Gene-Wise Burden of Coding Variants Correlates to Noncoding Pharmacogenetic Risk Variants

 and
and
Abstract
:1. Introduction
2. Results
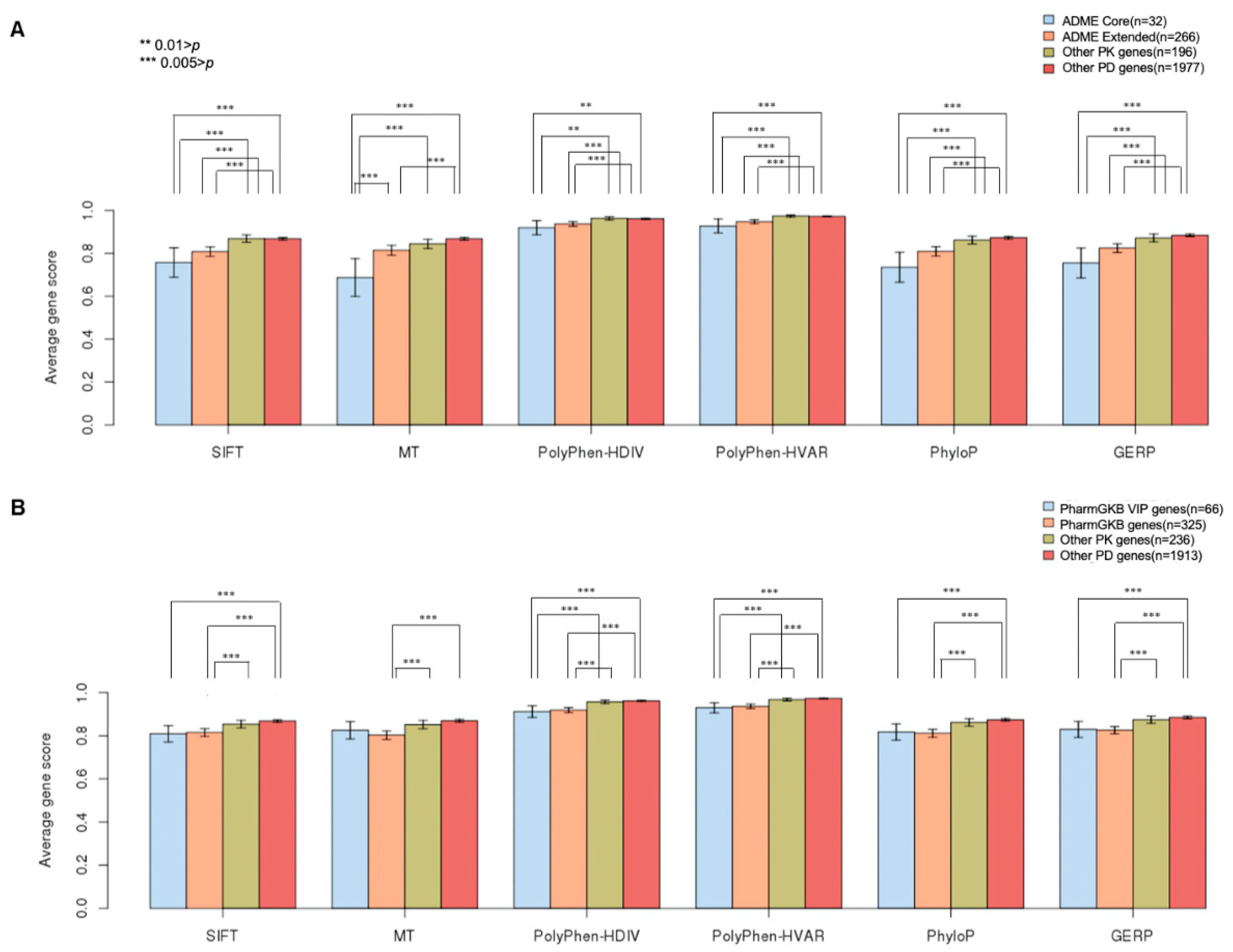
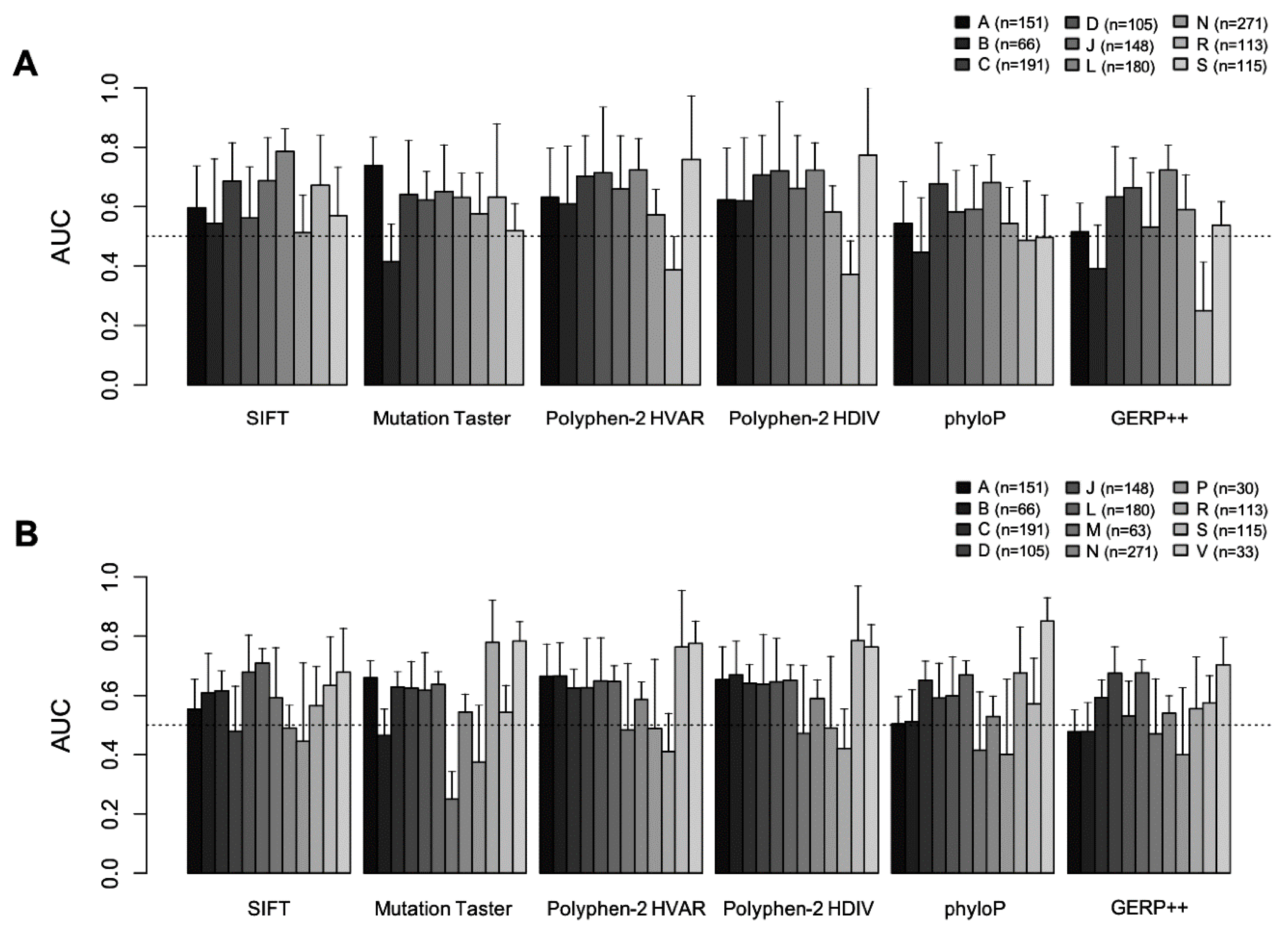
2.1. GVB Score Distribution According to Pharmacogenomics Categories
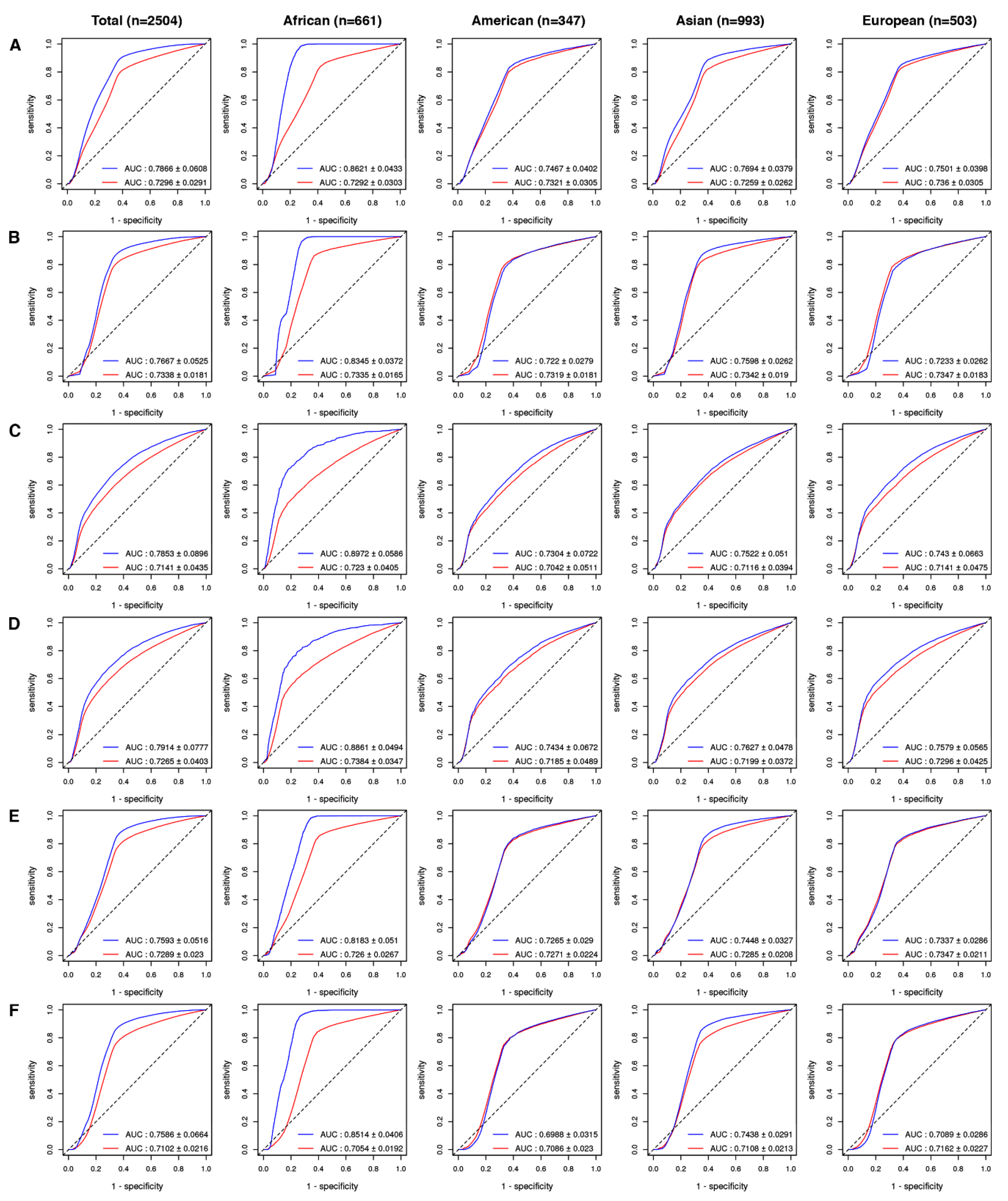
2.2. Drug-Level GVB Association with the ‘Gold Standard’ Variant-Drug Associations in PharmGKB
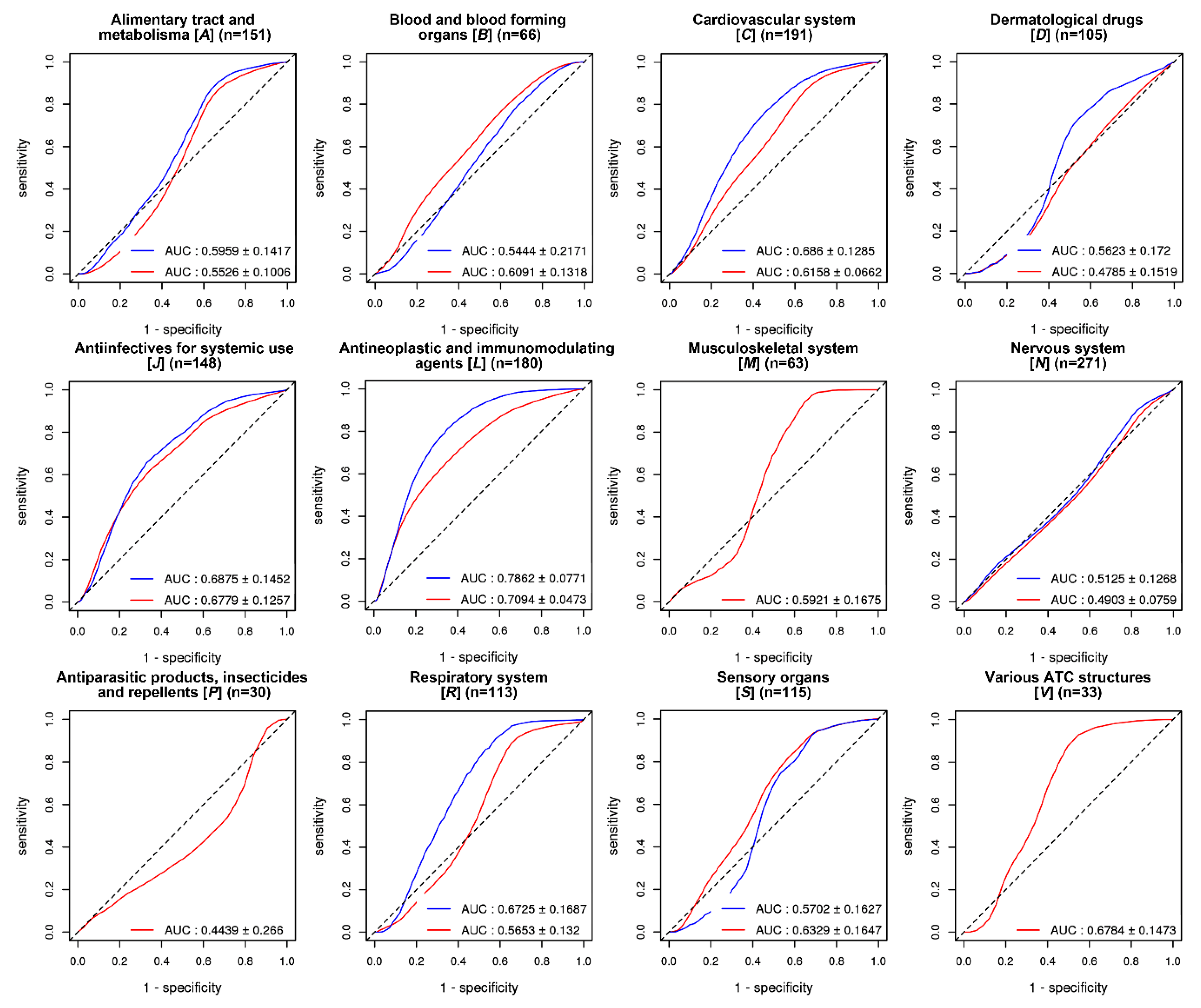
2.3. Evaluation of GVB Prediction Accuracies for Drugs in Different Drug Categories
3. Discussion
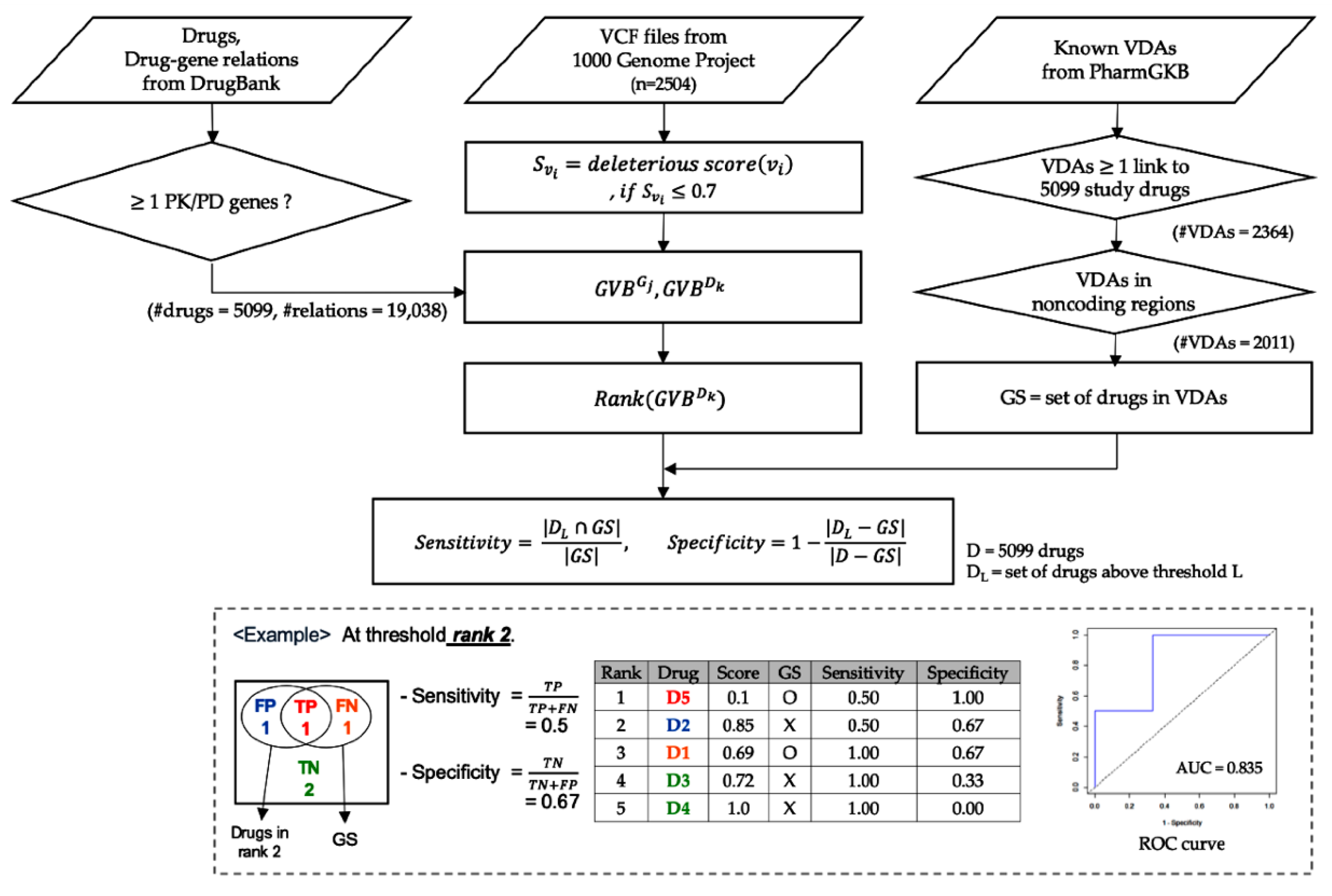
4. Materials and Methods
4.1. Data Sets
4.2. Calculation of Gene-Wise Variant Burden (GVB) Scores for Genes and Drugs
4.3. Validation Using Known VDAs in PharmGKB
4.4. Evaluation of GVB Derived from Other in Silico Variant Scoring Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADR | Adverse drug reaction |
| GWAS | Genome-wide association study |
| VDAs | Variant-drug associations |
| NGS | Next generation sequencing |
| GVB | Gene-wise variant burden |
| AUC | Area under the receiver operating characteristics curve |
| PK | Pharmacokinetics |
| PD | Pharmacodynamics |
| ROC | Receiver operating characteristic |
| TP | True-positive |
| TN | True-negative |
| FP | False-positive |
| FN | False-negative |
| GS | Gold standard |
| 5′-UTR | 5′-untranslated region |
| 3′-UTR | 3′-untranslated region |
| VKORC | Vitamin K epoxide reductase complex |
| BAR2 | Beta2-adrenergic receptor |
| OMB | Office of management and budget |
References
- Meyer, U.A. Genotype or phenotype: The definition of a pharmacogenetic polymorphism. Pharmacogenetics 1991, 1, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.E.; Relling, M.V. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science 1999, 286, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2017, 19, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, L.D.; Kellis, M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012, 30, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Baik, S.Y.; Lee, S.Y.; Park, C.H.; Park, P.J.; Kim, J.H. Genome Sequence Variability Predicts Drug Precautions and Withdrawals from the Market. PLoS ONE 2016, 11, e0162135. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Kim, H.; Choi, J.Y.; Yun, S.; Min, B.J.; Seo, M.E.; Im, H.J.; Kang, H.J.; Kim, J.H. Star Allele-Based Haplotyping versus Gene-Wise Variant Burden Scoring for Predicting 6-Mercaptopurine Intolerance in Pediatric Acute Lymphoblastic Leukemia Patients. Front. Pharmacol. 2019, 10, 654. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.; Kwon, E.J.; You, Y.A.; Park, Y.; Min, B.J.; Yoo, K.; Hwang, H.S.; Kim, J.H.; Kim, Y.J. Deleterious genetic variants in ciliopathy genes increase risk of ritodrine-induced cardiac and pulmonary side effects. BMC Med. Genom. 2018, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Kim, S.H.; Kim, C.H.; Min, B.J.; Kim, G.J.; Lim, Y.; Kim, H.S.; Ahn, K.M.; Kim, J.H. Identifying genetic variants underlying medication-induced osteonecrosis of the jaw in cancer and osteoporosis: A case control study. J. Transl. Med. 2019, 17, 381. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Dickson, S.P.; Wang, K.; Krantz, I.; Hakonarson, H.; Goldstein, D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010, 8, e1000294. [Google Scholar] [CrossRef] [Green Version]
- Genomes Project, C.; Abecasis, G.R.; Altshuler, D.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Gibbs, R.A.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramensky, V.; Bork, P.; Sunyaev, S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [Green Version]
- Pharma ADME. The Montreal Heart Institute Pharmacogenomics Center. Available online: http://www.pharmaadme.org/ (accessed on 4 September 2015).
- Schwartz, A.G.; Cote, M.L.; Wenzlaff, A.S.; Land, S.; Amos, C.I. Racial differences in the association between SNPs on 15q25.1, smoking behavior, and risk of non-small cell lung cancer. J. Thorac. Oncol. 2009, 4, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen-Torvik, L.J.; Guo, X.; Bowden, D.W.; Bertoni, A.G.; Sale, M.M.; Yao, J.; Bluemke, D.A.; Goodarzi, M.O.; Chen, Y.I.; Vaidya, D.; et al. Fasting glucose GWAS candidate region analysis across ethnic groups in the Multiethnic Study of Atherosclerosis (MESA). Genet. Epidemiol. 2012, 36, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Adeyemo, A.; Rotimi, C. Genetic variants associated with complex human diseases show wide variation across multiple populations. Public Health Genom. 2010, 13, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Desai, A.A.; Innocenti, F.; Ratain, M.J. Pharmacogenomics: Road to anticancer therapeutics nirvana? Oncogene 2003, 22, 6621–6628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.S.; Ratain, M.J. Pharmacogenetics and pharmacogenomics of anticancer agents. CA Cancer J. Clin. 2009, 59, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.E.; Johnson, J.A. Pharmacogenomics: The inherited basis for interindividual differences in drug response. Annu. Rev. Genom. Hum. Genet. 2001, 2, 9–39. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Hanson, C.; Sinha, S. Mechanistic interpretation of non-coding variants for discovering transcriptional regulators of drug response. BMC Biol. 2019, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.J.; Yang, P.; Xie, H.G. Gene variants in noncoding regions and their possible consequences. Pharmacogenomics 2006, 7, 203–209. [Google Scholar] [CrossRef]
- Rieder, M.J.; Reiner, A.P.; Gage, B.F.; Nickerson, D.A.; Eby, C.S.; McLeod, H.L.; Blough, D.K.; Thummel, K.E.; Veenstra, D.L.; Rettie, A.E. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005, 352, 2285–2293. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, W.; Danahey, K.; Pei, X.; Yeo, K.J.; Leung, E.; Volchenboum, S.L.; Ratain, M.J.; Meltzer, D.O.; Stranger, B.E.; Perera, M.A.; et al. Pharmacogenomic genotypes define genetic ancestry in patients and enable population-specific genomic implementation. Pharm. J. 2020, 20, 126–135. [Google Scholar] [CrossRef]
- Comparison of propranolol and hydrochlorothiazide for the initial treatment of hypertension. I. Results of short-term titration with emphasis on racial differences in response. Veterans Administration Cooperative Study Group on Antihypertensive agents. JAMA 1982, 248, 1996–2003.
- Xie, H.G.; Stein, C.M.; Kim, R.B.; Xiao, Z.S.; He, N.; Zhou, H.H.; Gainer, J.V.; Brown, N.J.; Haines, J.L.; Wood, A.J. Frequency of functionally important beta-2 adrenoceptor polymorphisms varies markedly among African-American, Caucasian and Chinese individuals. Pharmacogenetics 1999, 9, 511–516. [Google Scholar]
- Wang, Y.; Suzek, T.; Zhang, J.; Wang, J.; He, S.; Cheng, T.; Shoemaker, B.A.; Gindulyte, A.; Bryant, S.H. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2014, 42, D1075–D1082. [Google Scholar] [CrossRef] [Green Version]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the enzyme database: Updates and major new developments. Nucleic Acids Res. 2004, 32, D431–D433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittig, U.; Kania, R.; Golebiewski, M.; Rey, M.; Shi, L.; Jong, L.; Algaa, E.; Weidemann, A.; Sauer-Danzwith, H.; Mir, S.; et al. SABIO-RK--database for biochemical reaction kinetics. Nucleic Acids Res. 2012, 40, D790–D796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, R.; Altman, T.; Dale, J.M.; Dreher, K.; Fulcher, C.A.; Gilham, F.; Kaipa, P.; Karthikeyan, A.S.; Kothari, A.; Krummenacker, M.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2010, 38, D473–D479. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.A.; Sidow, A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005, 15, 978–986. [Google Scholar] [CrossRef] [Green Version]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Muller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Cooper, G.M.; Goode, D.L.; Ng, S.B.; Sidow, A.; Bamshad, M.J.; Shendure, J.; Nickerson, D.A. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat. Methods 2010, 7, 250–251. [Google Scholar] [CrossRef] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef] [Green Version]
- Garber, M.; Guttman, M.; Clamp, M.; Zody, M.C.; Friedman, N.; Xie, X. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 2009, 25, i54–i62. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Count | |
|---|---|---|
| Total drugs | 5099 | |
| Relations | Enzyme-drug relations | 3477 |
| Transporter-drug relations | 1772 | |
| Carrier-drug relations | 318 | |
| Target-drug relations | 13,471 | |
| Total gene-drug relations | 19,038 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Lee, S.Y.; Baik, S.Y.; Park, C.H.; Yoon, J.H.; Ryu, B.Y.; Kim, J.H. Gene-Wise Burden of Coding Variants Correlates to Noncoding Pharmacogenetic Risk Variants. Int. J. Mol. Sci. 2020, 21, 3091. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093091
Park J, Lee SY, Baik SY, Park CH, Yoon JH, Ryu BY, Kim JH. Gene-Wise Burden of Coding Variants Correlates to Noncoding Pharmacogenetic Risk Variants. International Journal of Molecular Sciences. 2020; 21(9):3091. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093091
Chicago/Turabian StylePark, Jihye, Soo Youn Lee, Su Youn Baik, Chan Hee Park, Jun Hee Yoon, Brian Y. Ryu, and Ju Han Kim. 2020. "Gene-Wise Burden of Coding Variants Correlates to Noncoding Pharmacogenetic Risk Variants" International Journal of Molecular Sciences 21, no. 9: 3091. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093091