Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis

{kind=link}
{kind=link}
Abstract
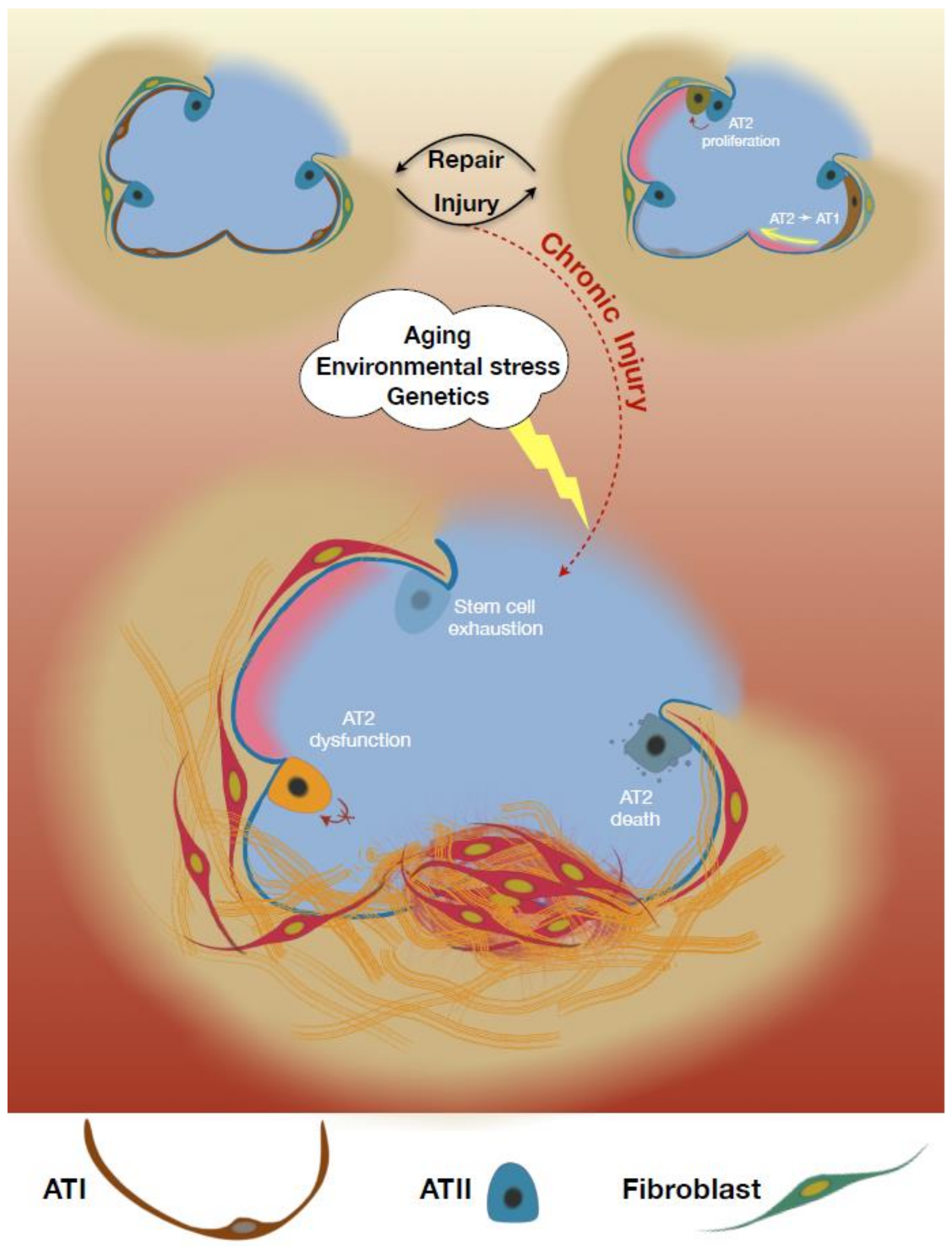
:1. Introduction
2. Alveolar Epithelial Type II Cells (AT2) of the Normal Mammalian Lung
3. AT2 Depletion in Idiopathic Pulmonary Fibrosis (IPF)
Apoptotic Death of AT2 Cells
4. ER Stress Contributes to AT2 Apoptosis
5. Mitochondrial Dysfunction Causes AT2 Death
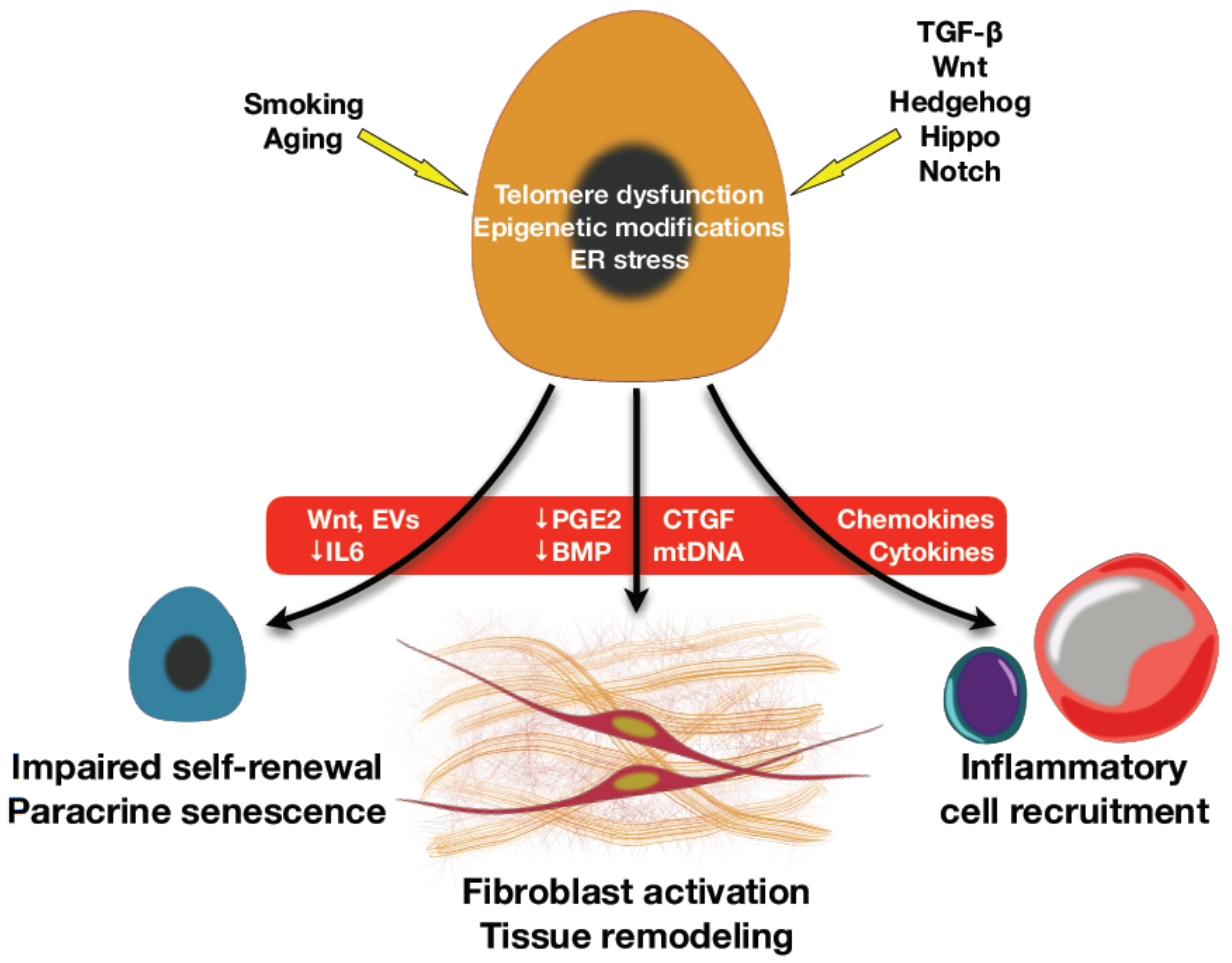
6. AT2 Dysfunction in IPF
7. Impaired AT2 Self-Renewal in IPF
8. Dysregulated Signaling Pathways Causing Intrinsic AT2 Dysfunction in IPF
9. Altered AT2-Fibroblast Signaling in IPF
10. The Senescence-associated Secretory Phenotype Has Pro-fibrotic Effects
11. Epigenetic Changes in the AT2 Promote Lung Fibrosis
12. Conclusions
Funding
Conflicts of Interest
Abbreviations
| IPF | Idiopathic Pulmonary Fibrosis |
| FDA | U.S. Food and Drug Administration |
| U.S. | The United States of America |
| AT2 | Alveolar Epithelial Type II Cells |
References
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, J.P.; McKeever, T.M.; Fogarty, A.W.; Navaratnam, V.; Hubbard, R.B. Increasing Global Mortality from Idiopathic Pulmonary Fibrosis in the Twenty-First Century. Ann. ATS 2014, 11, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbie, H.; Daccord, C.; Chua, F.; Devaraj, A. Evaluating disease severity in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2017, 26, 170051. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Shah, B.A.; Chowdhury, B.A. Forced vital capacity in idiopathic pulmonary fibrosis—FDA review of pirfenidone and nintedanib. N. Engl. J. Med. 2015, 372, 1189–1191. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Du Bois, R.M.; Fagan, E.A.; Fishman, R.S.; Glaspole, I.; Glassberg, M.K.; Lancaster, L.; et al. Pirfenidone for idiopathic pulmonary fibrosis: Analysis of pooled data from three multinational phase 3 trials. Eur. Respir. J. 2016, 47, 243–253. [Google Scholar] [CrossRef]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, A.; Nukiwa, T.; Tsuboi, E.; Suga, M.; Abe, S.; Nakata, K.; Taguchi, Y.; Nagai, S.; Itoh, H.; Ohi, M.; et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Corte, T.; Bonella, F.; Crestani, B.; Demedts, M.G.; Richeldi, L.; Coeck, C.; Pelling, K.; Quaresma, M.; Lasky, J.A. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costabel, U.; Inoue, Y.; Richeldi, L.; Collard, H.R.; Tschoepe, I.; Stowasser, S.; Azuma, A. Efficacy of Nintedanib in Idiopathic Pulmonary Fibrosis across Prespecified Subgroups in INPULSIS. Am. J. Respir. Crit. Care Med. 2016, 193, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkauskas, C.E.; Noble, P.W. Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am. J. Physiol. Cell Physiol. 2014, 306, C987–C996. [Google Scholar] [CrossRef] [Green Version]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. Molecular mechanisms of pulmonary fibrosis. Front. Biosci. 2002, 7, d1743–d1761. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.W.; Johnson, J.E.; Browning, P.J.; Cruz-Gervis, R.A.; Davis, A.; Graham, B.S.; Brigham, K.L.; Oates, J.A., Jr.; Loyd, J.E.; Stecenko, A.A. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J. Clin. Microbiol. 2003, 41, 2633–2640. [Google Scholar] [CrossRef] [Green Version]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Meiners, S.; Eickelberg, O.; Konigshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.K. Insights from human genetic studies of lung and organ fibrosis. J. Clin. Investig. 2018, 128, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell Signal. 2019, 66, 109482. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.L.; Barkauskas, C.E.; Chapman, H.A.; Epstein, J.A.; Jain, R.; Hsia, C.C.; Niklason, L.; Calle, E.; Le, A.; Randell, S.H.; et al. Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 2014, 15, 123–138. [Google Scholar] [CrossRef] [Green Version]
- Rackley, C.R.; Stripp, B.R. Building and maintaining the epithelium of the lung. J. Clin. Investig. 2012, 122, 2724–2730. [Google Scholar] [CrossRef] [Green Version]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Salwig, I.; Spitznagel, B.; Vazquez-Armendariz, A.I.; Khalooghi, K.; Guenther, S.; Herold, S.; Szibor, M.; Braun, T. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, K.; Cui, G.; Huang, X.; Yao, S.; Guo, W.; Qin, Z.; Li, Y.; Yang, R.; Pu, W.; et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat. Genet. 2019, 51, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.H.; Ryeom, S.; Kim, C.F. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.; Barkauskas, C.E.; Takeda, N.; Bowie, E.J.; Aghajanian, H.; Wang, Q.; Padmanabhan, A.; Manderfield, L.J.; Gupta, M.; Li, D.; et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat. Commun. 2015, 6, 6727. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisson, T.H.; Mendez, M.; Choi, K.; Subbotina, N.; Courey, A.; Cunningham, A.; Dave, A.; Engelhardt, J.F.; Liu, X.; White, E.S.; et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 181, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Soukiasian, H.J.; David, G.; Weigt, S.S.; Belperio, J.A.; et al. Senescence of alveolar stem cells drives progressive pulmonary fibrosis. Cell Stem Cell 2019, 59, 00545. [Google Scholar] [CrossRef]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Zhang, Y.; Xie, T.; Liu, N.; Chen, H.; Geng, Y.; Kurkciyan, A.; Mena, J.M.; Stripp, B.R.; Jiang, D.; et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 2016, 22, 1285–1293. [Google Scholar] [CrossRef]
- Uhal, B.D.; Joshi, I.; Hughes, W.F.; Ramos, C.; Pardo, A.; Selman, M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am. J. Physiol. 1998, 275, L1192–L1199. [Google Scholar] [CrossRef]
- Barbas-Filho, J.V.; Ferreira, M.A.; Sesso, A.; Kairalla, R.A.; Carvalho, C.R.; Capelozzi, V.L. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J. Clin. Pathol. 2001, 54, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeyama, T.; Kuwano, K.; Kawasaki, M.; Kunitake, R.; Hagimoto, N.; Matsuba, T.; Yoshimi, M.; Inoshima, I.; Yoshida, K.; Hara, N. Upregulation of Fas-signalling molecules in lung epithelial cells from patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2001, 17, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plataki, M.; Koutsopoulos, A.V.; Darivianaki, K.; Delides, G.; Siafakas, N.M.; Bouros, D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest 2005, 127, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.G.; Cho, S.J.; Kang, M.J.; Chapoval, S.P.; Lee, P.J.; Noble, P.W.; Yehualaeshet, T.; Lu, B.; Flavell, R.A.; Milbrandt, J.; et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J. Exp. Med. 2004, 200, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budinger, G.R.; Mutlu, G.M.; Eisenbart, J.; Fuller, A.C.; Bellmeyer, A.A.; Baker, C.M.; Wilson, M.; Ridge, K.; Barrett, T.A.; Lee, V.Y.; et al. Proapoptotic Bid is required for pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4604–4609. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kuwano, K.; Miyazaki, H.; Hagimoto, N.; Kawasaki, M.; Fujita, M.; Kunitake, R.; Kaneko, Y.; Hara, N. The involvement of Fas-Fas ligand pathway in fibrosing lung diseases. Am. J. Respir. Cell Mol. Biol. 1999, 20, 53–60. [Google Scholar] [CrossRef]
- Hagimoto, N.; Kuwano, K.; Nomoto, Y.; Kunitake, R.; Hara, N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 1997, 16, 91–101. [Google Scholar] [CrossRef]
- Wang, R.; Ibarra-Sunga, O.; Verlinski, L.; Pick, R.; Uhal, B.D. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L143–L151. [Google Scholar] [CrossRef]
- Kuwano, K.; Kunitake, R.; Maeyama, T.; Hagimoto, N.; Kawasaki, M.; Matsuba, T.; Yoshimi, M.; Inoshima, I.; Yoshida, K.; Hara, N. Attenuation of bleomycin-induced pneumopathy in mice by a caspase inhibitor. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L316–L325. [Google Scholar] [CrossRef]
- Kuwano, K.; Hagimoto, N.; Kawasaki, M.; Yatomi, T.; Nakamura, N.; Nagata, S.; Suda, T.; Kunitake, R.; Maeyama, T.; Miyazaki, H.; et al. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J. Clin. Investig. 1999, 104, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagimoto, N.; Kuwano, K.; Miyazaki, H.; Kunitake, R.; Fujita, M.; Kawasaki, M.; Kaneko, Y.; Hara, N. Induction of apoptosis and pulmonary fibrosis in mice in response to ligation of Fas antigen. Am. J. Respir. Cell Mol. Biol. 1997, 17, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Schutze, S.; Tchikov, V.; Schneider-Brachert, W. Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Investig. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.M.; Seeger, W.; et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef] [Green Version]
- Lawson, W.E.; Cheng, D.S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [Green Version]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [Green Version]
- Katzen, J.; Wagner, B.D.; Venosa, A.; Kopp, M.; Tomer, Y.; Russo, S.J.; Headen, A.C.; Basil, M.C.; Stark, J.M.; Mulugeta, S.; et al. An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nureki, S.I.; Tomer, Y.; Venosa, A.; Katzen, J.; Russo, S.J.; Jamil, S.; Barrett, M.; Nguyen, V.; Kopp, M.; Mulugeta, S.; et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Investig. 2018, 128, 4008–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borok, Z.; Horie, M.; Flodby, P.; Wang, H.; Liu, Y.; Ganesh, S.; Ryan Firth, A.L.; Minoo, P.; Li, C.; Beers, M.F.; et al. Grp78 Loss in Epithelial Progenitors Reveals an Age-Linked Role for ER Stress in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019. [Google Scholar] [CrossRef]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef]
- Jorgensen, E.; Stinson, A.; Shan, L.; Yang, J.; Gietl, D.; Albino, A.P. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 2008, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Kropski, J.A.; Pritchett, J.M.; Zoz, D.F.; Crossno, P.F.; Markin, C.; Garnett, E.T.; Degryse, A.L.; Mitchell, D.B.; Polosukhin, V.V.; Rickman, O.B.; et al. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 417–426. [Google Scholar] [CrossRef]
- McMillan, T.R.; Moore, B.B.; Weinberg, J.B.; Vannella, K.M.; Fields, W.B.; Christensen, P.J.; Van Dyk, L.F.; Toews, G.B. Exacerbation of established pulmonary fibrosis in a murine model by gammaherpesvirus. Am. J. Respir. Crit. Care Med. 2008, 177, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Vannella, K.M.; Luckhardt, T.R.; Wilke, C.A.; Van Dyk, L.F.; Toews, G.B.; Moore, B.B. Latent herpesvirus infection augments experimental pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 181, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.L.; Woods, C.R.; Garcia, A.; Xu, J.; Rojas, M.; Speck, S.H.; Roman, J.; Brigham, K.L.; Stecenko, A.A. Lung infection with gamma-herpesvirus induces progressive pulmonary fibrosis in Th2-biased mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L711–L721. [Google Scholar] [CrossRef]
- Lok, S.S.; Haider, Y.; Howell, D.; Stewart, J.P.; Hasleton, P.S.; Egan, J.J. Murine gammaherpes virus as a cofactor in the development of pulmonary fibrosis in bleomycin resistant mice. Eur. Respir. J. 2002, 20, 1228–1232. [Google Scholar] [CrossRef]
- Torres-Gonzalez, E.; Bueno, M.; Tanaka, A.; Krug, L.T.; Cheng, D.S.; Polosukhin, V.V.; Sorescu, D.; Lawson, W.E.; Blackwell, T.S.; Rojas, M.; et al. Role of endoplasmic reticulum stress in age-related susceptibility to lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 46, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Suliman, H.B. Mitochondrial Dysfunction in Lung Pathogenesis. Annu. Rev. Physiol. 2017, 79, 495–515. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bernard, K.; Thannickal, V.J. Mitochondrial Dysfunction in Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S383–S388. [Google Scholar] [CrossRef]
- Bueno, M.; Zank, D.; Buendia-Roldan, I.; Fiedler, K.; Mays, B.G.; Alvarez, D.; Sembrat, J.; Kimball, B.; Bullock, J.K.; Martin, J.L.; et al. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PLoS ONE 2019, 14, e0218003. [Google Scholar] [CrossRef]
- Patel, A.S.; Song, J.W.; Chu, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., 2nd. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef]
- Chung, K.-P.; Hsu, C.-L.; Fan, L.-C.; Huang, Z.; Bhatia, D.; Chen, Y.-J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390. [Google Scholar] [CrossRef]
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Morales-Nebreda, L.; Cheng, Y.; Hogan, E.; Yeldandi, A.; Chi, M.; Piseaux, R.; Ridge, K.; et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic. Biol. Med. 2016, 101, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, R.P.; Kim, S.-J.; Cheresh, P.; Williams, D.B.; Morales-Nebreda, L.; Cheng, Y.; Yeldandi, A.; Bhorade, S.; Pardo, A.; Selman, M.; et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. 2017, 31, 2520–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; St Croix, C.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; De Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ganzleben, I.; He, G.W.; Gunther, C.; Prigge, E.S.; Richter, K.; Rieker, R.J.; Mougiakakos, D.; Neurath, M.F.; Becker, C. PGAM5 is a key driver of mitochondrial dysfunction in experimental lung fibrosis. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Kulkarni, T.; De Andrade, J.; Zhou, Y.; Luckhardt, T.; Thannickal, V.J. Alveolar epithelial disintegrity in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L185–L191. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kugler, M.C.; Wei, Y.; Kim, K.K.; Li, X.; Brumwell, A.N.; Chapman, H.A. Integrin alpha3beta1-dependent beta-catenin phosphorylation links epithelial Smad signaling to cell contacts. J. Cell Biol. 2009, 184, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [Green Version]
- Borok, Z.; Whitsett, J.A.; Bitterman, P.B.; Thannickal, V.J.; Kotton, D.N.; Reynolds, S.D.; Krasnow, M.A.; Bianchi, D.W.; Morrisey, E.E.; Hogan, B.L.; et al. Cell plasticity in lung injury and repair: Report from an NHLBI workshop, April 19–20, 2010. Proc. Am. Thorac. Soc. 2011, 8, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2019, 180, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cheung, H.H.; Tu, J.; Miu, K.K.; Chan, W.Y. New insights into the unfolded protein response in stem cells. Oncotarget 2016, 7, 54010–54027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Gonzalez De Los Santos, F.; Zhao, Y.; Wu, Z.; Rinke, A.E.; Kim, K.K.; Phan, S.H. Telomerase reverse transcriptase ameliorates lung fibrosis by protecting alveolar epithelial cells against senescence. J. Biol. Chem. 2019, 294, 8861–8871. [Google Scholar] [CrossRef]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [Green Version]
- Povedano, J.M.; Martinez, P.; Serrano, R.; Tejera, A.; Gomez-Lopez, G.; Bobadilla, M.; Flores, J.M.; Bosch, F.; Blasco, M.A. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Thannickal, V.J. The Aging Lung and Idiopathic Pulmonary Fibrosis. Am. J. Med. Sci. 2019, 357, 384–389. [Google Scholar] [CrossRef]
- Selman, M.; Buendia-Roldan, I.; Pardo, A. Aging and Pulmonary Fibrosis. Rev. Investig. Clin. 2016, 68, 75–83. [Google Scholar]
- Thannickal, V.J. Mechanistic links between aging and lung fibrosis. Biogerontology 2013, 14, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Chilosi, M.; Carloni, A.; Rossi, A.; Poletti, V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl. Res. 2013, 162, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Arish, N.; Petukhov, D.; Wallach-Dayan, S.B. The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Snetselaar, R.; Van Batenburg, A.A.; Van Oosterhout, M.F.M.; Kazemier, K.M.; Roothaan, S.M.; Peeters, T.; Van der Vis, J.J.; Goldschmeding, R.; Grutters, J.C.; Van Moorsel, C.H.M. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS ONE 2017, 12, e0189467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [Green Version]
- Morla, M.; Busquets, X.; Pons, J.; Sauleda, J.; MacNee, W.; Agusti, A.G. Telomere shortening in smokers with and without COPD. Eur. Respir. J. 2006, 27, 525–528. [Google Scholar] [CrossRef] [Green Version]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz de Leon, A.; Cronkhite, J.T.; Katzenstein, A.L.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE 2010, 5, e10680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannengiesser, C.; Borie, R.; Menard, C.; Reocreux, M.; Nitschke, P.; Gazal, S.; Mal, H.; Taille, C.; Cadranel, J.; Nunes, H.; et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur. Respir. J. 2015, 46, 474–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogan, J.D.; Kropski, J.A.; Zhao, M.; Mitchell, D.B.; Rives, L.; Markin, C.; Garnett, E.T.; Montgomery, K.H.; Mason, W.R.; McKean, D.F.; et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2015, 191, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Petrovski, S.; Todd, J.L.; Durheim, M.T.; Wang, Q.; Chien, J.W.; Kelly, F.L.; Frankel, C.; Mebane, C.M.; Ren, Z.; Bridgers, J.; et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 82–93. [Google Scholar] [CrossRef]
- Kropski, J.A.; Mitchell, D.B.; Markin, C.; Polosukhin, V.V.; Choi, L.; Johnson, J.E.; Lawson, W.E.; Phillips, J.A., 3rd; Cogan, J.D.; Blackwell, T.S.; et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest 2014, 146, e1–e7. [Google Scholar] [CrossRef] [Green Version]
- Stanley, S.E.; Gable, D.L.; Wagner, C.L.; Carlile, T.M.; Hanumanthu, V.S.; Podlevsky, J.D.; Khalil, S.E.; DeZern, A.E.; Rojas-Duran, M.F.; Applegate, C.D.; et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci. Transl. Med. 2016, 8, 351ra107. [Google Scholar] [CrossRef] [Green Version]
- Alder, J.K.; Stanley, S.E.; Wagner, C.L.; Hamilton, M.; Hanumanthu, V.S.; Armanios, M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest 2015, 147, 1361–1368. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, T.W.; Van der Vis, J.J.; Van Oosterhout, M.F.; Van Es, H.W.; Van Kessel, D.A.; Grutters, J.C.; Van Moorsel, C.H. TINF2 Gene Mutation in a Patient with Pulmonary Fibrosis. Case Rep. Pulmonol. 2016, 2016, 1310862. [Google Scholar] [CrossRef] [Green Version]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamson, I.Y.; King, G.M.; Young, L. Influence of extracellular matrix and collagen components on alveolar type 2 cell morphology and function. In Vitro Cell Dev. Biol. 1989, 25, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjermer, L.; Lundgren, R.; Hallgren, R. Hyaluronan and type III procollagen peptide concentrations in bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. Thorax 1989, 44, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Habiel, D.M.; Ge, L.; Bora, S.A.; Brauer, R.; Evans, C.M.; Xie, T.; Alonso-Valenteen, F.; Medina-Kauwe, L.K.; et al. Syndecan-1 promotes lung fibrosis by regulating epithelial reprogramming through extracellular vesicles. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Raredon, M.S.B.; Adams, T.S.; Suhail, Y.; Schupp, J.C.; Poli, S.; Neumark, N.; Leiby, K.L.; Greaney, A.M.; Yuan, Y.; Horien, C.; et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 2019, 5, eaaw3851. [Google Scholar] [CrossRef] [Green Version]
- Konigshoff, M.; Kramer, M.; Balsara, N.; Wilhelm, J.; Amarie, O.V.; Jahn, A.; Rose, F.; Fink, L.; Seeger, W.; Schaefer, L.; et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Investig. 2009, 119, 772–787. [Google Scholar] [CrossRef] [Green Version]
- Konigshoff, M.; Balsara, N.; Pfaff, E.M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef] [Green Version]
- Chilosi, M.; Poletti, V.; Zamo, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Frank, D.B.; Peng, T.; Zepp, J.A.; Snitow, M.; Vincent, T.L.; Penkala, I.J.; Cui, Z.; Herriges, M.J.; Morley, M.P.; Zhou, S.; et al. Emergence of a Wave of Wnt Signaling that Regulates Lung Alveologenesis by Controlling Epithelial Self-Renewal and Differentiation. Cell Rep. 2016, 17, 2312–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, T.; Csongei, V.; Feller, D.; Ernszt, D.; Smuk, G.; Sarosi, V.; Jakab, L.; Kvell, K.; Bartis, D.; Pongracz, J.E. Alteration in the Wnt microenvironment directly regulates molecular events leading to pulmonary senescence. Aging Cell 2014, 13, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.P.; Herazo-Maya, J.D.; Sennello, J.A.; Flozak, A.S.; Russell, S.; Mutlu, G.M.; Budinger, G.R.; DasGupta, R.; Varga, J.; Kaminski, N.; et al. Wnt coreceptor Lrp5 is a driver of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Henderson, W.R., Jr.; Chi, E.Y.; Ye, X.; Nguyen, C.; Tien, Y.T.; Zhou, B.; Borok, Z.; Knight, D.A.; Kahn, M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14309–14314. [Google Scholar] [CrossRef] [Green Version]
- Ulsamer, A.; Wei, Y.; Kim, K.K.; Tan, K.; Wheeler, S.; Xi, Y.; Thies, R.S.; Chapman, H.A. Axin pathway activity regulates in vivo pY654-beta-catenin accumulation and pulmonary fibrosis. J. Biol. Chem. 2012, 287, 5164–5172. [Google Scholar] [CrossRef] [Green Version]
- Olajuyin, A.M.; Zhang, X.; Ji, H.L. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. 2019, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Mutze, K.; Vierkotten, S.; Milosevic, J.; Eickelberg, O.; Konigshoff, M. Enolase 1 (ENO1) and protein disulfide-isomerase associated 3 (PDIA3) regulate Wnt/beta-catenin-driven trans-differentiation of murine alveolar epithelial cells. Dis. Model. Mech. 2015, 8, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Konigshoff, M.; Eickelberg, O. WNT signaling in lung disease: A failure or a regeneration signal? Am. J. Respir. Cell Mol. Biol. 2010, 42, 21–31. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Azzolin, L.; Zanconato, F.; Bresolin, S.; Forcato, M.; Basso, G.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Role of TAZ as mediator of Wnt signaling. Cell 2012, 151, 1443–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitani, A.; Nagase, T.; Fukuchi, K.; Aburatani, H.; Makita, R.; Kurihara, H. Transcriptional coactivator with PDZ-binding motif is essential for normal alveolarization in mice. Am. J. Respir. Crit. Care Med. 2009, 180, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Whitsett, J.A.; Di Palma, T.; Hong, J.H.; Yaffe, M.B.; Zannini, M. TAZ interacts with TTF-1 and regulates expression of surfactant protein-C. J. Biol. Chem. 2004, 279, 17384–17390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makita, R.; Uchijima, Y.; Nishiyama, K.; Amano, T.; Chen, Q.; Takeuchi, T.; Mitani, A.; Nagase, T.; Yatomi, Y.; Aburatani, H.; et al. Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am. J. Physiol. Ren. Physiol. 2008, 294, F542–F553. [Google Scholar] [CrossRef]
- Lin, C.; Yao, E.; Zhang, K.; Jiang, X.; Croll, S.; Thompson-Peer, K.; Chuang, P.T. YAP is essential for mechanical force production and epithelial cell proliferation during lung branching morphogenesis. Elife 2017, 6. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Huang, Z.; Zhang, H.; Posner, C.; Jia, G.; Ramalingam, T.R.; Xu, M.; Brightbill, H.; Egen, J.G.; Dey, A.; et al. TAZ is required for lung alveolar epithelial cell differentiation after injury. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Tschumperlin, D.J.; Ligresti, G.; Hilscher, M.B.; Shah, V.H. Mechanosensing and fibrosis. J. Clin. Investig. 2018, 128, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [Green Version]
- Piersma, B.; Bank, R.A. Keeping fibroblasts in suspense: TAZ-mediated signaling activates a context-dependent profibrotic phenotype. Focus on “TAZ activation drives fibroblast spheroid growth, expression of profibrotic paracrine signals, and context-dependent ECM gene expression”. Am. J. Physiol. Cell Physiol. 2017, 312, C274–C276. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, S.; Saito, A.; Mikami, Y.; Urushiyama, H.; Horie, M.; Matsuzaki, H.; Takeshima, H.; Makita, K.; Miyashita, N.; Mitani, A.; et al. TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci. Rep. 2017, 7, 42595. [Google Scholar] [CrossRef]
- Gokey, J.J.; Sridharan, A.; Xu, Y.; Green, J.; Carraro, G.; Stripp, B.R.; Perl, A.-K.T.; Whitsett, J.A. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight 2018, 3, e98738. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Xu, C.; Ahmad, M.; Yang, Y.; Lu, M.; Wu, X.; Tang, L.; Wu, X. Notch Signaling: Linking Embryonic Lung Development and Asthmatic Airway Remodeling. Mol. Pharmacol. 2017, 92, 676–693. [Google Scholar] [CrossRef] [Green Version]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Tsao, P.N.; Matsuoka, C.; Wei, S.C.; Sato, A.; Sato, S.; Hasegawa, K.; Chen, H.K.; Ling, T.Y.; Mori, M.; Cardoso, W.V.; et al. Epithelial Notch signaling regulates lung alveolar morphogenesis and airway epithelial integrity. Proc. Natl. Acad. Sci. USA 2016, 113, 8242–8247. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Nieuwenhuis, E.; Cohen, B.L.; Wang, W.; Canty, A.J.; Danska, J.S.; Coultas, L.; Rossant, J.; Wu, M.Y.; Piscione, T.D.; et al. Lunatic Fringe-mediated Notch signaling is required for lung alveogenesis. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 298, L45–L56. [Google Scholar] [CrossRef] [Green Version]
- Kavian, N.; Servettaz, A.; Weill, B.; Batteux, F. New insights into the mechanism of notch signalling in fibrosis. Open Rheumatol. J. 2012, 6, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Wasnick, R.M.; Korfei, M.; Piskulak, K.; Henneke, I.; Wilhelm, J.; Mahavadi, P.; Von der Beck, D.; Koch, M.; Shalashova, I.; Klymenko, O.; et al. Restored alveolar epithelial differentiation and reversed human lung fibrosis upon Notch inhibition. BioRxiv 2019, 580498. [Google Scholar] [CrossRef]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef]
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.S.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature 2015, 526, 578–582. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Krishnaveni, M.S.; Li, C.; Zhou, B.; Xing, Y.; Banfalvi, A.; Li, A.; Lombardi, V.; Akbari, O.; Borok, Z.; et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Investig. 2011, 121, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Coker, R.K.; Laurent, G.J.; Jeffery, P.K.; Du Bois, R.M.; Black, C.M.; McAnulty, R.J. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax 2001, 56, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Bonniaud, P.; Martin, G.; Margetts, P.J.; Ask, K.; Robertson, J.; Gauldie, J.; Kolb, M. Connective tissue growth factor is crucial to inducing a profibrotic environment in “fibrosis-resistant” BALB/c mouse lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Bonniaud, P.; Margetts, P.J.; Kolb, M.; Haberberger, T.; Kelly, M.; Robertson, J.; Gauldie, J. Adenoviral gene transfer of connective tissue growth factor in the lung induces transient fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Watts, K.L.; Spiteri, M.A. Connective tissue growth factor expression and induction by transforming growth factor-beta is abrogated by simvastatin via a Rho signaling mechanism. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L1323–L1332. [Google Scholar] [CrossRef] [Green Version]
- Lasky, J.A.; Ortiz, L.A.; Tonthat, B.; Hoyle, G.W.; Corti, M.; Athas, G.; Lungarella, G.; Brody, A.; Friedman, M. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am. J. Physiol. 1998, 275, L365–L371. [Google Scholar] [CrossRef]
- Pan, L.H.; Yamauchi, K.; Uzuki, M.; Nakanishi, T.; Takigawa, M.; Inoue, H.; Sawai, T. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur. Respir. J. 2001, 17, 1220–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Cassandras, M.; Peng, T. The Role of Hedgehog Signaling in Adult Lung Regeneration and Maintenance. J. Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Kugler, M.C.; Joyner, A.L.; Loomis, C.A.; Munger, J.S. Sonic hedgehog signaling in the lung. From development to disease. Am. J. Respir. Cell Mol. Biol. 2015, 52, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Liu, J.; Wu, Z.; Liu, T.; Ullenbruch, M.R.; Ding, L.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Reemergence of hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolanos, A.L.; Milla, C.M.; Lira, J.C.; Ramirez, R.; Checa, M.; Barrera, L.; Garcia-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M.; et al. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L978–L990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, G.A.; Hoyne, G.F.; Ahmad, S.A.; Jarman, E.; Wallace, W.A.; Harrison, D.J.; Haslett, C.; Lamb, J.R.; Howie, S.E. Expression of the developmental Sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. J. Pathol. 2003, 199, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Coon, D.R.; Roberts, D.J.; Loscertales, M.; Kradin, R. Differential epithelial expression of SHH and FOXF1 in usual and nonspecific interstitial pneumonia. Exp. Mol. Pathol. 2006, 80, 119–123. [Google Scholar] [CrossRef]
- Fitch, P.M.; Howie, S.E.; Wallace, W.A. Oxidative damage and TGF-beta differentially induce lung epithelial cell sonic hedgehog and tenascin-C expression: Implications for the regulation of lung remodelling in idiopathic interstitial lung disease. Int. J. Exp. Pathol. 2011, 92, 8–17. [Google Scholar] [CrossRef]
- Moshai, E.F.; Wemeau-Stervinou, L.; Cigna, N.; Brayer, S.; Somme, J.M.; Crestani, B.; Mailleux, A.A. Targeting the hedgehog-glioma-associated oncogene homolog pathway inhibits bleomycin-induced lung fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 2014, 51, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Kugler, M.C.; Loomis, C.A.; Samdani, R.; Zhao, Z.; Chen, G.J.; Brandt, J.P.; Brownell, I.; Joyner, A.L.; Rom, W.N.; et al. Hedgehog signaling in neonatal and adult lung. Am. J. Respir. Cell Mol. Biol. 2013, 48, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Kugler, M.C.; Yie, T.A.; Cai, Y.; Berger, J.Z.; Loomis, C.A.; Munger, J.S. The Hedgehog target Gli1 is not required for bleomycin-induced lung fibrosis. Exp. Lung Res. 2019, 45, 22–29. [Google Scholar] [CrossRef]
- Ge, L.; Habiel, D.M.; Hansbro, P.M.; Kim, R.Y.; Gharib, S.A.; Edelman, J.D.; Konigshoff, M.; Parimon, T.; Brauer, R.; Huang, Y.; et al. miR-323a-3p regulates lung fibrosis by targeting multiple profibrotic pathways. JCI Insight 2016, 1, e90301. [Google Scholar] [CrossRef]
- Pan, T.; Mason, R.J.; Westcott, J.Y.; Shannon, J.M. Rat alveolar type II cells inhibit lung fibroblast proliferation in vitro. Am. J. Respir. Cell Mol. Biol. 2001, 25, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portnoy, J.; Pan, T.; Dinarello, C.A.; Shannon, J.M.; Westcott, J.Y.; Zhang, L.; Mason, R.J. Alveolar type II cells inhibit fibroblast proliferation: Role of IL-1alpha. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L307–L316. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Peters-Golden, M.; Christensen, P.J.; Lama, V.; Kuziel, W.A.; Paine, R., 3rd; Toews, G.B. Alveolar epithelial cell inhibition of fibroblast proliferation is regulated by MCP-1/CCR2 and mediated by PGE2. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L342–L349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borok, Z.; Gillissen, A.; Buhl, R.; Hoyt, R.F.; Hubbard, R.C.; Ozaki, T.; Rennard, S.I.; Crystal, R.G. Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am. Rev. Respir. Dis. 1991, 144, 1080–1084. [Google Scholar] [CrossRef]
- Lama, V.; Moore, B.B.; Christensen, P.; Toews, G.B.; Peters-Golden, M. Prostaglandin E2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am. J. Respir. Cell Mol. Biol. 2002, 27, 752–758. [Google Scholar] [CrossRef]
- Tan, Q.; Ma, X.Y.; Liu, W.; Meridew, J.A.; Jones, D.L.; Haak, A.J.; Sicard, D.; Ligresti, G.; Tschumperlin, D.J. Nascent Lung Organoids Reveal Epithelium- and Bone Morphogenetic Protein-mediated Suppression of Fibroblast Activation. Am. J. Respir. Cell Mol. Biol. 2019, 61, 607–619. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Gahl, W.A.; Brantly, M.; Kaiser-Kupfer, M.I.; Iwata, F.; Hazelwood, S.; Shotelersuk, V.; Duffy, L.F.; Kuehl, E.M.; Troendle, J.; Bernardini, I. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome). N. Engl. J. Med. 1998, 338, 1258–1264. [Google Scholar] [CrossRef]
- Young, L.R.; Gulleman, P.M.; Short, C.W.; Tanjore, H.; Sherrill, T.; Qi, A.; McBride, A.P.; Zaynagetdinov, R.; Benjamin, J.T.; Lawson, W.E.; et al. Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky-Pudlak syndrome. JCI Insight 2016, 1, e88947. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; Von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFbeta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging (Albany N.Y.) 2012, 4, 932–951. [Google Scholar] [CrossRef]
- Hoare, M.; Narita, M. Notch and Senescence. Adv. Exp. Med. Biol. 2018, 1066, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Narita, M. NOTCH and the 2 SASPs of senescence. Cell Cycle 2017, 16, 239–240. [Google Scholar] [CrossRef] [Green Version]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17. [Google Scholar] [CrossRef]
- Borghesan, M.; Fafian-Labora, J.; Eleftheriadou, O.; Carpintero-Fernandez, P.; Paez-Ribes, M.; Vizcay-Barrena, G.; Swisa, A.; Kolodkin-Gal, D.; Ximenez-Embun, P.; Lowe, R.; et al. Small Extracellular Vesicles Are Key Regulators of Non-cell Autonomous Intercellular Communication in Senescence via the Interferon Protein IFITM3. Cell Rep. 2019, 27, 3956–3971. [Google Scholar] [CrossRef] [Green Version]
- Martin-Medina, A.; Lehmann, M.; Burgy, O.; Hermann, S.; Baarsma, H.A.; Wagner, D.E.; De Santis, M.M.; Ciolek, F.; Hofer, T.P.; Frankenberger, M.; et al. Increased Extracellular Vesicles Mediate WNT-5A Signaling in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018. [Google Scholar] [CrossRef]
- Yang, I.V.; Schwartz, D.A. Epigenetics of idiopathic pulmonary fibrosis. Transl. Res. 2015, 165, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Li, S.; Subramaniam, S.; Shyy, J.Y.; Chien, S. Epigenetic Regulation: A New Frontier for Biomedical Engineers. Annu. Rev. Biomed. Eng. 2017, 19, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.R.; Zhang, Y.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef] [Green Version]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyn, H.; Li, N.; Ferreira, H.J.; Moran, S.; Pisano, D.G.; Gomez, A.; Diez, J.; Sanchez-Mut, J.V.; Setien, F.; Carmona, F.J.; et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA 2012, 109, 10522–10527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, J.P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestri, G.A.; Vachani, A.; Whitney, D.; Elashoff, M.; Porta Smith, K.; Ferguson, J.S.; Parsons, E.; Mitra, N.; Brody, J.; Lenburg, M.E.; et al. A Bronchial Genomic Classifier for the Diagnostic Evaluation of Lung Cancer. N. Engl. J. Med. 2015, 373, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Steiling, K.; Kadar, A.Y.; Bergerat, A.; Flanigon, J.; Sridhar, S.; Shah, V.; Ahmad, Q.R.; Brody, J.S.; Lenburg, M.E.; Steffen, M.; et al. Comparison of proteomic and transcriptomic profiles in the bronchial airway epithelium of current and never smokers. PLoS ONE 2009, 4, e5043. [Google Scholar] [CrossRef]
- Sridhar, S.; Schembri, F.; Zeskind, J.; Shah, V.; Gustafson, A.M.; Steiling, K.; Liu, G.; Dumas, Y.M.; Zhang, X.; Brody, J.S.; et al. Smoking-induced gene expression changes in the bronchial airway are reflected in nasal and buccal epithelium. BMC Genom. 2008, 9, 259. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.; Sridhar, S.; Beane, J.; Brody, J.S.; Spira, A. SIEGE: Smoking Induced Epithelial Gene Expression Database. Nucleic Acids Res. 2005, 33, D573–D579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buro-Auriemma, L.J.; Salit, J.; Hackett, N.R.; Walters, M.S.; Strulovici-Barel, Y.; Staudt, M.R.; Fuller, J.; Mahmoud, M.; Stevenson, C.S.; Hilton, H.; et al. Cigarette smoking induces small airway epithelial epigenetic changes with corresponding modulation of gene expression. Hum. Mol. Genet. 2013, 22, 4726–4738. [Google Scholar] [CrossRef] [PubMed]
- Cotton, A.M.; Lam, L.; Affleck, J.G.; Wilson, I.M.; Penaherrera, M.S.; McFadden, D.E.; Kobor, M.S.; Lam, W.L.; Robinson, W.P.; Brown, C.J. Chromosome-wide DNA methylation analysis predicts human tissue-specific X inactivation. Hum. Genet. 2011, 130, 187–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boks, M.P.; Derks, E.M.; Weisenberger, D.J.; Strengman, E.; Janson, E.; Sommer, I.E.; Kahn, R.S.; Ophoff, R.A. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS ONE 2009, 4, e6767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarter, B.; Long, T.I.; Tsong, W.H.; Koh, W.P.; Yu, M.C.; Laird, P.W. Sex differential in methylation patterns of selected genes in Singapore Chinese. Hum. Genet. 2005, 117, 402–403. [Google Scholar] [CrossRef]
- El-Maarri, O.; Becker, T.; Junen, J.; Manzoor, S.S.; Diaz-Lacava, A.; Schwaab, R.; Wienker, T.; Oldenburg, J. Gender specific differences in levels of DNA methylation at selected loci from human total blood: A tendency toward higher methylation levels in males. Hum. Genet. 2007, 122, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dakhlallah, D.; Batte, K.; Wang, Y.; Cantemir-Stone, C.Z.; Yan, P.; Nuovo, G.; Mikhail, A.; Hitchcock, C.L.; Wright, V.P.; Nana-Sinkam, S.P.; et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 187, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, K.; Singh, I.; Dobersch, S.; Sarvari, P.; Gunther, S.; Cordero, J.; Mehta, A.; Wujak, L.; Cabrera-Fuentes, H.; Chao, C.M.; et al. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019, 10, 2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korfei, M.; Skwarna, S.; Henneke, I.; MacKenzie, B.; Klymenko, O.; Saito, S.; Ruppert, C.; Von der Beck, D.; Mahavadi, P.; Klepetko, W.; et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 2015, 70, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Sanders, Y.Y.; Hagood, J.S.; Liu, H.; Zhang, W.; Ambalavanan, N.; Thannickal, V.J. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur. Respir. J. 2014, 43, 1448–1458. [Google Scholar] [CrossRef]
- Conforti, F.; Davies, E.R.; Calderwood, C.J.; Thatcher, T.H.; Jones, M.G.; Smart, D.E.; Mahajan, S.; Alzetani, A.; Havelock, T.; Maher, T.M.; et al. The histone deacetylase inhibitor, romidepsin, as a potential treatment for pulmonary fibrosis. Oncotarget 2017, 8, 48737–48754. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Peng, R.; Phillips, J.E.; Deguzman, J.; Ren, Y.; Apparsundaram, S.; Luo, Q.; Bauer, C.M.; Fuentes, M.E.; DeMartino, J.A.; et al. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am. J. Pathol. 2013, 183, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Xiong, Y.; Zhang, G.; Chang, J. MicroRNAs in idiopathic pulmonary fibrosis, new research progress and their pathophysiological implication. Exp. Lung Res. 2018, 44, 178–190. [Google Scholar] [CrossRef]
- Wang, Y.; Frank, D.B.; Morley, M.P.; Zhou, S.; Wang, X.; Lu, M.M.; Lazar, M.A.; Morrisey, E.E. HDAC3-Dependent Epigenetic Pathway Controls Lung Alveolar Epithelial Cell Remodeling and Spreading via miR-17-92 and TGF-beta Signaling Regulation. Dev. Cell 2016, 36, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008, 24, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.; Royo, H.; Bortolin, M.L.; Lin, S.P.; Ferguson-Smith, A.C.; Cavaille, J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004, 14, 1741–1748. [Google Scholar] [CrossRef] [Green Version]
- Snyder, C.M.; Rice, A.L.; Estrella, N.L.; Held, A.; Kandarian, S.C.; Naya, F.J. MEF2A regulates the Gtl2-Dio3 microRNA mega-cluster to modulate WNT signaling in skeletal muscle regeneration. Development 2013, 140, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Milosevic, J.; Pandit, K.; Magister, M.; Rabinovich, E.; Ellwanger, D.C.; Yu, G.; Vuga, L.J.; Weksler, B.; Benos, P.V.; Gibson, K.F.; et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 879–887. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072269
Parimon T, Yao C, Stripp BR, Noble PW, Chen P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences. 2020; 21(7):2269. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072269
Chicago/Turabian StyleParimon, Tanyalak, Changfu Yao, Barry R Stripp, Paul W Noble, and Peter Chen. 2020. "Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis" International Journal of Molecular Sciences 21, no. 7: 2269. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072269