Synovial Fluid Interleukin-16 Contributes to Osteoclast Activation and Bone Loss through the JNK/NFATc1 Signaling Cascade in Patients with Periprosthetic Joint Infection

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
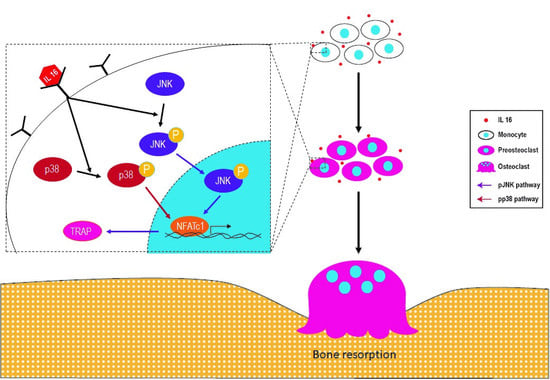
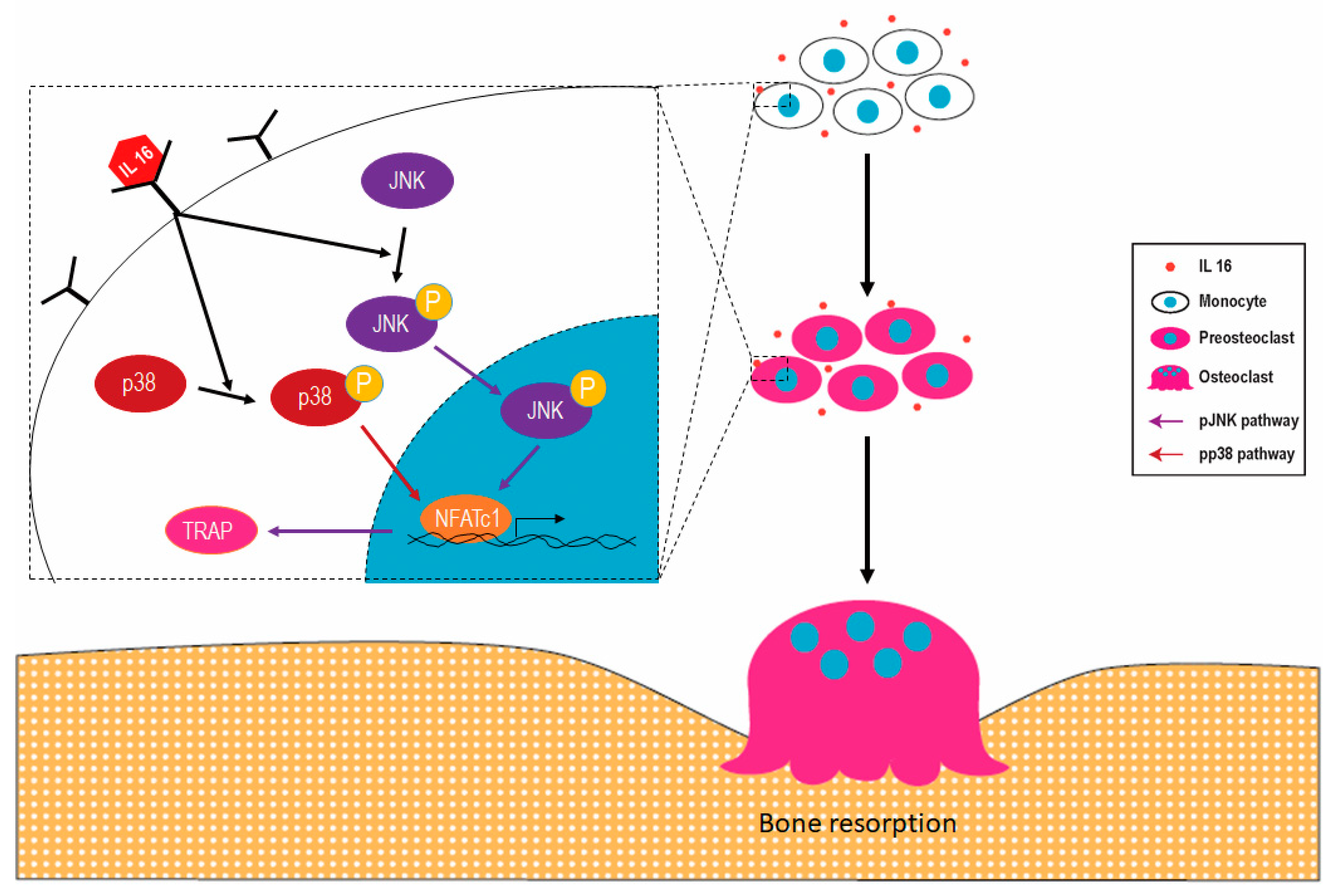
2.1. Synovial Fluid Interleukin-16 in Patients with Periprosthetic Joint Infection Contributes to Osteoclast Activation
2.2. Effect of IL-16 on Osteoclast Activation through p38 and JNK MAPK Signaling
2.3. Effect of IL-16 on TRAP-Positive Osteoclast Activation through JNK/NFATc1 Signaling Cascade
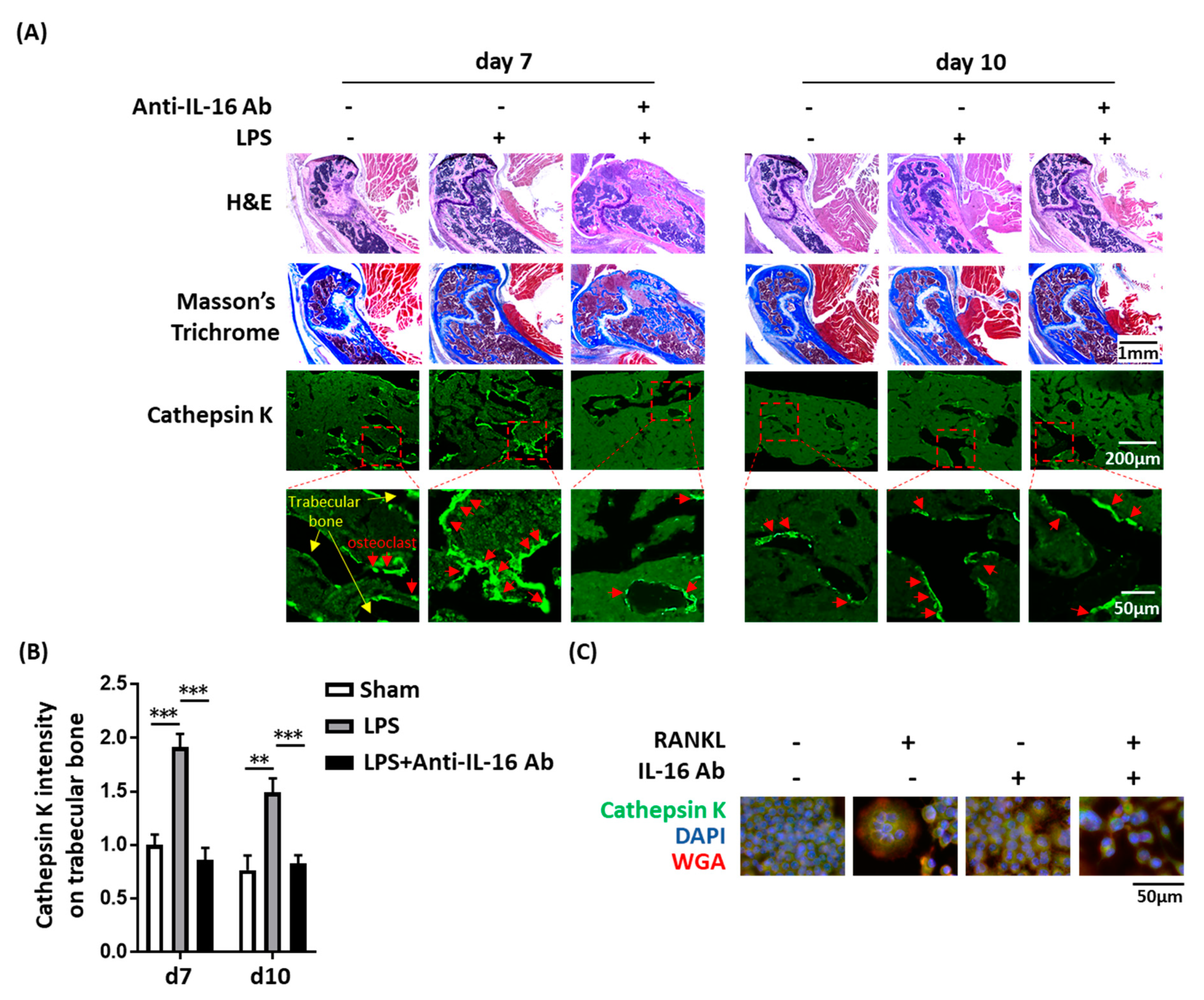
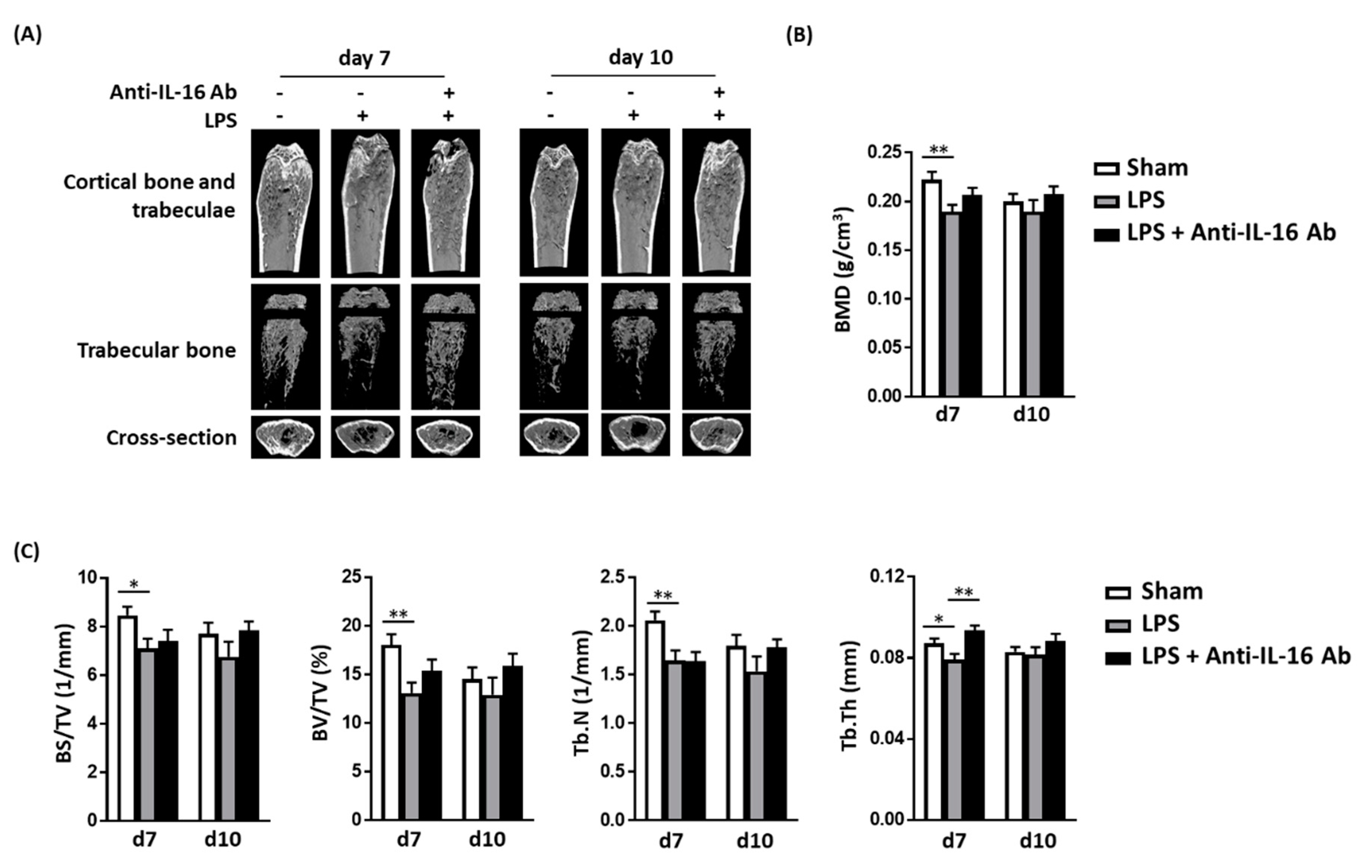
2.4. Effect of Anti-IL-16 Antibody on LPS-Induced Cathepsin K Expression and Bone Loss In Vivo
3. Discussion
4. Materials and Methods
4.1. Patients and Sampling
4.2. Enzyme-Linked Immunosorbent Assay
4.3. Osteoclast and Osteoblast Differentiation
4.4. Protein Extraction and Western Blot Analysis
4.5. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction
4.6. siRNA Transfection
4.7. Experimental Animal Studies
4.8. Anti-IL-16 Antibody Treatment
4.9. Histochemistry and Immunofluorescence Staining
4.10. Micro-CT Bone Imaging
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LPS | lipopolysaccharide |
| PJI | periprosthetic joint infection |
| IL-16 | interleukin-16 |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| TRAP | tartrate-resistant acid phosphatase |
| NFATc1 | nuclear factor of activated T cells 1 |
| SF | synovial fluid |
| SAPKs | stress-activated protein kinases |
| ERK | extracellular signal-regulated kinase |
| TNF | tumor necrosis factor |
| GP | Gram-positive |
| GN | Gram-negative |
| LTA | lipoteichoic acid |
| M-CSF | macrophage colony-stimulating factor |
| RANKL | receptor activator of the nuclear factor-kappa B ligand |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| ANOVA | analysis of variance |
| H&E | hematoxylin and eosin |
| Tb.Th | trabecular thickness |
| ALP | alkaline phosphatase |
References
- Hooper, G. The challenge of the increasing demand for joint replacement. N. Z. Med. J. 2016, 129, 8–9. [Google Scholar]
- Lombardi, A.V.; Berend, K.R.; Adams, J.B. Why knee replacements fail in 2013: Patient, surgeon, or implant? Bone Jt. J. B 2014, 96, 101–104. [Google Scholar] [CrossRef]
- Weston, J.T.; Watts, C.D.; Mabry, T.M.; Hanssen, A.D.; Berry, D.J.; Abdel, M.P. Irrigation and debridement with chronic antibiotic suppression for the management of infected total knee arthroplasty: A Contemporary Analysis. Bone Jt. J. B 2018, 100, 1471–1476. [Google Scholar] [CrossRef]
- Ahmed, S.S.; Haddad, F.S. Prosthetic joint infection. Bone Jt. Res. 2019, 8, 570–572. [Google Scholar] [CrossRef]
- Gwam, C.U.; Mistry, J.B.; Mohamed, N.S.; Thomas, M.; Bigart, K.C.; Mont, M.A.; Delanois, R.E. Current Epidemiology of Revision Total Hip Arthroplasty in the United States: National Inpatient Sample 2009 to 2013. J. Arthroplast. 2017, 32, 2088–2092. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Elbuluk, A.; Dundon, J.; Herrero, C.; Hernandez, C.; Vigdorchik, J.M.; Schwarzkopf, R.; Iorio, R.; Long, W.J. Surgical approach significantly affects the complication rates associated with total hip arthroplasty. Bone Jt. J. B 2019, 101, 646–651. [Google Scholar] [CrossRef]
- Khan, N.; Parmar, D.; Ibrahim, M.S.; Kayani, B.; Haddad, F.S. Outcomes of repeat two-stage exchange hip arthroplasty for prosthetic joint infection. Bone Jt. J. B 2019, 101, 110–115. [Google Scholar] [CrossRef]
- Tsang, S.J.; Ting, J.; Simpson, A.; Gaston, P. Outcomes following debridement, antibiotics and implant retention in the management of periprosthetic infections of the hip: A review of cohort studies. Bone Jt. J. B 2017, 99, 1458–1466. [Google Scholar] [CrossRef]
- Hexter, A.T.; Hislop, S.M.; Blunn, G.W.; Liddle, A.D. The effect of bearing surface on risk of periprosthetic joint infection in total hip arthroplasty: A systematic review and meta-analysis. Bone Jt. J. B 2018, 100, 134–142. [Google Scholar] [CrossRef]
- Chen, M.F.; Chang, C.H.; Yang, L.Y.; Hsieh, P.H.; Shih, H.N.; Ueng, S.W.N.; Chang, Y. Synovial fluid interleukin-16, interleukin-18, and CRELD2 as novel biomarkers of prosthetic joint infections. Bone Jt. Res. 2019, 8, 179–188. [Google Scholar] [CrossRef]
- Ren, F.; Zhan, X.; Martens, G.; Lee, J.; Center, D.; Hanson, S.K.; Kornfeld, H. Pro-IL-16 regulation in activated murine CD4+ lymphocytes. J. Immunol. 2005, 174, 2738–2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaser, A.; Dunzendorfer, S.; Offner, F.A.; Ryan, T.; Schwabegger, A.; Cruikshank, W.W.; Wiedermann, C.J.; Tilg, H. A role for IL-16 in the cross-talk between dendritic cells and T cells. J. Immunol. 1999, 163, 3232–3238. [Google Scholar] [PubMed]
- Sciaky, D.; Brazer, W.; Center, D.M.; Cruikshank, W.W.; Smith, T.J. Cultured human fibroblasts express constitutive IL-16 mRNA: Cytokine induction of active IL-16 protein synthesis through a caspase-3-dependent mechanism. J. Immunol. 2000, 164, 3806–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krautwald, S. IL-16 activates the SAPK signaling pathway in CD4+ macrophages. J. Immunol. 1998, 160, 5874–5879. [Google Scholar] [PubMed]
- McFadden, C.; Morgan, R.; Rahangdale, S.; Green, D.; Yamasaki, H.; Center, D.; Cruikshank, W. Preferential migration of T regulatory cells induced by IL-16. J. Immunol. 2007, 179, 6439–6445. [Google Scholar] [CrossRef] [Green Version]
- Bandeira-Melo, C.; Sugiyama, K.; Woods, L.J.; Phoofolo, M.; Center, D.M.; Cruikshank, W.W.; Weller, P.F. IL-16 promotes leukotriene C(4) and IL-4 release from human eosinophils via CD4- and autocrine CCR3-chemokine-mediated signaling. J. Immunol. 2002, 168, 4756–4763. [Google Scholar] [CrossRef]
- Campbell, I.K.; Leong, D.; Edwards, K.M.; Rayzman, V.; Ng, M.; Goldberg, G.L.; Wilson, N.J.; Scalzo-Inguanti, K.; Mackenzie-Kludas, C.; Lawlor, K.E.; et al. Therapeutic Targeting of the G-CSF Receptor Reduces Neutrophil Trafficking and Joint Inflammation in Antibody-Mediated Inflammatory Arthritis. J. Immunol. 2016, 197, 4392–4402. [Google Scholar] [CrossRef]
- Shimizu, K.; Nakajima, A.; Sudo, K.; Liu, Y.; Mizoroki, A.; Ikarashi, T.; Horai, R.; Kakuta, S.; Watanabe, T.; Iwakura, Y. IL-1 receptor type 2 suppresses collagen-induced arthritis by inhibiting IL-1 signal on macrophages. J. Immunol. 2015, 194, 3156–3168. [Google Scholar] [CrossRef] [Green Version]
- Gwyer Findlay, E.; Danks, L.; Madden, J.; Cavanagh, M.M.; McNamee, K.; McCann, F.; Snelgrove, R.J.; Shaw, S.; Feldmann, M.; Taylor, P.C.; et al. OX40L blockade is therapeutic in arthritis, despite promoting osteoclastogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 2289–2294. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.F.; Chang, C.H.; Hu, C.C.; Wu, Y.Y.; Chang, Y.; Ueng, S.W.N. Periprosthetic Joint Infection Caused by Gram-Positive Versus Gram-Negative Bacteria: Lipopolysaccharide, but not Lipoteichoic Acid, Exerts Adverse Osteoclast-Mediated Effects on the Bone. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.E.; Horswill, A.R. Staphylococcus aureus osteomyelitis: Bad to the bone. Cell Host Microbe 2013, 13, 629–631. [Google Scholar] [CrossRef] [Green Version]
- Kasonga, A.; Kruger, M.C.; Coetzee, M. Activation of PPARs Modulates Signalling Pathways and Expression of Regulatory Genes in Osteoclasts Derived from Human CD14+ Monocytes. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, G.; Roger, P.M. From Crosstalk between Immune and Bone Cells to Bone Erosion in Infection. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Cho, E.; Lee, J.; Lee, S.; Lee, T.H. Inhibitory Effects of N-[2-(4-acetyl-1-piperazinyl) phenyl]-2-(2-chlorophenoxy) acetamide on Osteoclast Differentiation In Vitro via the Downregulation of TRAF6. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Chung, Y.H.; Ahn, H.; Kim, H.; Rho, J.; Jeong, D. Selective Regulation of MAPK Signaling Mediates RANKL-dependent Osteoclast Differentiation. Int. J. Biol. Sci. 2016, 12, 235–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.M.; Bronisz, A.; Hu, R.; Patel, K.; Mansky, K.C.; Sif, S.; Ostrowski, M.C. MITF and PU.1 recruit p38 MAPK and NFATc1 to target genes during osteoclast differentiation. J. Biol. Chem. 2007, 282, 15921–15929. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Gao, J.; Ng, P.Y.; Qin, A.; Steer, J.H.; Pavlos, N.J.; Zheng, M.H.; Dong, Y.; Cheng, T.S. Alexidine Dihydrochloride Attenuates Osteoclast Formation and Bone Resorption and Protects Against LPS-Induced Osteolysis. J. Bone Min. Res. 2016, 31, 560–572. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, T.; Mokuda, S.; Kohno, H.; Yukawa, K.; Kuranobu, T.; Oi, K.; Yoshida, Y.; Hirata, S.; Sugiyama, E. TGFbeta1 Regulates Human RANKL-Induced Osteoclastogenesis via Suppression of NFATc1 Expression. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.J.; Lee, H.; Lee, J.; Lee, K.; Kim, J.; Kim, Y.; Park, J.I.; Kim, K. Bone Remodeling: Histone Modifications as Fate Determinants of Bone Cell Differentiation. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.H.; Yang, M.Y. The Role of Macrophage in the Pathogenesis of Osteoporosis. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.H. Osteoporosis: From Molecular Mechanisms to Therapies. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winquist, R.A.; Hansen, S.T., Jr.; Clawson, D.K. Closed intramedullary nailing of femoral fractures. A report of five hundred and twenty cases. J. Bone Jt. Surg. Am. 1984, 66, 529–539. [Google Scholar] [CrossRef]
- Yang, C.; Wang, J.; Yin, Z.; Wang, Q.; Zhang, X.; Jiang, Y.; Shen, H. A sophisticated antibiotic-loading protocol in articulating cement spacers for the treatment of prosthetic joint infection: A retrospective cohort study. Bone Jt. Res. 2019, 8, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Romano, C.L.; Tsuchiya, H.; Morelli, I.; Battaglia, A.G.; Drago, L. Antibacterial coating of implants: Are we missing something? Bone Jt. Res. 2019, 8, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Verdrengh, M.; Carlsten, H.; Ohlsson, C.; Tarkowski, A. Addition of bisphosphonate to antibiotic and anti-inflammatory treatment reduces bone resorption in experimental Staphylococcus aureus-induced arthritis. J. Orthop. Res. 2007, 25, 304–310. [Google Scholar] [CrossRef]
- Rochford, E.T.; Sabate Bresco, M.; Zeiter, S.; Kluge, K.; Poulsson, A.; Ziegler, M.; Richards, R.G.; O’Mahony, L.; Moriarty, T.F. Monitoring immune responses in a mouse model of fracture fixation with and without Staphylococcus aureus osteomyelitis. Bone 2016, 83, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Wang, W.; Kwiecinski, J.; Josefsson, E.; Pullerits, R.; Jonsson, I.M.; Magnusson, M.; Jin, T. The combination of a tumor necrosis factor inhibitor and antibiotic alleviates staphylococcal arthritis and sepsis in mice. J. Infect. Dis. 2011, 204, 348–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomomatsu, N.; Aoki, K.; Alles, N.; Soysa, N.S.; Hussain, A.; Nakachi, H.; Kita, S.; Shimokawa, H.; Ohya, K.; Amagasa, T. LPS-induced inhibition of osteogenesis is TNF-alpha dependent in a murine tooth extraction model. J. Bone Min. Res. 2009, 24, 1770–1781. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Virdi, A.S.; Sena, K.; Hughes, W.F.; Sumner, D.R. Bone turnover markers correlate with implant fixation in a rat model using LPS-doped particles to induced implant loosening. J. Biomed. Mater. Res. A 2012, 100, 918–928. [Google Scholar] [CrossRef] [Green Version]
- Abu-Amer, Y.; Ross, F.P.; Edwards, J.; Teitelbaum, S.L. Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its P55 receptor. J. Clin. Investig. 1997, 100, 1557–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvizi, J.; Zmistowski, B.; Berbari, E.F.; Bauer, T.W.; Springer, B.D.; Della Valle, C.J.; Garvin, K.L.; Mont, M.A.; Wongworawat, M.D.; Zalavras, C.G. New definition for periprosthetic joint infection: From the Workgroup of the Musculoskeletal Infection Society. Clin. Orthop. Relat. Res. 2011, 469, 2992–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.F.; Chang, C.H.; Chiang-Ni, C.; Hsieh, P.H.; Shih, H.N.; Ueng, S.W.N.; Chang, Y. Rapid analysis of bacterial composition in prosthetic joint infection by 16S rRNA metagenomic sequencing. Bone Jt. Res. 2019, 8, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Tsuda, H.; Akiyama, Y.; Honda, M.; Shimizu, N.; Suzuki, N.; Komiyama, K. Alkaline phosphatase determines polyphosphate-induced mineralization in a cell-type independent manner. J. Bone Min. Metab. 2016, 34, 627–637. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.; Hsiao, Y.-m.; Hu, C.-C.; Chang, C.-H.; Li, C.-Y.; Ueng, S.W.N.; Chen, M.-F. Synovial Fluid Interleukin-16 Contributes to Osteoclast Activation and Bone Loss through the JNK/NFATc1 Signaling Cascade in Patients with Periprosthetic Joint Infection. Int. J. Mol. Sci. 2020, 21, 2904. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082904
Chang Y, Hsiao Y-m, Hu C-C, Chang C-H, Li C-Y, Ueng SWN, Chen M-F. Synovial Fluid Interleukin-16 Contributes to Osteoclast Activation and Bone Loss through the JNK/NFATc1 Signaling Cascade in Patients with Periprosthetic Joint Infection. International Journal of Molecular Sciences. 2020; 21(8):2904. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082904
Chicago/Turabian StyleChang, Yuhan, Yi-min Hsiao, Chih-Chien Hu, Chih-Hsiang Chang, Cai-Yan Li, Steve W. N. Ueng, and Mei-Feng Chen. 2020. "Synovial Fluid Interleukin-16 Contributes to Osteoclast Activation and Bone Loss through the JNK/NFATc1 Signaling Cascade in Patients with Periprosthetic Joint Infection" International Journal of Molecular Sciences 21, no. 8: 2904. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082904