathy | Recombinant human erythropoietin | NCT01783847 | 1 and 2 | 2013 |
| Optic Nerve Diseases (methanol associated optic neuropathy) | Erythropoietin | NCT02376881 | 3 | 2015 |
| Leber’s Hereditary Optic Neuropathy | Idebenone | NCT02774005 | 4 | 2016 |
| Leber’s Hereditary Optic Neuropathy | Cyclosporine | NCT02176733 | 2 | 2014 |
| Leber’s Hereditary Optic Neuropathy | Idebenone | NCT00747487 | 2 | 2008 |
| Condition or Disease | Intervention | ClinicalTrials.gov Identifier | Phase | Starting Date |
| Optic Neuropathy | Transplantation of autologous purified stem cells | NCT02638714 | 1 and 2 | 2015 |
| Non-arteritic Ischemic Optic Neuropathy | Intravitreal injection of mesenchymal stem cells | NCT03173638 | 2 | 2017 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boia, R.; Ruzafa, N.; Aires, I.D.; Pereiro, X.; Ambrósio, A.F.; Vecino, E.; Santiago, A.R. Neuroprotective Strategies for Retinal Ganglion Cell Degeneration: Current Status and Challenges Ahead. Int. J. Mol. Sci. 2020, 21, 2262. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072262
Boia R, Ruzafa N, Aires ID, Pereiro X, Ambrósio AF, Vecino E, Santiago AR. Neuroprotective Strategies for Retinal Ganglion Cell Degeneration: Current Status and Challenges Ahead. International Journal of Molecular Sciences. 2020; 21(7):2262. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072262
Chicago/Turabian StyleBoia, Raquel, Noelia Ruzafa, Inês Dinis Aires, Xandra Pereiro, António Francisco Ambrósio, Elena Vecino, and Ana Raquel Santiago. 2020. "Neuroprotective Strategies for Retinal Ganglion Cell Degeneration: Current Status and Challenges Ahead" International Journal of Molecular Sciences 21, no. 7: 2262. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072262