Genetic and Clinical Heterogeneity in Thirteen New Cases with Aceruloplasminemia. Atypical Anemia as a Clue for an Early Diagnosis

 ,
,  , , , , , , , and add
Show full author list
, , , , , , , and add
Show full author list
Abstract
:
1. Introduction
2. Patients and Methods
2.1. Patients
2.2. DNA Extraction, PCR Amplification, and DNA Sequencing
2.3. Bioinformatics and Computational Analysis
3. Results
3.1. Clinical and Biochemical Profile
3.2. Genetic Spectrum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miyajima, H. Aceruloplasminemia. Neuropathology 2015, 35, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Marchi, G.; Busti, F.; Lira Zidanes, A.; Castagna, A.; Girelli, D. Aceruloplasminemia: A Severe Neurodegenerative Disorder Deserving an Early Diagnosis. Front. Neurosci. 2019, 13, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyajima, H.; Nishimura, Y.; Mimguchi, K.; Sakamoto, M.; Shimizu, T.; Honda, N. Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology 1987, 37, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H.; Takahashi, Y.; Kamata, T.; Shimizu, H.; Sakai, N.; Gitlin, J.D. Use of desferrioxamine in the treatment of aceruloplasminemia. Ann. Neurol. 1997, 41, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Skidmore, F.M.; Drago, V.; Foster, P.; Schmalfuss, I.M.; Heilman, K.M.; Streiff, R.R. Aceruloplasminaemia with progressive atrophy without brain iron overload: Treatment with oral chelation. J. Neurol. Neurosurg. Psychiatry 2008, 79, 467–470. [Google Scholar] [CrossRef]
- Piperno, A.; Alessio, M. Aceruloplasminemia: Waiting for an Efficient Therapy. Front. Neurosci. 2018, 12, 903. [Google Scholar] [CrossRef] [Green Version]
- Yonekawa, M.; Okabe, T.; Asamoto, Y.; Ohta, M. A case of hereditary ceruloplasmin deficiency with iron deposition in the brain associated with chorea, dementia, diabetes mellitus and retinal pigmentation: Administration of fresh-frozen human plasma. Eur. Neurol. 1999, 42, 157–162. [Google Scholar] [CrossRef]
- Kuhn, J.; Bewermeyer, H.; Miyajima, H.; Takahashi, Y.; Kuhn, K.F.; Hoogenraad, T.U. Treatment of symptomatic heterozygous aceruloplasminemia with oral zinc sulphate. Brain Dev. 2007, 29, 450–453. [Google Scholar] [CrossRef]
- Zanardi, A.; Conti, A.; Cremonesi, M.; D’Adamo, P.; Gilberti, E.; Apostoli, P.; Cannistraci, C.C.; Piperno, A.; David, S.; Alessio, M. Ceruloplasmin replacement therapy ameliorates neurological symptoms in a preclinical model of aceruloplasminemia. EMBO Mol. Med. 2018, 10, 91–106. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.P. Does Ceruloplasmin Defend Against Neurodegenerative Diseases? Curr. Neuropharmacol. 2019, 17, 539–549. [Google Scholar] [CrossRef]
- Bento, I.; Peixoto, C.; Zaitsev, V.N.; Lindley, P.F. Ceruloplasmin revisited: Structural and functional roles of various metal cation-binding sites. Acta Crystallogr. D Biol. Crystallogr. 2007, 63 Pt 2, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Gitlin, J.D. Mechanisms of copper incorporation during the biosynthesis of human ceruloplasmin. J. Biol. Chem. 1991, 266, 5128–5134. [Google Scholar]
- Patel, B.N.; Dunn, R.J.; David, S. Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J. Biol. Chem. 2000, 275, 4305–4310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, S.; Yoshida, K.; Tomosugi, N.; Terada, T.; Hamaya, Y.; Kanaoka, S.; Miyajima, H. Biological effects of mutant ceruloplasmin on hepcidin-mediated internalization of ferroportin. Biochim. Biophys. Acta 2010, 1802, 968–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenawi, M.; Rouger, E.; Island, M.L.; Leroyer, P.; Robin, F.; Remy, S.; Tesson, L.; Anegon, I.; Nay, K.; Derbre, F.; et al. Ceruloplasmin deficiency does not induce macrophagic iron overload: Lessons from a new rat model of hereditary aceruloplasminemia. FASEB J. 2019, 33, 13492–13502. [Google Scholar] [CrossRef] [Green Version]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.B.; Jung, G.; Carla, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef]
- Kono, S. Aceruloplasminemia: An update. Int. Rev. Neurobiol. 2013, 110, 125–151. [Google Scholar]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Piubelli, C.; Castagna, A.; Marchi, G.; Rizzi, M.; Busti, F.; Badar, S.; Marchetti, M.; de Gobbi, M.; Roetto, A.; Xumerle, L.; et al. Identification of new BMP6 pro-peptide mutations in patients with iron overload. Am. J. Hematol. 2017, 92, 562–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badar, S.; Busti, F.; Ferrarini, A.; Xumerle, L.; Bozzini, P.; Capelli, P.; Giorgetti, A. Identification of novel mutations in hemochromatosis genes by targeted next generation sequencing in Italian patients with unexplained iron overload. Am. J. Hematol. 2016, 91, 420–425. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Geody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Sakamoto, H.; Inoue, K.; Higuchi, I.; Ono, Y.; Shimura, Y. Control of Drosophila Sex-lethal pre-mRNA splicing by its own female-specific product. Nucleic Acids Res. 1992, 20, 5533–5540. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.; Shvartsman, M.; Morán, E.; Lois, S.; Aranda, J.; Barqué, A.; de la Cruz, X.; Bruguera, M.; Vagace, J.M.; Gervasini, G.; et al. Functional consequences of transferrin receptor-2 mutations causing hereditary hemochromatosis type 3. Mol. Genet. Genom. Med. 2015, 3, 221–232. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2010, 38, D211–D222. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Samygina, V.R.; Sokolov, A.V.; Bourenkov, G.; Petoukhov, M.V.; Pulina, M.O.; Zakharova, E.T.; Vasilyev, V.B.; Bartunik, H.; Svergun, D.I. Ceruloplasmin: Macromolecular assemblies with iron-containing acute phase proteins. PLoS ONE 2013, 8, e67145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Pelucchi, S.; Mariani, R.; Ravasi, G.; Pelloni, I.; Marano, M.; Tremolizzo, L.; Alessio, M.; Piperno, A. Phenotypic heterogeneity in seven Italian cases of aceruloplasminemia. Parkinsonism Relat. Disord. 2018, 51, 36–42. [Google Scholar] [CrossRef]
- Miyajima, H.; Takahashi, Y.; Kono, S. Aceruloplasminemia, an inherited disorder of iron metabolism. Biometals 2003, 16, 205–213. [Google Scholar] [CrossRef]
- Brissot, P.; Bernard, D.G.; Brissot, E.; Loréal, O.; Troadec, M.B. Rare anemias due to genetic iron metabolism defects. Mutat. Res. 2018, 777, 52–63. [Google Scholar] [CrossRef]
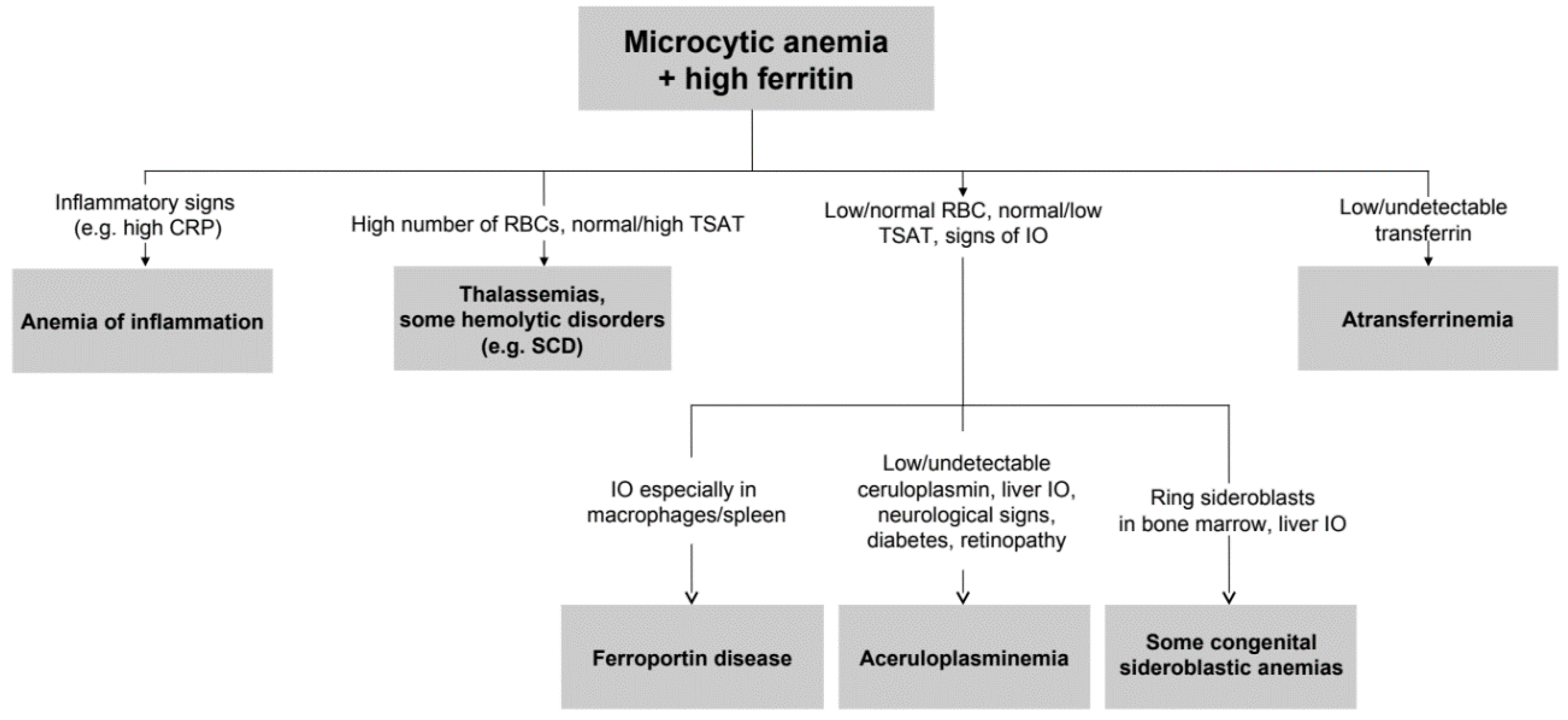
- Camaschella, C. How I manage patients with atypical microcytic anemia. Br. J. Haematol. 2013, 160, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Donker, A.E.; Raymakers, R.A.; Vlasveld, L.T.; van Barneveld, T.; Terink, R.; Dors, N.; Brons, P.P.; Knoers, N.V.; Swinkels, D.W. Practice guidelines for the diagnosis and management of microcytic anemias due to genetic disorders of iron metabolism or heme synthesis. Blood 2014, 123, 3873–3886. [Google Scholar] [CrossRef] [PubMed]
- Vroegindeweij, L.H.P.; Langendonk, J.G.; Langeveld, M.; Hoogendoorn, M.; Kievit, A.J.; Di Raimondo, D.; Wilson, J.H.P.; Boon, A.J.W. New insights in the neurological phenotype of aceruloplasminemia in Caucasian patients. Parkinsonism Relat. Disord. 2017, 36, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H.; Hosoi, Y. Aceruloplasminemia. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Family 1 | Family 2 | Family 3 | Family 4 | Family 4 | Family 5 | Family 6 | Family 7 | Family 8 | Family 8 | Family 9 | Family 10 | Family 11 | Normal Values |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II.3 | II.3 | II.3 | II.2 (proband) | II.3 | II.1 | II.2 | II.3 | II.3 (proband) | II.2 | II.2 | II.1 | II.1 | ||
| Country of Origin | Lithuania | Spain | India | India | India | Poland | Italy | Italy | Brazil | Brazil | Brazil | India | Pakistan | |
| Sex | F | F | M | M | M | F | M | M | F | F | F | F | M | |
| Age at Dx (years) | 75 | 33 | 40 | 66 | 61 | 40 | 46 | 62 | 37 | 29 | 46 | 25 | 16 | |
| Hb (g/dl) | 11.1 | 10.9 | 12.1 | 11.6 | 10.7 | 11.1 | 12.5 | 12.2 | 11.7 | 12.3 | 9.4 | 9.2 | 13.4 | 12–16 |
| MCV (fl) | 82,0 | 85.1 | 77 | 66.8 | 81 | 88.0 | 84 | 70.6 | 75.2 | 71.4 | 64.5 | 71.5 | 69 | 79–99 |
| Retyculocytes (%) | 0.5 | 0.8 | n/a | 0.9 | 1.2 | n/a | 0.58 | 1 | 1.77 | 1.1 | 1.77 | 0.51 | n.a. | 1.1–2.7 |
| RDW (%) | 16.8 | 15.2 | 14.3 | 17.3 | 15.5 | 15.5 | 15.4 | 16.1 | 16 | 17.2 | 18 | 17.1 | 18 | 11.3–14.5 |
| Serum iron (µg/dl) | 82.8 | 15 | 28 | 19 | 39.1 | 81.0 | 33 | 215 | 23 | 23 | 22 | 9.5 | 33.5 | 37–170 |
| Ferritin (ng/ml) | 12159 | 355.4 | 1077 | 1112 | 3845 | 1143 | 2100 | 3650 | 791 | 732 | 1060 | 757 | 1065 | 10–290 |
| Transferrin Saturation (%) | 12.4 | 4.3 | 10 | 5 | 12 | 39 | 9 | 88 | 9.24 | 9.83 | 8.2 | 4 | 8.8 | 20–55 |
| CP (mg/dl) | < 0.02 | < 2.0 | < 0.03 | < 0.03 | < 0.03 | 0.12 | undetectable | 0.12 | 11 | 9 | < 2 | n.a. | < 0.02 | 17–65 |
| ALT (U/l) | 55 | 12 | 32 | 24 | 36 | 19 | 37 | 113 | 26 | 179 | 37 | 24 | 87 | 14–36 |
| AST (U/l) | 43 | 17 | n.a | n.a | 24 | 16 | 20 | 80 | 12 | 82 | 29 | n/a | n/a | 8–40 |
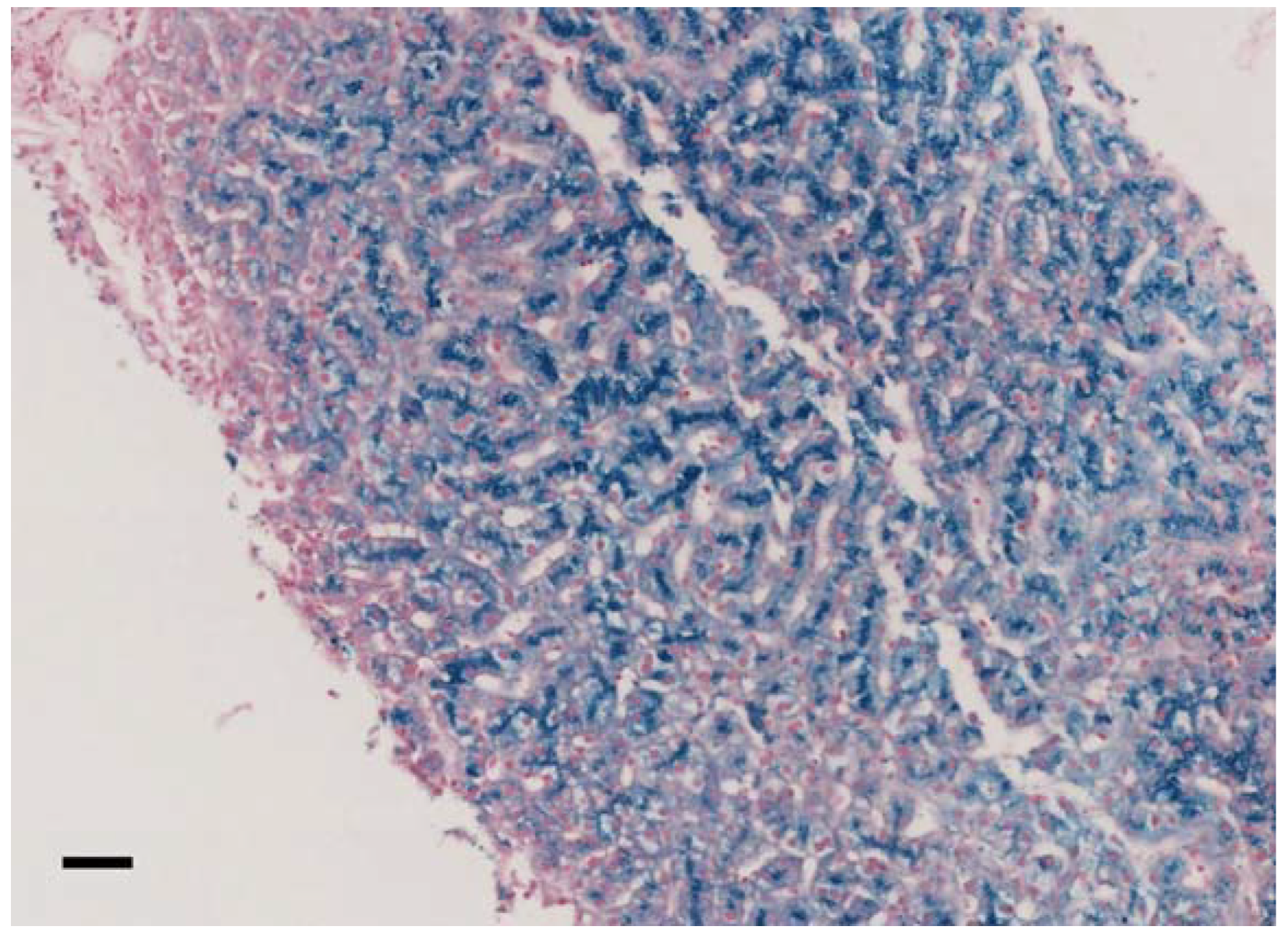
| Clue to ACP diagnosis | Low level of Cp. No Cu urine excretion Hepatocellular siderosis. Iron deposition in basal ganglia. | Low level of Cp. Low serum Cu and low urine Cu. Hepatic iron overload. Iron deposit in basal ganglia. | Iron deposition at brain MRI, Unexplained hyperferritinemia, low CP | Iron deposition at brain MRI, Unexplained hyperferritinemia, low CP | Iron deposition at brain MRI, Unexplained hyperferritinemia, low CP | Symptoms (including tremor) and very low CP | unexplained hyperferritinemia | overexpressed HFE Hemochromatosis, supposed additional non-HFE mutation(s) tested with NGS | Iron deposition at brain MRI | Familial investigation | Low CP in investigation of iron-refractory anaemia | Hair loss, mild executive dysfunction on formal neurocognitive assessment | low CP level | |
| Clinical presentation (symptoms and signs) | Moderate dementia with prevalent frontal features, cerebellar ataxia, oromandibular dystonia, torsion of the trunk, severe chorea-athetosis with choreiform movements. Mild type-2 DM. Retinal degeneration. Mild anaemi | Unspecific symptoms: Fatigue; abdominal discomfort and chronic anaemia. Liver ultrasonografy showed hepatic lesions that justified a MRI, that showed iron overload | Neurological symptoms: progressive cognitive decline, diabetes, mild bradykinesia with mild finger nose ataxia and dysdiadochokinesia .Unexplained anaemia | Neurological symptoms: progressive(over 5 years) cognitive decline, diabetes, tremor left hand Unexplained anaemia | Concentration and intellectual ability decline. Unexplained anaemia, DM. | Head and postural tremor of upper and lower limbs, slight dysmetria, ataxia, proximal weakness of lower extremities, horizontal nystagmus, and tunnel vision. | Liver iron overload and mild anaemia | Iron overload. Mild anaemia was consistent also with b-thalassemia trait | Choreiform movement disorder, mild anaemia, DM-2, asymptomatic retina pigmentation | Asymptomatic. Familial investigation | Mild anaemia, DM-2, asthenia, mild movement disorder | April 2015 acute psychotic episode with catatonia, hair loss, mild executive dysfunction on formal neurocognitive assessment | Concentration/memory lapses | |
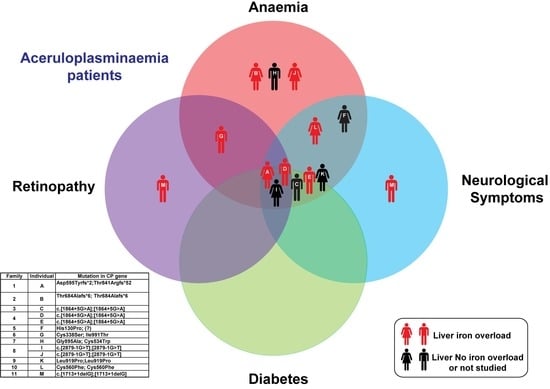
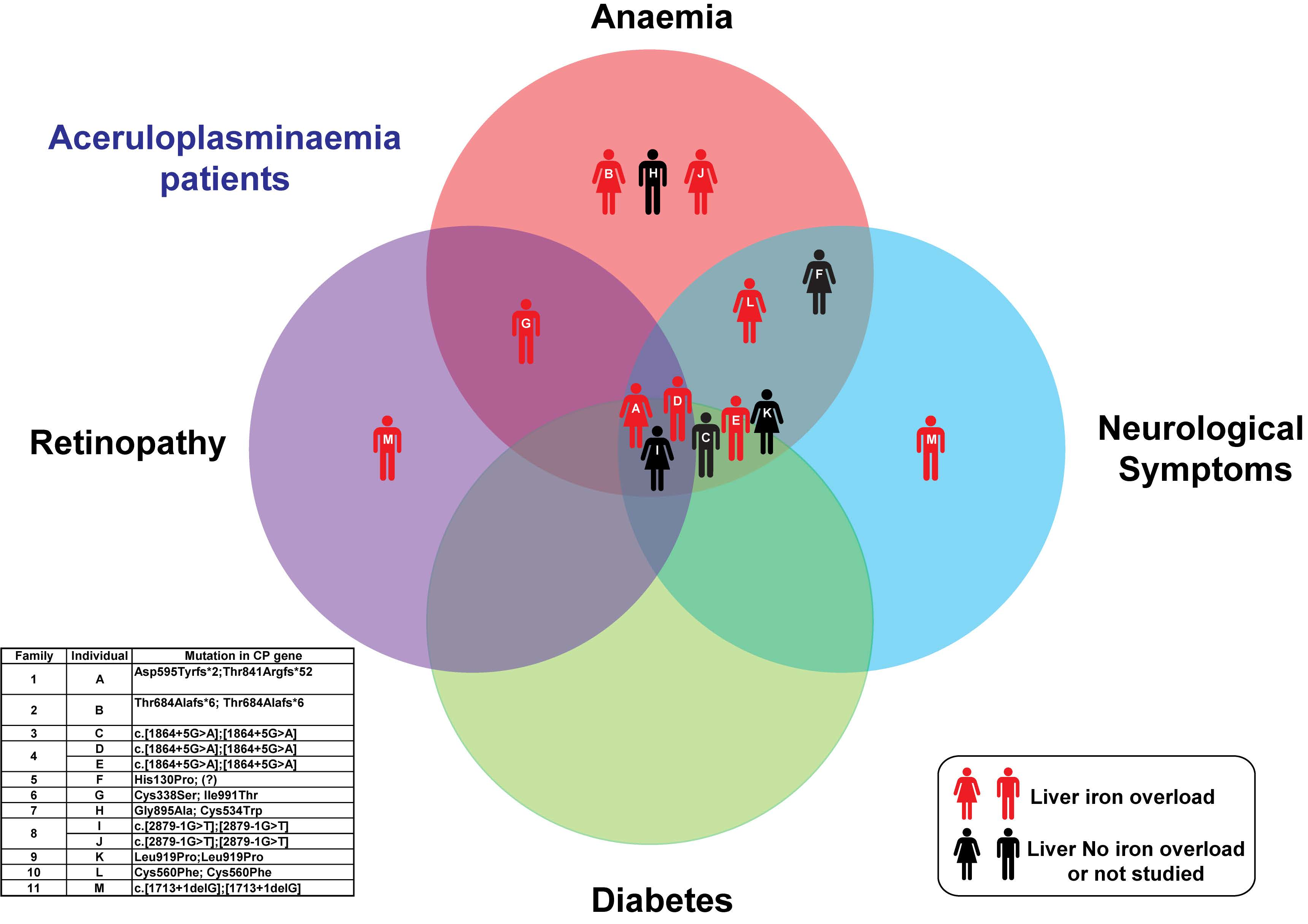
| Anaemia | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | No | |
| Neurological symptoms | Yes | No | Yes | Yes | Yes | Yes | No | No | Yes | No | Yes | Yes | Yes | |
| Liver Iron overload | Yes | Yes | n.a. | Yes | Yes | No | Yes | n.a. | n.a. | Yes | n.a | Yes | Yes | |
| Diabetes | Yes | No | Yes | Yes | Yes | No | No | No | Yes | No | Yes | No | No | |
| Retinopathy | Yes | No | No | No | No | Not evaluated | Yes but not typical for aceruloplasminemia | No | Yes | Not evaluated | Not evaluated | No | Yes | |
| Brain MRI (sites of iron accumulation, in brief) | Iron overload in putamen | Iron overload in lenticular, dentate and thalamus | SWI increased susceptibility involving the cerebelum, basal ganglia, thalami, red and dentate nuclei. | MRI SWI with marked susceptibility predominantly involving the lateral putamen, red nucleus, striaum, thalamic, pulvinar, cerebellar dentate nucleus | Iron overload in lentiform caudate, dorsal lateral thalami and dentate nuclei | FLAIR and T2 hypointenseties in the putamen and substantia nigra | no iron overload | no iron overload | Iron overload in thalami, basal ganglia, and cerebellum | Iron overload in thalami, basal ganglia, red nuclei, dentate, cerebellum and brain cortex | Iron overload in thalami, basal ganglia, dentate, and cerebellum | Iron overload in choroid plexus, bilateral dentate nuclei thalamic and basal ganglia | n.a. | |
| Liver MRI or (biopsy) | hepatocellular siderosis grade III | HII = 9.58, severe iron overload | n.a. | Feriscan LIC: 9.4 mg/g dw | LIC by Ferriscan: 6.3 mg/g dw | Biopsy: small depositions of yellow-brown pigment (stain for ferrum - negative) - probably lipofuscin | LIC 340 µM/g = HII 7.9 (severe iron overload) | n.a. | not performed | Iron overload in liver and pancreas (qualitative) | not performed | LIC by Ferriscan 9.0 mg/g/dw | LIC by Ferriscan: 5.3 mg/g dw | HII < 1.9 |
| Diagnostic delay | 20 years | 1.5 years | 5 years | 2 years | 4 years | 13 years | 3 years | 30 years | 1 year | Not applicable | 32 years | 3 years | Not known | |
| Iron chelation or other therapy | No Iron chelation therapy. | Deferasirox from 10/2014 to 11/2015; Deferiprone from 11/2015; Vitamin E. | Started on FFP OctaplasLG every 2 weeks on diagnosis. Later added Deferiprone | Deferiprone started on diagnosis (25 mg/kg/d) | Deferiprone started 2011 | Zinc | Deferasirox and desferoxamine both suspended for renal insufficiency, actually on deferiprone | Desferoxamine and “micro”-phlebotomies | Deferiprone | Desferoxamine, combination with deferiprone | Deferiprone | Deferiprone | ||
| Other clinical data | Bilateral cataracts | Psoriasis | None | hypertension, previous CVA, low Vitamin B12, bilateral cataracts | 2013: DM, 2011: hearing loss | Hypertension | 3-4 alcoholic units/day, arterial hypertension, overweight, central serous chorioretinopathy | Beta-Thal trait; fully penetrant HFE-related HH (C282Y homozygous) in the 3rd decade of life | Hypercholesterolemia, macroalbuminuria | Hypothyroidism, lower limb venous thrombosis, migraine | Hypothyroidism, glaucoma, kidney stones, chronic diarrhea | Iatrogenic iron overload due to oral iron supplementation for microcytic anaemia, hypothyroid, amenorhoea (thought to be due to polycystic ovaries), microcytic anaemia, hypothyroid, borderline oral glucose tolerance test. | White nails, Acquired leukopenia, Bilateral lattice degeneration of fundi-risk of retinal detachment. Vit D depletion | |
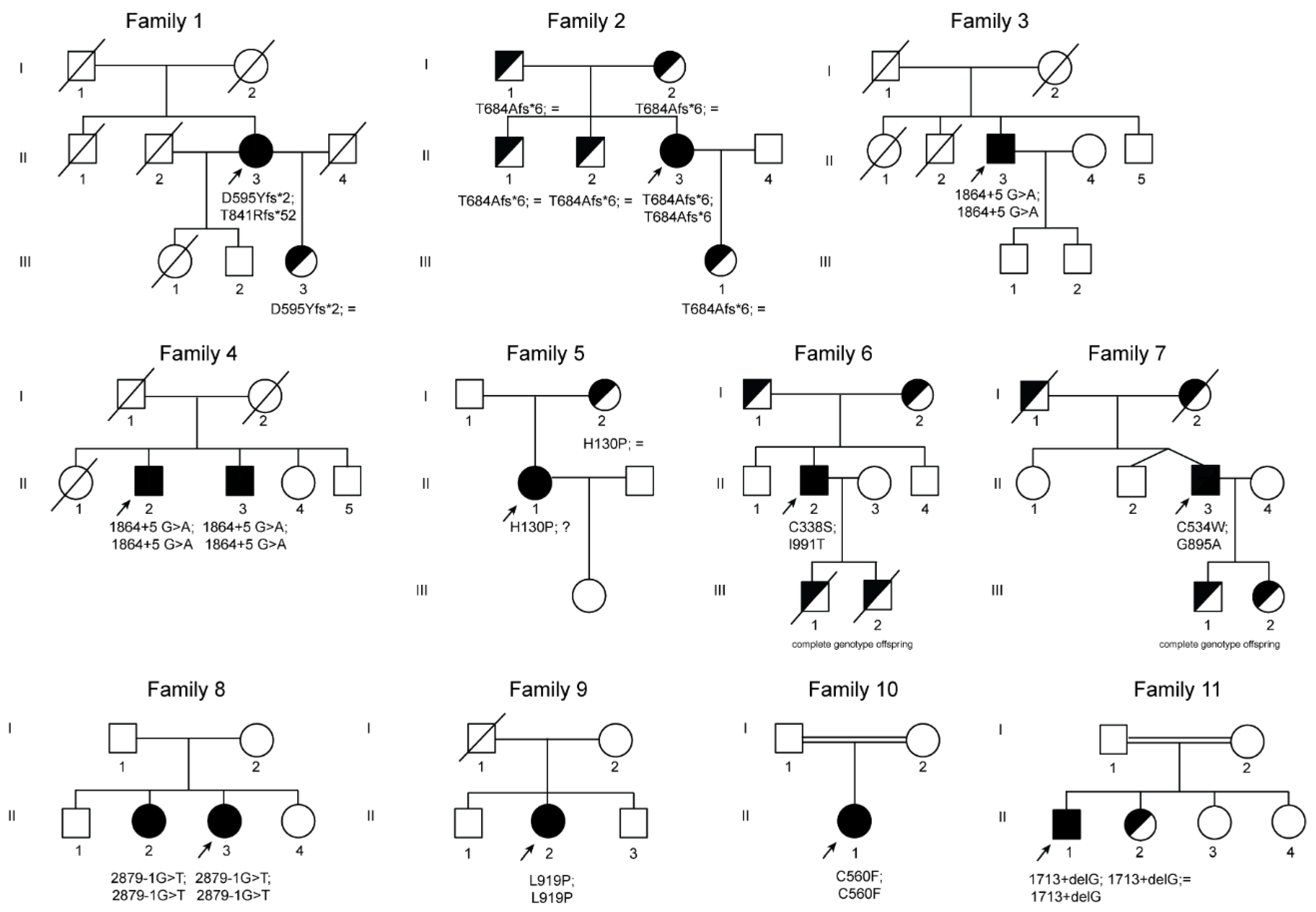
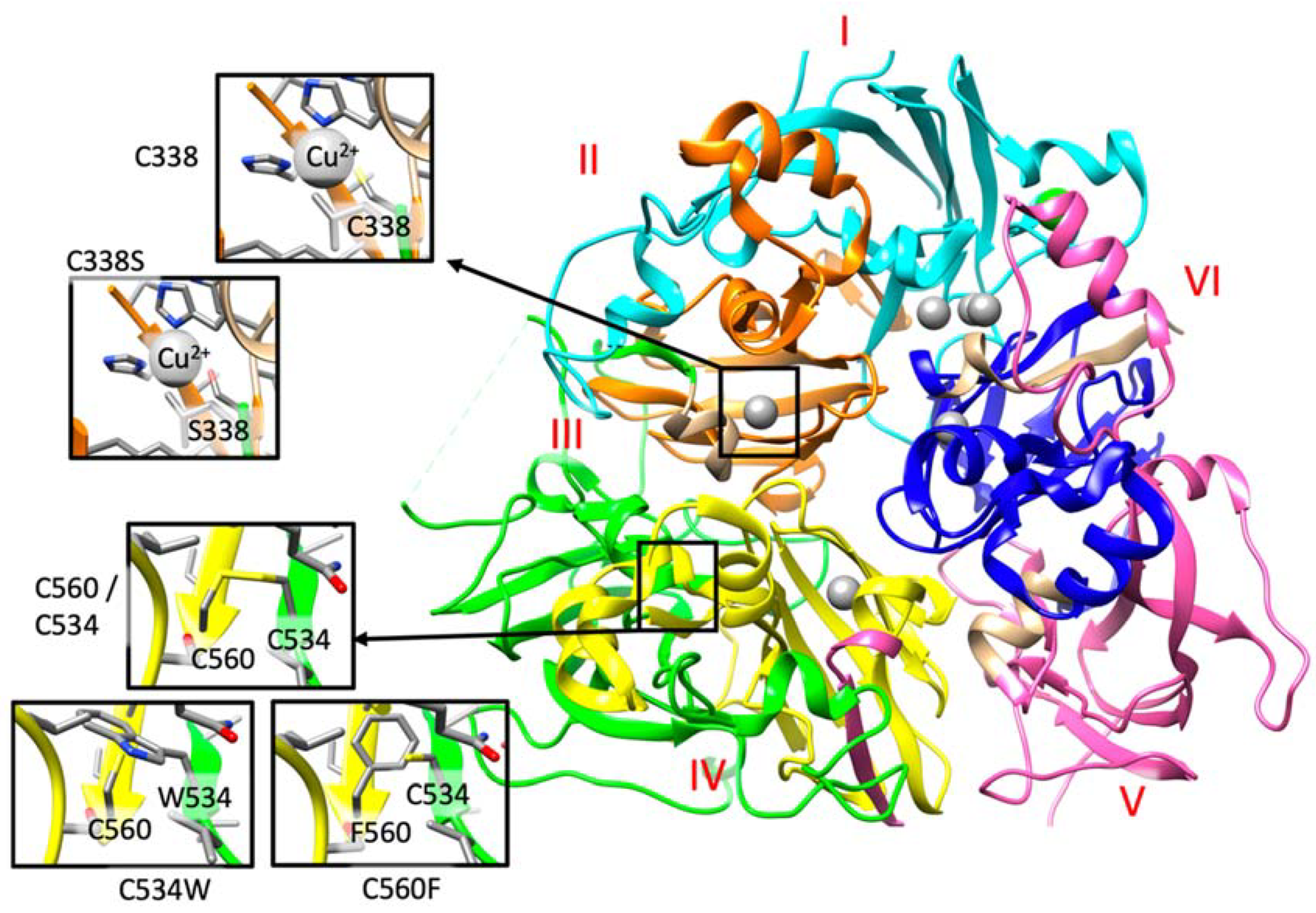
| Genetics CP gene NM_000096.3; NP_000087.1 | c.[1783_1787delGATAA(;)2520_2523delAACA] p.(Asp595Tyrfs*2;Thr841Argfs*52) | c.[2050_2051delAC]; [2050_2051delAC] p.(Thr684Alafs*6); (Thr684Alafs*6) | c.[1864+5G>A];[1864+5G>A] | c.[1864+5G>A];[1864+5G>A] | c.[1864+5G>A];[1864+5G>A] | c.[389A>C]; [=] p.(His130Pro); (=) | c.[1012T>A(;)2972T>C] p. (Cys338Ser);(Ile991Thr) | c.[2684G>C(;)1602T>G] p. (Gly895Ala);(Cys534Trp) | c.[2879-1G>T];[2879-1G>T] | c.[2879-1G>T];[2879-1G>T] | c.[2756T>C];[2756T>C] p.(Leu919Pro);(Leu919Pro) | c.[1679G>T];[1679G>T] p.(Cys560Phe);(Cys560Phe) | c.[1713+1delG];[1713+1delG] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vila Cuenca, M.; Marchi, G.; Barqué, A.; Esteban-Jurado, C.; Marchetto, A.; Giorgetti, A.; Chelban, V.; Houlden, H.; Wood, N.W.; Piubelli, C.; et al. Genetic and Clinical Heterogeneity in Thirteen New Cases with Aceruloplasminemia. Atypical Anemia as a Clue for an Early Diagnosis. Int. J. Mol. Sci. 2020, 21, 2374. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072374
Vila Cuenca M, Marchi G, Barqué A, Esteban-Jurado C, Marchetto A, Giorgetti A, Chelban V, Houlden H, Wood NW, Piubelli C, et al. Genetic and Clinical Heterogeneity in Thirteen New Cases with Aceruloplasminemia. Atypical Anemia as a Clue for an Early Diagnosis. International Journal of Molecular Sciences. 2020; 21(7):2374. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072374
Chicago/Turabian StyleVila Cuenca, Marc, Giacomo Marchi, Anna Barqué, Clara Esteban-Jurado, Alessandro Marchetto, Alejandro Giorgetti, Viorica Chelban, Henry Houlden, Nicholas W Wood, Chiara Piubelli, and et al. 2020. "Genetic and Clinical Heterogeneity in Thirteen New Cases with Aceruloplasminemia. Atypical Anemia as a Clue for an Early Diagnosis" International Journal of Molecular Sciences 21, no. 7: 2374. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072374