Checkpoint Kinase 1 (CHK1) Inhibition Enhances the Sensitivity of Triple-Negative Breast Cancer Cells to Proton Irradiation via Rad51 Downregulation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
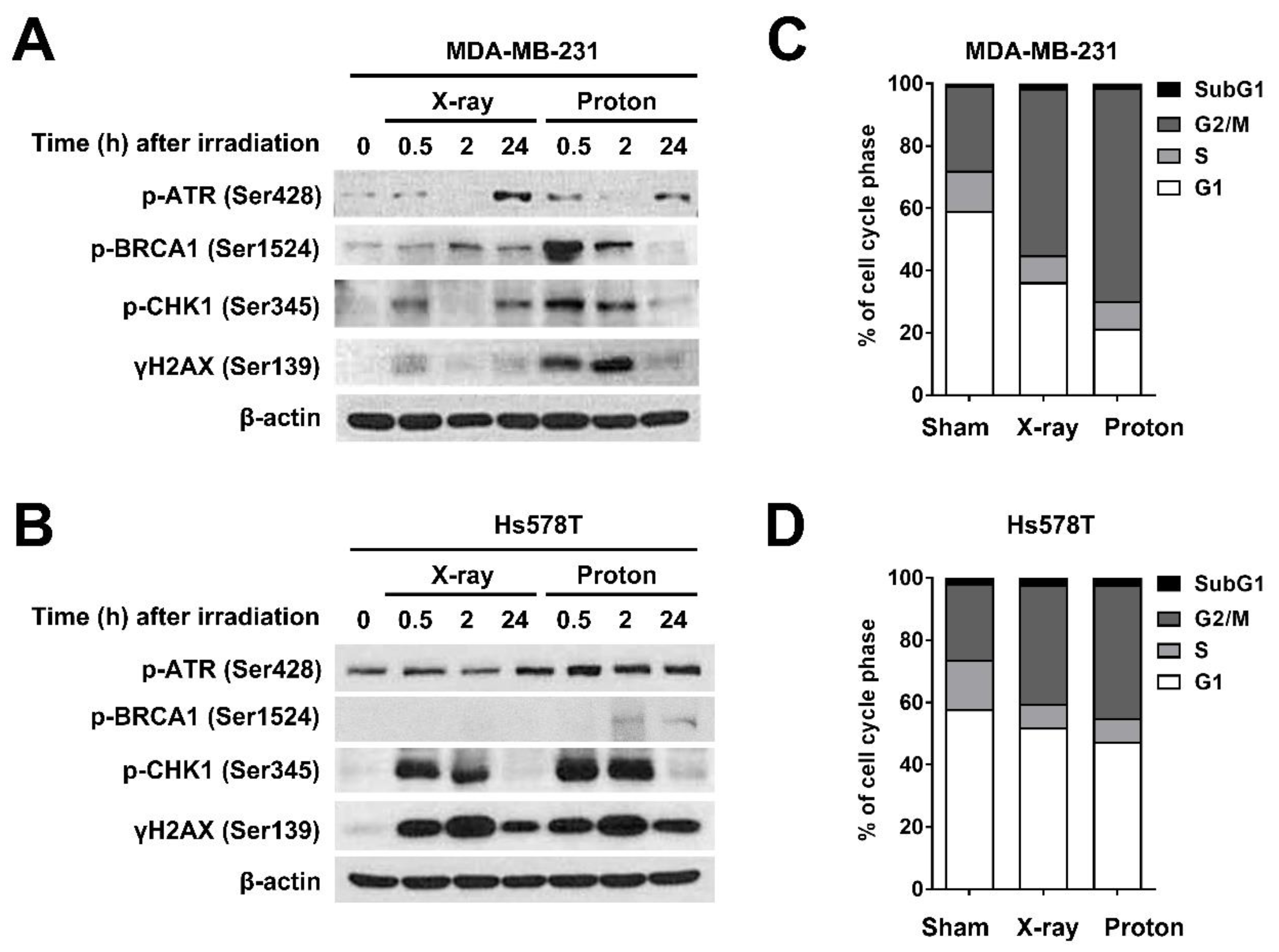
2.1. X-Rays and Protons Differently Affect ATR/CHK1 Activation in Human TNBC Cell Lines
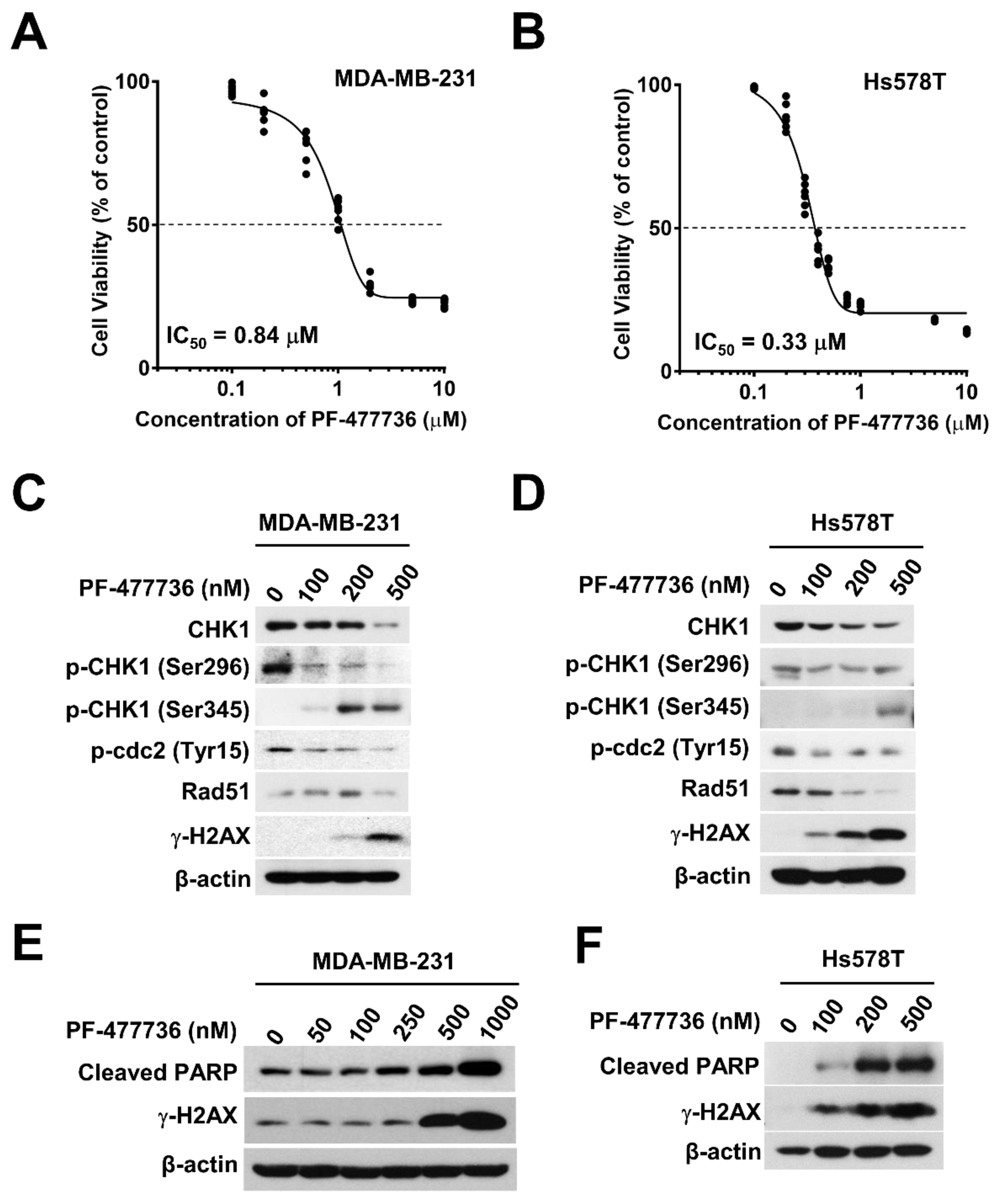
2.2. Pharmacological Inhibition of CHK1 Induces DNA Damage and Apoptosis in TNBC Cells
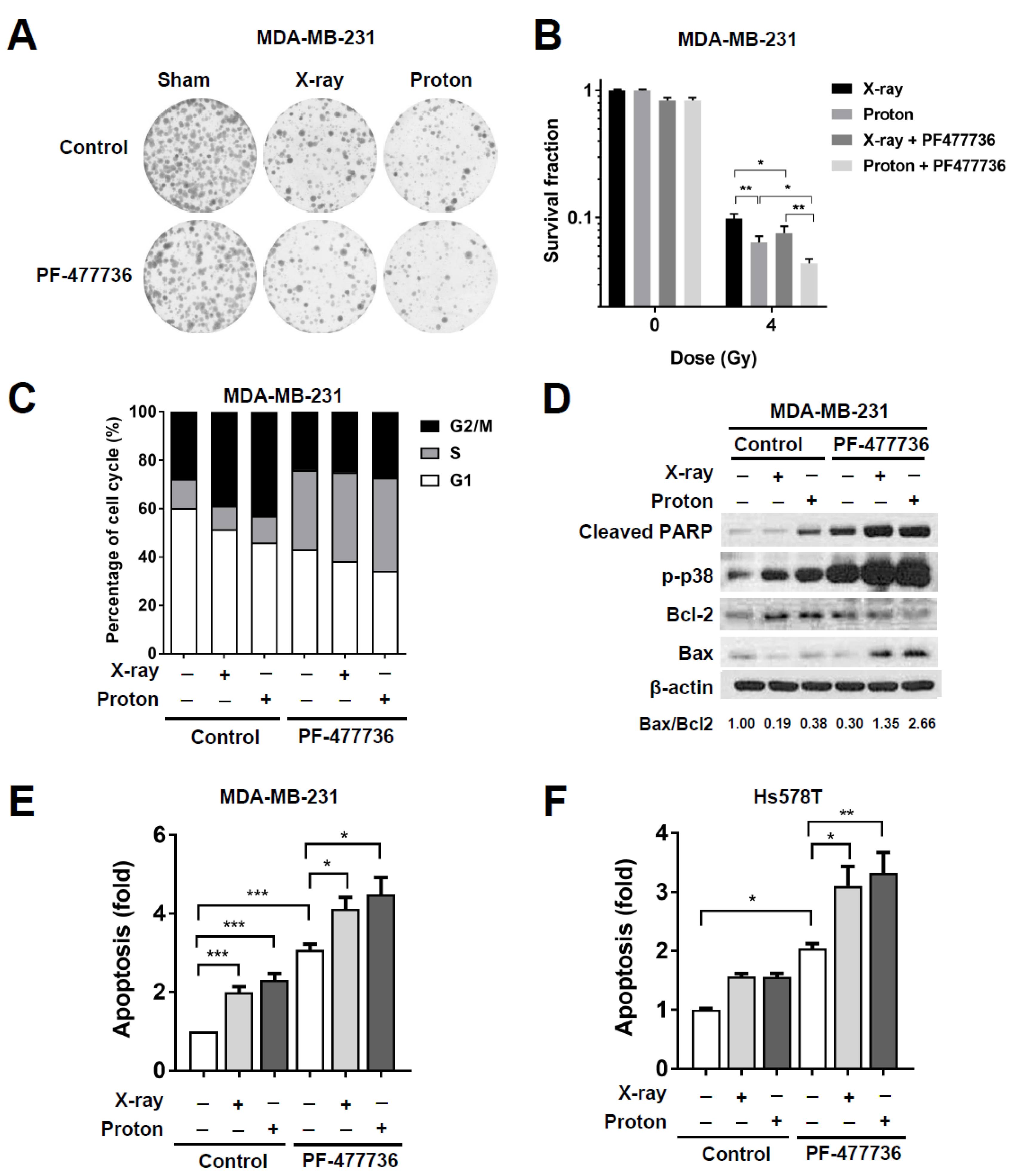
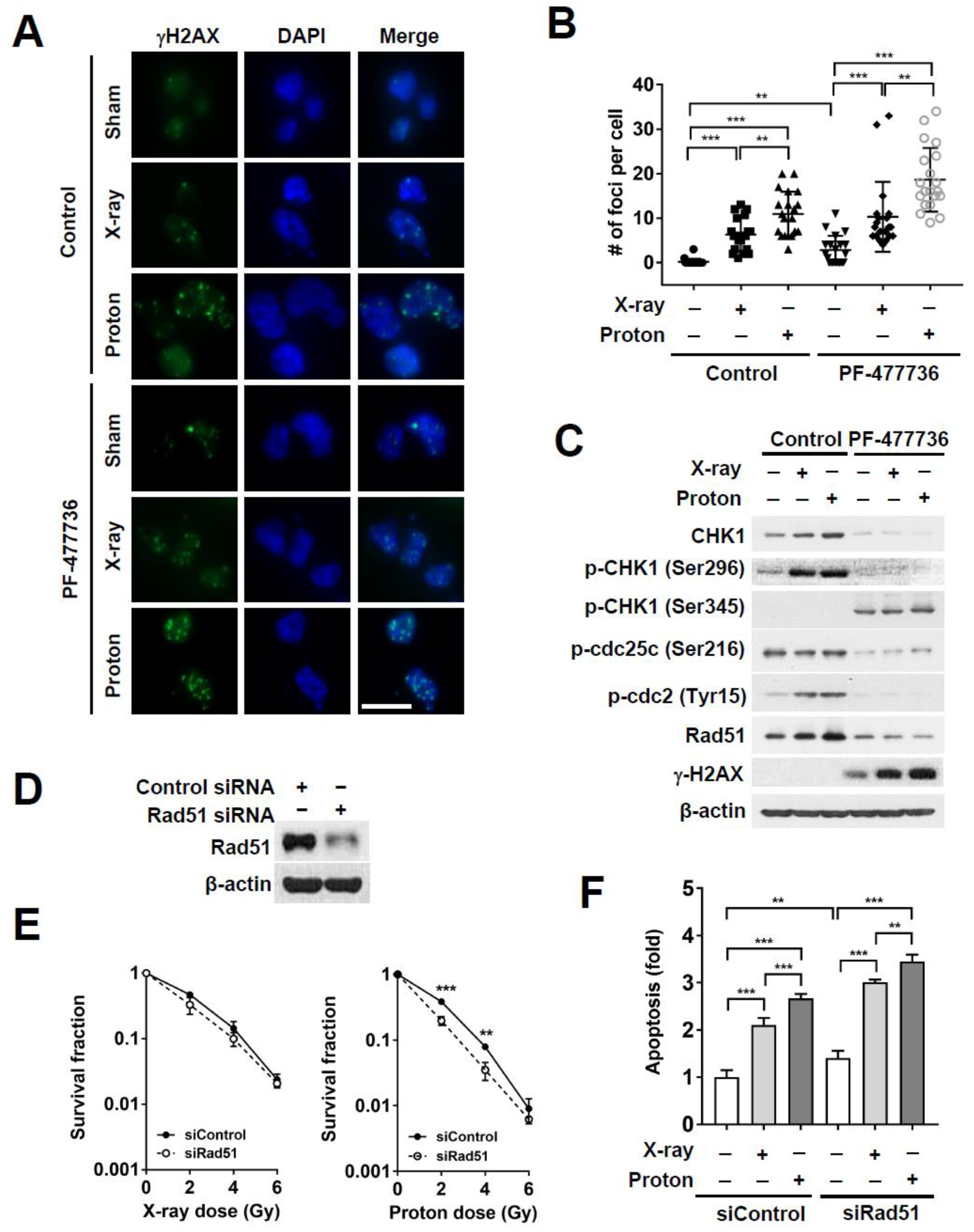
2.3. CHK1 Inhibition in Response to PF-477736 Treatment Sensitizes TNBC Cells to Proton Irradiation
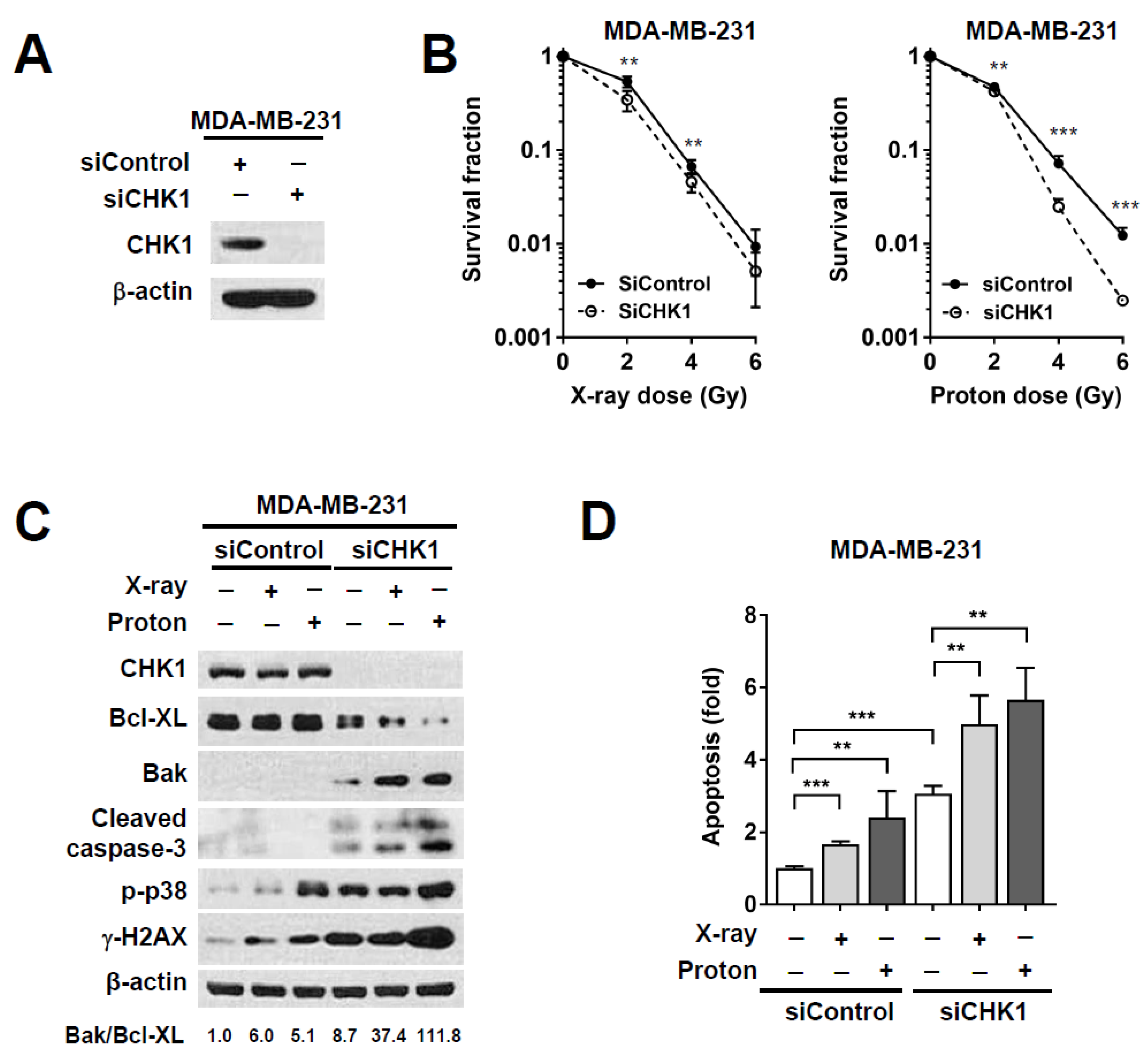
2.4. CHK1 Knockdown via Small Interfering RNA (siRNA) Treatment Sensitizes MDA-MB-231 Cells to Proton Irradiation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents and Antibodies
4.3. Cell Viability Assay
4.4. Irradiation Experiments
4.5. Clonogenic Survival Assay
4.6. Immunoblotting
4.7. Flow Cytometry
4.8. Immunofluorescence
4.9. Gene Knockdown Experiments
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| ATR | Ataxia telangiectasia and rad3-related |
| Bax | Bcl-2-associated X |
| Bcl-2 | B-cell lymphoma 2 |
| BSA | Bovine serum albumin |
| CHK1 | Checkpoint kinase 1 |
| DAPI | 4′,6-diamino-2-phenylindole |
| FITC | Fluorescein isothiocyanate |
| PBT | Proton beam therapy |
| DSB | Double strand break |
| HRR | Homologous recombination repair |
| NHEJ | Non-homologous end joining |
| PARP | Poly(ADP-ribose)polymerase |
| RT | Radiation therapy |
| siRNA | Small interfering ribonucleic acid |
| TNBC | Triple negative breast cancer |
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. New Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. New Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Turk, A.A.; Wisinski, K.B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef]
- Fatehi, D.; Soltani, A.; Ghatrehsamani, M. SRT1720, a potential sensitizer for radiotherapy and cytotoxicity effects of NVB-BEZ235 in metastatic breast cancer cells. Pathol. Res. Pract. 2018, 214, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.K.; Jagsi, R.; Woodward, W.A.; Ho, A. Breast Cancer Biology: Clinical Implications for Breast Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 23–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, N.Y.; Kim, D.H.; Cho, B.J.; Choi, E.J.; Lee, J.S.; Wu, H.G.; Chie, E.K.; Kim, I.A. Radiosensitization with combined use of olaparib and PI-103 in triple-negative breast cancer. BMC Cancer 2015, 15, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Leteur, C.; Yang, C.; Zhang, P.; Castedo, M.; Pierre, A.; Golsteyn, R.M.; Bourhis, J.; Kroemer, G.; Deutsch, E. Radiosensitization by Chir-124, a selective CHK1 inhibitor: Effects of p53 and cell cycle checkpoints. Cell Cycle 2009, 8, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.; Barker, H.E.; Kyula, J.; McLaughlin, M.; Dillon, M.T.; Schick, U.; Hafsi, H.; Thompson, A.; Khoo, V.; Harrington, K.; et al. An orally bioavailable Chk1 inhibitor, CCT244747, sensitizes bladder and head and neck cancer cell lines to radiation. Radiother. Oncol. 2017, 122, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.R.; Yang, Z.Z.; Wang, S.J.; Zhang, L.; Luo, J.R.; Feng, Y.; Yu, X.L.; Chen, X.X.; Guo, X.M. The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy. Acta Pharm. Sin. 2017, 38, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunstein, L.Z.; Cahlon, O. Potential Morbidity Reduction with Proton Radiation Therapy for Breast Cancer. Semin. Radiat. Oncol. 2018, 28, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Ghosh, P.; Magpayo, N.; Testa, M.; Tang, S.; Gheorghiu, L.; Biggs, P.; Paganetti, H.; Efstathiou, J.A.; Lu, H.M.; et al. Lung cancer cell line screen links fanconi anemia/BRCA pathway defects to increased relative biological effectiveness of proton radiation. Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 1081–1089. [Google Scholar] [CrossRef]
- Liu, Q.; Underwood, T.S.; Kung, J.; Wang, M.; Lu, H.M.; Paganetti, H.; Held, K.D.; Hong, T.S.; Efstathiou, J.A.; Willers, H. Disruption of SLX4-MUS81 Function Increases the Relative Biological Effectiveness of Proton Radiation. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.; Son, A.; Lee, G.H.; Shin, S.W.; Park, S.; Ahn, S.H.; Chung, Y.; Yu, J.I.; Park, H.C. Targeting DNA-dependent protein kinase sensitizes hepatocellular carcinoma cells to proton beam irradiation through apoptosis induction. PLoS ONE 2019, 14, e0218049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.; Park, S.; Cho, W.K.; Choi, D.H. Cyclin D1 is Associated with Radiosensitivity of Triple-Negative Breast Cancer Cells to Proton Beam Irradiation. Int. J. Mol. Sci. 2019, 20, 4943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Complex DNA Damage Induced by High Linear Energy Transfer Alpha-Particles and Protons Triggers a Specific Cellular DNA Damage Response. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Fontana, A.O.; Augsburger, M.A.; Grosse, N.; Guckenberger, M.; Lomax, A.J.; Sartori, A.A.; Pruschy, M.N. Differential DNA repair pathway choice in cancer cells after proton- and photon-irradiation. Radiother. Oncol. 2015, 116, 374–380. [Google Scholar] [CrossRef] [Green Version]
- Konings, K.; Vandevoorde, C.; Baselet, B.; Baatout, S.; Moreels, M. Combination Therapy With Charged Particles and Molecular Targeting: A Promising Avenue to Overcome Radioresistance. Front. Oncol. 2020, 10, 128. [Google Scholar] [CrossRef]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Janetka, J.W.; Piwnica-Worms, H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011, 17, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, Z.; Xiao, Z.; Liu, H.; Dou, Z.; Feng, X.; Shi, H. Chk1 knockdown confers radiosensitization in prostate cancer stem cells. Oncol. Rep. 2012, 28, 2247–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, C.J.; Kriegs, M.; Laban, S.; Tribius, S.; Knecht, R.; Petersen, C.; Dikomey, E.; Rieckmann, T. HPV-positive HNSCC cell lines but not primary human fibroblasts are radiosensitized by the inhibition of Chk1. Radiother. Oncol. 2013, 108, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Bertheau, P.; Lehmann-Che, J.; Varna, M.; Dumay, A.; Poirot, B.; Porcher, R.; Turpin, E.; Plassa, L.F.; de Roquancourt, A.; Bourstyn, E.; et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast (Edinb. Scotl.) 2013, 22 (Suppl. 2), S27–S29. [Google Scholar] [CrossRef] [PubMed]
- Cuddihy, A.R.; Bristow, R.G. The p53 protein family and radiation sensitivity: Yes or no? Cancer Metastasis Rev. 2004, 23, 237–257. [Google Scholar] [CrossRef]
- Albiges, L.; Goubar, A.; Scott, V.; Vicier, C.; Lefebvre, C.; Alsafadi, S.; Commo, F.; Saghatchian, M.; Lazar, V.; Dessen, P.; et al. Chk1 as a new therapeutic target in triple-negative breast cancer. Breast (Edinb. Scotl.) 2014, 23, 250–258. [Google Scholar] [CrossRef]
- Paganetti, H. Relating proton treatments to photon treatments via the relative biological effectiveness-should we revise current clinical practice? Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 892–894. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Li, Y.; Han, S.; Zhu, J.; Wang, X.; Molkentine, D.P.; Blanchard, P.; Yang, Y.; Zhang, R.; et al. Human papillomavirus status and the relative biological effectiveness of proton radiotherapy in head and neck cancer cells. Head Neck 2017, 39, 708–715. [Google Scholar] [CrossRef]
- Choi, C.; Lee, C.; Shin, S.W.; Kim, S.Y.; Hong, S.N.; Park, H.C. Comparison of Proton and Photon Beam Irradiation in Radiation-Induced Intestinal Injury Using a Mouse Model. Int. J. Mol. Sci. 2019, 20, 1894. [Google Scholar] [CrossRef] [Green Version]
- De Summa, S.; Pinto, R.; Sambiasi, D.; Petriella, D.; Paradiso, V.; Paradiso, A.; Tommasi, S. BRCAness: A deeper insight into basal-like breast tumors. Ann. Oncol. 2013, 24 (Suppl. 8), viii13–viii21. [Google Scholar] [CrossRef]
- Xu, Y.; Diao, L.; Chen, Y.; Liu, Y.; Wang, C.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; et al. Promoter methylation of BRCA1 in triple-negative breast cancer predicts sensitivity to adjuvant chemotherapy. Ann. Oncol. 2013, 24, 1498–1505. [Google Scholar] [CrossRef]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- Chung, K.; Han, Y.; Kim, J.; Ahn, S.H.; Ju, S.G.; Jung, S.H.; Chung, Y.; Cho, S.; Jo, K.; Shin, E.H.; et al. The first private-hospital based proton therapy center in Korea; status of the Proton Therapy Center at Samsung Medical Center. Radiat. Oncol. J. 2015, 33, 337–343. [Google Scholar] [CrossRef]
- Yu, J.I.; Choi, C.; Shin, S.W.; Son, A.; Lee, G.H.; Kim, S.Y.; Park, H.C. Valproic Acid Sensitizes Hepatocellular Carcinoma Cells to Proton Therapy by Suppressing NRF2 Activation. Sci. Rep. 2017, 7, 14986. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Son, A.; Lee, H.S.; Lee, Y.J.; Park, H.C. Radiosensitization by Marine Sponge Agelas sp. Extracts in Hepatocellular Carcinoma Cells with Autophagy Induction. Sci. Rep. 2018, 8, 6317. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.W.; Choi, C.; Lee, G.H.; Son, A.; Kim, S.H.; Park, H.C.; Batinic-Haberle, I.; Park, W. Mechanism of the Antitumor and Radiosensitizing Effects of a Manganese Porphyrin, MnHex-2-PyP. Antioxid. Redox. Signal. 2017, 27, 1067–1082. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, C.; Cho, W.K.; Park, S.; Shin, S.-W.; Park, W.; Kim, H.; Choi, D.H. Checkpoint Kinase 1 (CHK1) Inhibition Enhances the Sensitivity of Triple-Negative Breast Cancer Cells to Proton Irradiation via Rad51 Downregulation. Int. J. Mol. Sci. 2020, 21, 2691. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082691
Choi C, Cho WK, Park S, Shin S-W, Park W, Kim H, Choi DH. Checkpoint Kinase 1 (CHK1) Inhibition Enhances the Sensitivity of Triple-Negative Breast Cancer Cells to Proton Irradiation via Rad51 Downregulation. International Journal of Molecular Sciences. 2020; 21(8):2691. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082691
Chicago/Turabian StyleChoi, Changhoon, Won Kyung Cho, Sohee Park, Sung-Won Shin, Won Park, Haeyoung Kim, and Doo Ho Choi. 2020. "Checkpoint Kinase 1 (CHK1) Inhibition Enhances the Sensitivity of Triple-Negative Breast Cancer Cells to Proton Irradiation via Rad51 Downregulation" International Journal of Molecular Sciences 21, no. 8: 2691. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082691