DFT Study of Molecular and Electronic Structure of Ca(II) and Zn(II) Complexes with Porphyrazine and tetrakis(1,2,5-thiadiazole)porphyrazine

Abstract
:1. Introduction
2. Results and Discussion
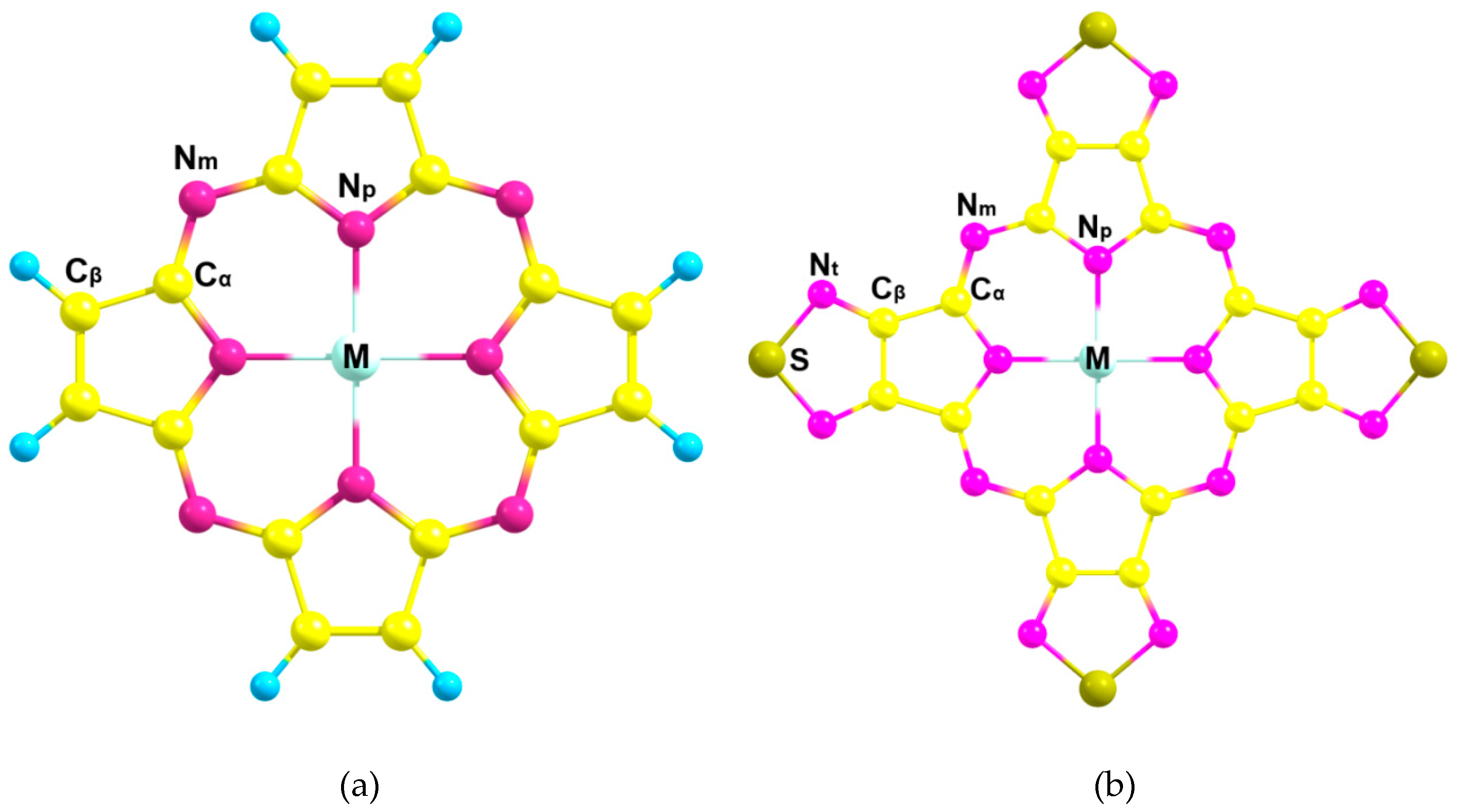
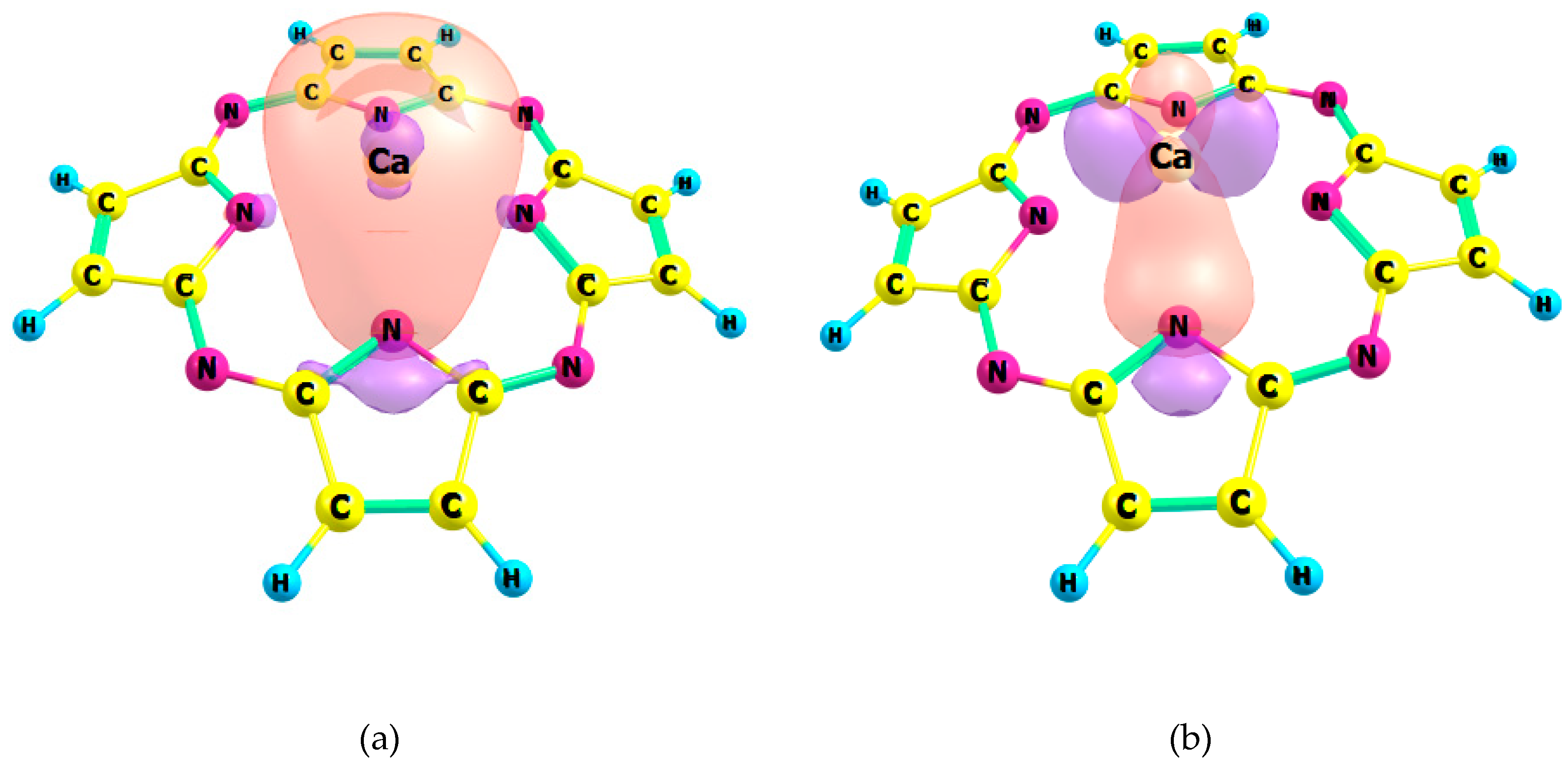
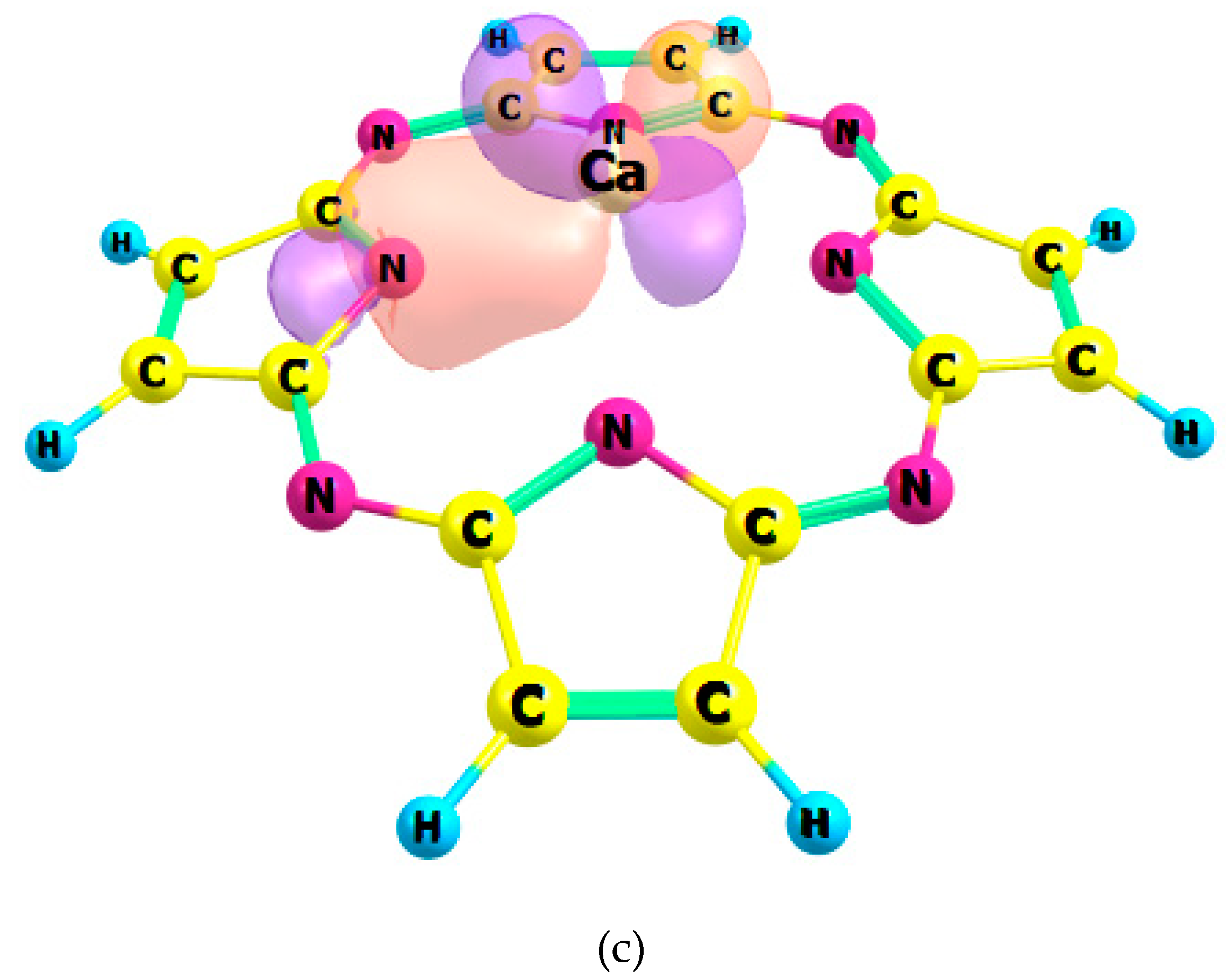
2.1. Chemical Bonding in MPz and MTTDPz
2.2. Molecular Orbitals
2.3. Electonic Absorption Spectra
3. Computational Methods
4. Experimental
Synthesis of CaTTDPz
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Pz | Porphyrazine |
| TTDPz | Tetrakis(1,2,5-thiadiazole) porphyrazine |
| DFT | Density Functional Theory |
| TDDFT | Time Dependent Density Functional Theory |
| NBO | Natural bond orbital |
| QTAIM | Quantum theory of atoms in molecules |
References
- Wöhrle, D.; Schnurpfeil, G.; Makarov, S.G.; Kazarin, A.; Suvorova, O.N. Practical Applications of Phthalocyanines—From Dyes and Pigments to Materials for Optical, Electronic and Photo-electronic Devices. Macroheterocycles 2012, 5, 191–202. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Stuzhin, P.A. Novel families of phthalocyanine-like macrocycles-Porphyrazines with annulated strongly electron-withdrawing 1,2,5-thia/selenodiazole rings. Coord. Chem. Rev. 2006, 250, 1530–1561. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Novakova, V.; Zimcik, P.; Stuzhin, P.A. Tetrapyrazinoporphyrazines and their metal derivatives. Part I: Synthesis and basic structural information. Coord. Chem. Rev. 2016, 309, 107–179. [Google Scholar] [CrossRef]
- Novakova, V.; Donzello, M.P.; Ercolani, C.; Zimcik, P.; Stuzhin, P.A. Tetrapyrazinoporphyrazines and their metal derivatives. Part II: Electronic structure, electrochemical, spectral, photophysical and other application related properties. Coord. Chem. Rev. 2018, 361, 1–73. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Z.; Sheng, N.; Jiang, J. Molecular structure, electronic structure and vibrational spectra of metal-free, N,N′-dideuterio, and magnesium tetrakis(thiadiazole)porphyrazines: Density functional calculations. J. Mol. Struct. THEOCHEM 2005, 755, 179–186. [Google Scholar] [CrossRef]
- Cai, X.; Zhang, Y.; Zhang, X.; Jiang, J. Structures and properties of metal-free and copper tetrakis(thiadiazole) porphyrazine and metal-free tetrakis(selenodiazole) porphyrazine based on density functional theory calculations. J. Mol. Struct. THEOCHEM 2007, 812, 63–70. [Google Scholar] [CrossRef]
- Donzello, M.P.; Ercolani, C.; Kadish, K.M.; Ricciardi, G.; Rosa, A.; Stuzhin, P.A. Tetrakis(thiadiazole)porphyrazines. 5. Electrochemical and DFT/TDDFT studies of the free-base macrocycle and its MgII ZnII, and CuII complexes. Inorg. Chem. 2007, 46, 4145–4157. [Google Scholar] [CrossRef]
- Tverdova, N.V.; Giricheva, N.I.; Savelyev, D.S.; Mikhailov, M.S.; Vogt, N.; Koifman, O.I.; Stuzhin, P.A.; Girichev, G.V. Molecular structure of tetrakis(1,2,5-thiadiazolo)- porphyrazinatozinc(II) in gaseous phase. Macroheterocycles 2017, 10, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.A.; Pachter, R. Ground state electronic structures and spectra of zinc complexes of porphyrin, tetraazaporphyrin, tetrabenzoporphyrin, and phthalocyanine: A density functional theory study. J. Chem. Phys. 2001, 114, 10757–10767. [Google Scholar] [CrossRef]
- Liao, M.S.; Scheiner, S. Comparative study of metal-porphyrins, -porphyrazines, and -phthalocyanines. J. Comput. Chem. 2002, 23, 1391–1403. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ricciardi, G.; Rosa, A.; Van Gisbergen, S.J.A. A DFT/TDDFT interpretation of the ground and excited states of porphyrin and porphyrazine complexes. Coord. Chem. Rev. 2002, 230, 5–27. [Google Scholar] [CrossRef]
- Rosa, A.; Ricciardi, G.; Baerends, E.J.; Van Gisbergen, S.J.A. The optical spectra of NiP, NiPz, NiTBP, and NiPc: Electronic effects of meso-tetraaza substitution and tetrabenzo annulation. J. Phys. Chem. A 2001, 105, 3311–3327. [Google Scholar] [CrossRef]
- Sliznev, V.V. Theoretical study of structures of alkaline-earth metal (M=Be, Mg) complexes of porphyrin (MP), porphyrazine (MPz) and phthalocyanine (MPc). Macroheterocycles 2013, 6, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, Y.; Kubo, M.; Fujinawa, T.; Suzuki, Y.; Yoshikawa, H.; Awaga, K. Electrochromism and stable n-type doping of highly oriented thin films of tetrakis(thiadiazole)porphyrazine. Angew. Chem.-Int. Ed. 2007, 46, 5532–5536. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Miyoshi, Y.; Matsushita, M.M.; Awaga, K. A complementary organic inverter of porphyrazine thin films: Low-voltage operation using ionic liquid gate dielectrics. Chem. Commun. 2011, 47, 5837–5839. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Fujimoto, T.; Yoshikawa, H.; Matsushita, M.M.; Awaga, K.; Yamada, T.; Ito, H. Photoconductivity and FET performance of an n-type porphyrazine semiconductor, tetrakis(thiadiazole)porphyrazine. Org. Electron. 2011, 12, 239–243. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Takahashi, K.; Fujimoto, T.; Yoshikawa, H.; Matsushita, M.M.; Ouchi, Y.; Kepenekian, M.; Robert, V.; Donzello, M.P.; Ercolani, C.; et al. Crystal structure, spin polarization, solid-state electrochemistry, and high n-type carrier mobility of a paramagnetic semiconductor: Vanadyl tetrakis(thiadiazole)porphyrazine. Inorg. Chem. 2012, 51, 456–462. [Google Scholar] [CrossRef]
- Stuzhin, P.; Mikhailov, M.; Travkin, V.; Gudkov, E.; Pakhomov, G. Multilayer Photovoltaic Structures Based on Tetrathiadiazoloporphyrazine/Subphthalocyanine Heterojunction. Macroheterocycles 2012, 5, 162–165. [Google Scholar] [CrossRef] [Green Version]
- Donzello, M.P.; Agostinetto, R.; Ivanova, S.S.; Fujimori, M.; Suzuki, Y.; Yoshikawa, H.; Shen, J.; Awaga, K.; Ercolani, C.; Kadish, K.M.; et al. Tetrakis(thiadiazole)porphyrazines. 4. Direct template synthesis, structure, general physicochemical behavior, and redox properties of Al III, GaIII, and InIII complexes. Inorg. Chem. 2005, 44, 8539–8551. [Google Scholar] [CrossRef]
- Pia Donzello, M.; Viola, E.; Giustini, M.; Ercolani, C.; Monacelli, F. Tetrakis(thiadiazole)porphyrazines. 8. Singlet oxygen production, fluorescence response and liposomal incorporation of tetrakis(thiadiazole) porphyrazine macrocycles [TTDPzM] (M = Mg II(H 2O), Zn II, Al IIICl, Ga IIICl, Cd II, Cu II, 2H I). Dalton Trans. 2012, 41, 6112–6121. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Tverdova, N.V.; Giricheva, N.I.; Girichev, G.V.; Stuzhin, P.A. DFT Study of molecular and electronic structure of magnesium (II) tetra(1,2,5-chalcogenadiazolo) porphyrazines, [TXDPzMg] (X = O, S, Se, Te). J. Porphyr. Phthalocyanines 2017, 21, 439–452. [Google Scholar] [CrossRef]
- Sliznev, V.V.; Girichev, G.V. Theoretical Study of Alkali Metal Complexes of Porphyrin (M2P), Porphyrazine (M2Pz) and Phthalocyanine (M2Pc), M=Li, Na, K. Macroheterocycles 2011, 4, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Bauer, E.M.; Cardarilli, D.; Ercolani, C.; Stuzhin, P.A.; Russo, U. Tetrakis(thiadiazole)porphyrazines. 2. Metal complexes with Mn(II), Fe(II), Co(II), Ni(II), and Zn(II). Inorg. Chem. 1999, 38, 6114–6120. [Google Scholar] [CrossRef]
- Zhabanov, Y.A.; Giricheva, N.I.; Zakharov, A.V.; Islyaikin, M.K. Distinctive features of the structure of hemihexaphyrazine complexes with Y, La, and Lu according to quantum chemical data. J. Mol. Struct. 2017, 1132, 28–33. [Google Scholar] [CrossRef]
- Gouterman, M.; Wagnière, G.H.; Snyder, L.C. Spectra of porphyrins. Part II. Four orbital model. J. Mol. Spectrosc. 1963, 11, 108–127. [Google Scholar] [CrossRef]
- Gouterman, M. Spectra of porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Berezin, B.D.; Khelevina, O.G.; Stuzhin, P.A. Kinetics of the formation of metal complexes of unsubstituted tetraazaporphine (porphyrazine) in pyridine. Russ. J. Phys. Chem. A 1985, 59, 1295–1299. [Google Scholar]
- Jensen, F. Unifying general and segmented contracted basis sets. segmented polarization consistent basis sets. J. Chem. Theory Comput. 2014, 10, 1074–1085. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Granovsky, A.A. Firefly Version 8. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 21 April 2020).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory, Oxford University Press; Clarendon Press: Oxford, UK, 1990; ISBN 9780198558651. [Google Scholar]
- Some References Related to AIMAll. Available online: http://aim.tkgristmill.com/references.html (accessed on 13 March 2020).
- Zhurko, G.A.; Zhurko, D.A. ChemCraft Version 1.6 (build 312); Version 1.6 (build 312) Ed. Available online: http://www.chemcraftprog.com/index.html (accessed on 21 April 2020).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CaPz | CaTTDPz | ZnPz | ZnTTDPz | |
|---|---|---|---|---|
| M-Np | 2.276 | 2.299 | 1.979 | 2.025 |
| M-X 2 | 1.079 | 1.020 | ||
| Np-Cα | 1.364 | 1.373 | 1.363 | 1.375 |
| Cα-Cβ | 1.458 | 1.462 | 1.457 | 1.458 |
| Cα-Nm | 1.333 | 1.322 | 1.331 | 1.317 |
| Cβ-Cβ | 1.354 | 1.424 | 1.457 | 1.421 |
| Cβ-Nt | 1.316 | 1.316 | ||
| Nt-S | 1.645 | 1.644 | ||
| (Np…Np)opp | 4.008 | 4.120 | 3.958 | 4.049 |
| (Np…Np)adj | 2.834 | 2.913 | 2.799 | 2.863 |
| ∠ (Np–M–Np) | 123.4 | 127.3 | 180.0 | 180.0 |
| ∠ (Np–Cα–Nm) | 127.6 | 128.1 | 127.2 | 128.0 |
| ∠ (Cα–Nm–Cα) | 124.6 | 126.7 | 124.4 | 125.8 |
| ∠ (Cα–Np–Cα) | 107.7 | 111.8 | 108.8 | 111.7 |
| ∠ (Nt–S–Nt) | 100.2 | 100.3 |
| CaPz | ZnPz | CaTTDPz | ZnTTDPz | |
|---|---|---|---|---|
| E(HOMO),eV | −5.73 | −5.99 | −6.07 | −6.19 |
| E(LUMO),eV | −3.10 | −3.33 | −3.78 | −3.91 |
| ∆E, eV | 2.64 | 2.66 | 2.29 | 2.29 |
| ∇2ρ, a.u. | 0.219 | 0.394 | 0.207 | 0.339 |
| δ(M|Np) | 0.270 | 0.464 | 0.262 | 0.446 |
| q(M) NPA | 1.754 | 1.198 | 1.768 | 1.234 |
| q(Np) NPA | −0.702 | −0.633 | −0.660 | −0.596 |
| configuration | 4s0.123d0.14 | 4s0.363d9.964p0.48 | 4s0.113d0.13 | 4s0.353d9.974p0.44 |
| ∑ E(d-a), kcal/mol | 18 | 116 | 17 | 103 |
| Q(M-Np) | 0.110 | 0.336 | 0.104 | 0.321 |
| r(M-Np) | 2.276 | 1.979 | 2.299 | 2.025 |
| State | Composition (%) | λ, nm | f | exp λ, nm |
|---|---|---|---|---|
| CaPz | ||||
| 1 1E | (18) (80) | 513 | 0.16 | |
| 4 1E | (33) (53) (9) | 344 | 0.21 | |
| 5 1E | (62) (25) (9) | 308 | 0.59 | |
| 10 1E | (99) | 238 | 0.06 | |
| CaTTDPz | ||||
| 1 1E | (7) (90) | 585 | 0.27 | 647 (Py) [this work] 641 (acetone) [this work] |
| 6 1E | (74) (8) (8) | 322 | 0.98 | |
| 16 1E | (9) (14) (67) | 254 | 0.28 | |
| 17 1E | (7) (77) | 251 | 0.15 | |
| 18 1E | (6) (30) (34) (5) (23) | 250 | 0.14 | |
| ZnPz | ||||
| 1 1Eu | (17) (82) | 505 | 0.17 | 584 (Py) [27] |
| 3 1Eu | (50) (6) (37) (6) | 329 | 0.15 | |
| 4 1Eu | (44) (42) (11) | 307 | 0.71 | 327 |
| 5 1Eu | (99) | 238 | 0.06 | |
| ZnTTDPz | ||||
| 1 1Eu | (5) (91) | 580 | 0.29 | 638 (DMSO) [23] 44 (DMF) [8] |
| 4 1Eu | (44) (42) (11) | 334 | 0.28 | 400 |
| 5 1Eu | (39) (42) (7) (6) | 312 | 0.81 | 372 |
| 8 1Eu | (6) (29) (8) (52) | 252 | 0.55 | 320 |
| 9 1Eu | (6) (86) | 246 | 0.05 | |
| 12 1Eu | (52) (17) (18) (6) | 230 | 0.10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otlyotov, A.A.; Ryzhov, I.V.; Kuzmin, I.A.; Zhabanov, Y.A.; Mikhailov, M.S.; Stuzhin, P.A. DFT Study of Molecular and Electronic Structure of Ca(II) and Zn(II) Complexes with Porphyrazine and tetrakis(1,2,5-thiadiazole)porphyrazine. Int. J. Mol. Sci. 2020, 21, 2923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082923
Otlyotov AA, Ryzhov IV, Kuzmin IA, Zhabanov YA, Mikhailov MS, Stuzhin PA. DFT Study of Molecular and Electronic Structure of Ca(II) and Zn(II) Complexes with Porphyrazine and tetrakis(1,2,5-thiadiazole)porphyrazine. International Journal of Molecular Sciences. 2020; 21(8):2923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082923
Chicago/Turabian StyleOtlyotov, Arseniy A., Igor V. Ryzhov, Ilya A. Kuzmin, Yuriy A. Zhabanov, Maxim S. Mikhailov, and Pavel A. Stuzhin. 2020. "DFT Study of Molecular and Electronic Structure of Ca(II) and Zn(II) Complexes with Porphyrazine and tetrakis(1,2,5-thiadiazole)porphyrazine" International Journal of Molecular Sciences 21, no. 8: 2923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082923