Functional Role of N-Terminal Extension of Human AP Endonuclease 1 In Coordination of Base Excision DNA Repair via Protein–Protein Interactions



Abstract
:1. Introduction
2. Results
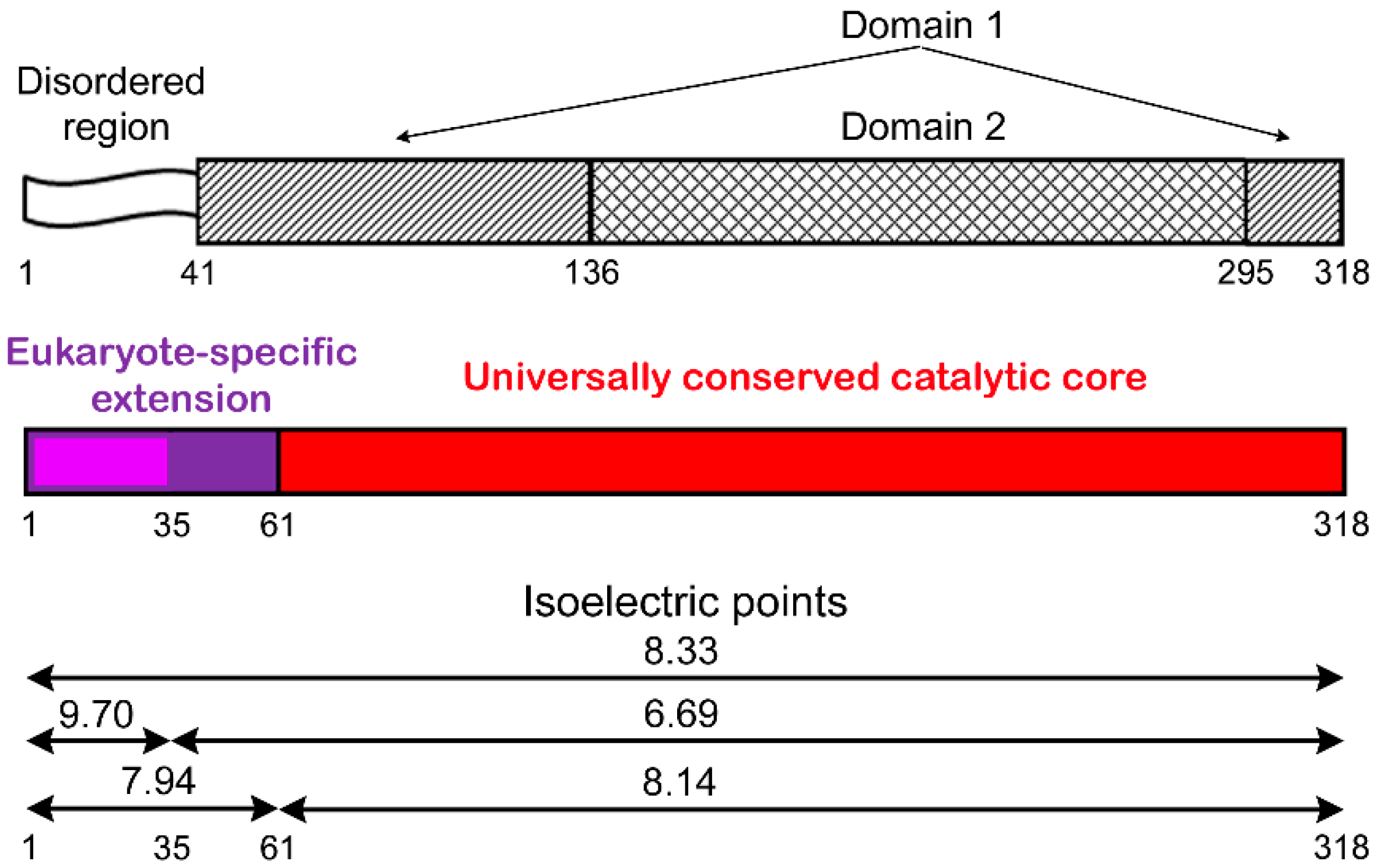
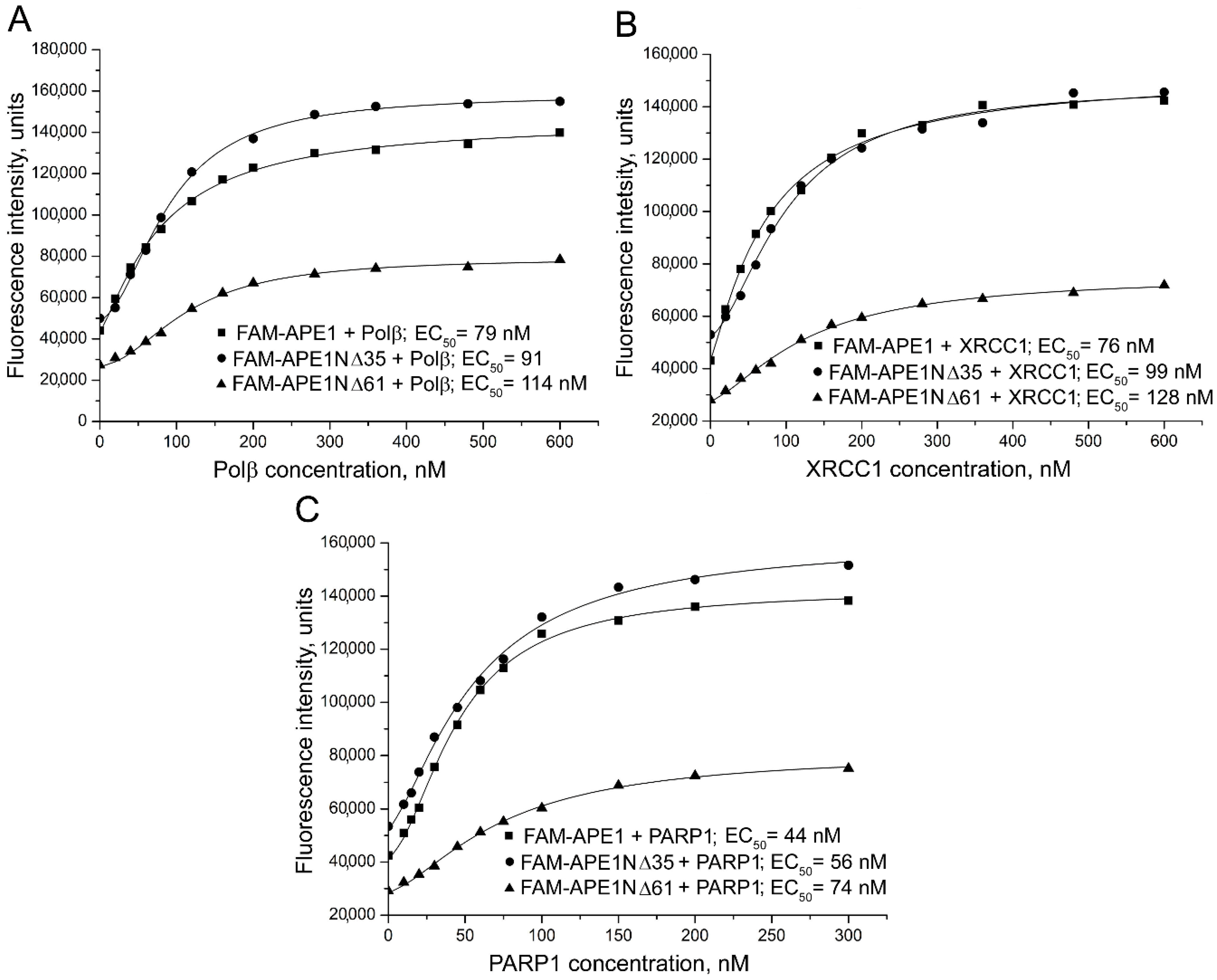
2.1. Contribution of N-terminal Extension of Human APE1 to the Interaction with Canonical BER Proteins
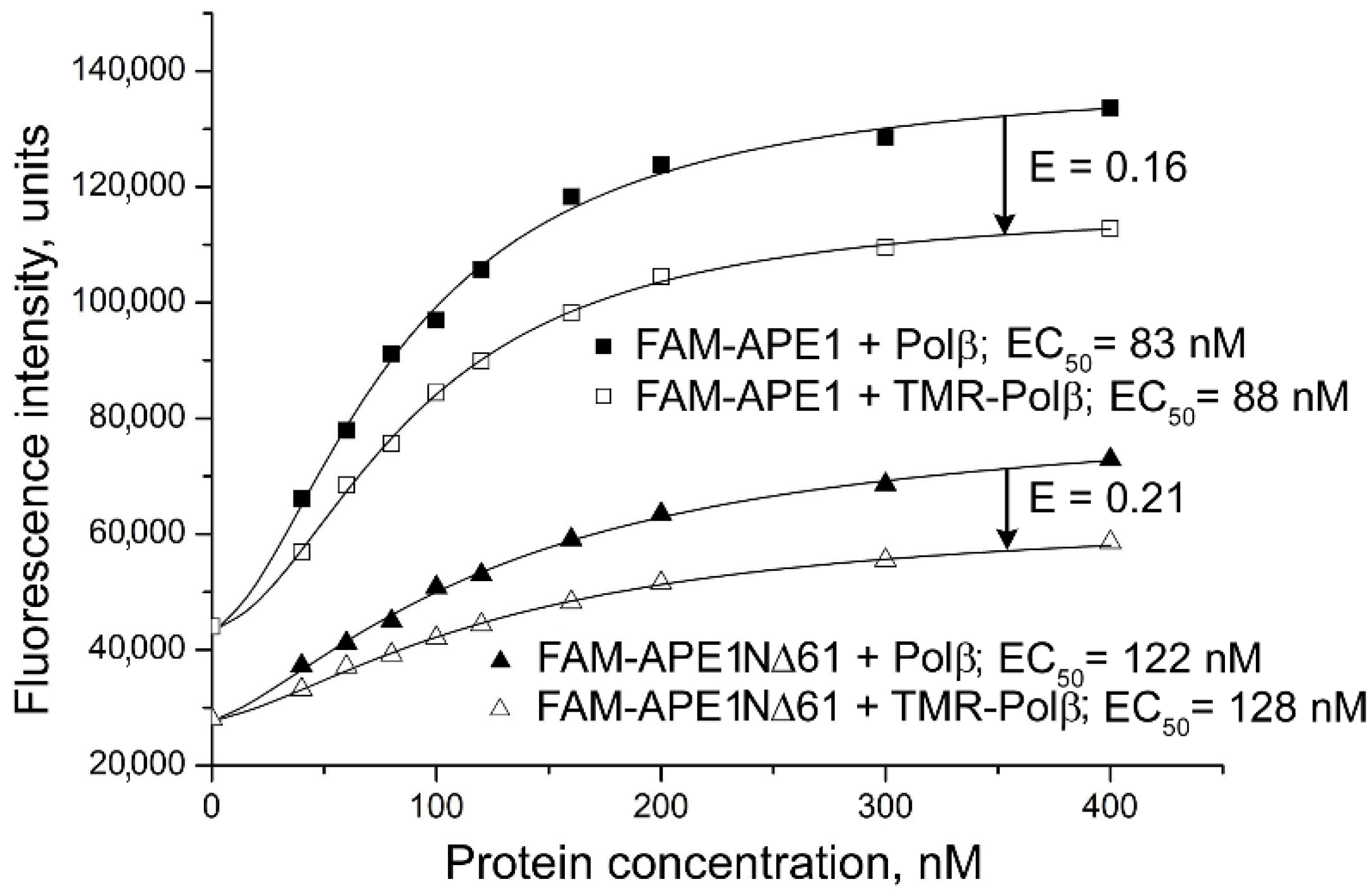
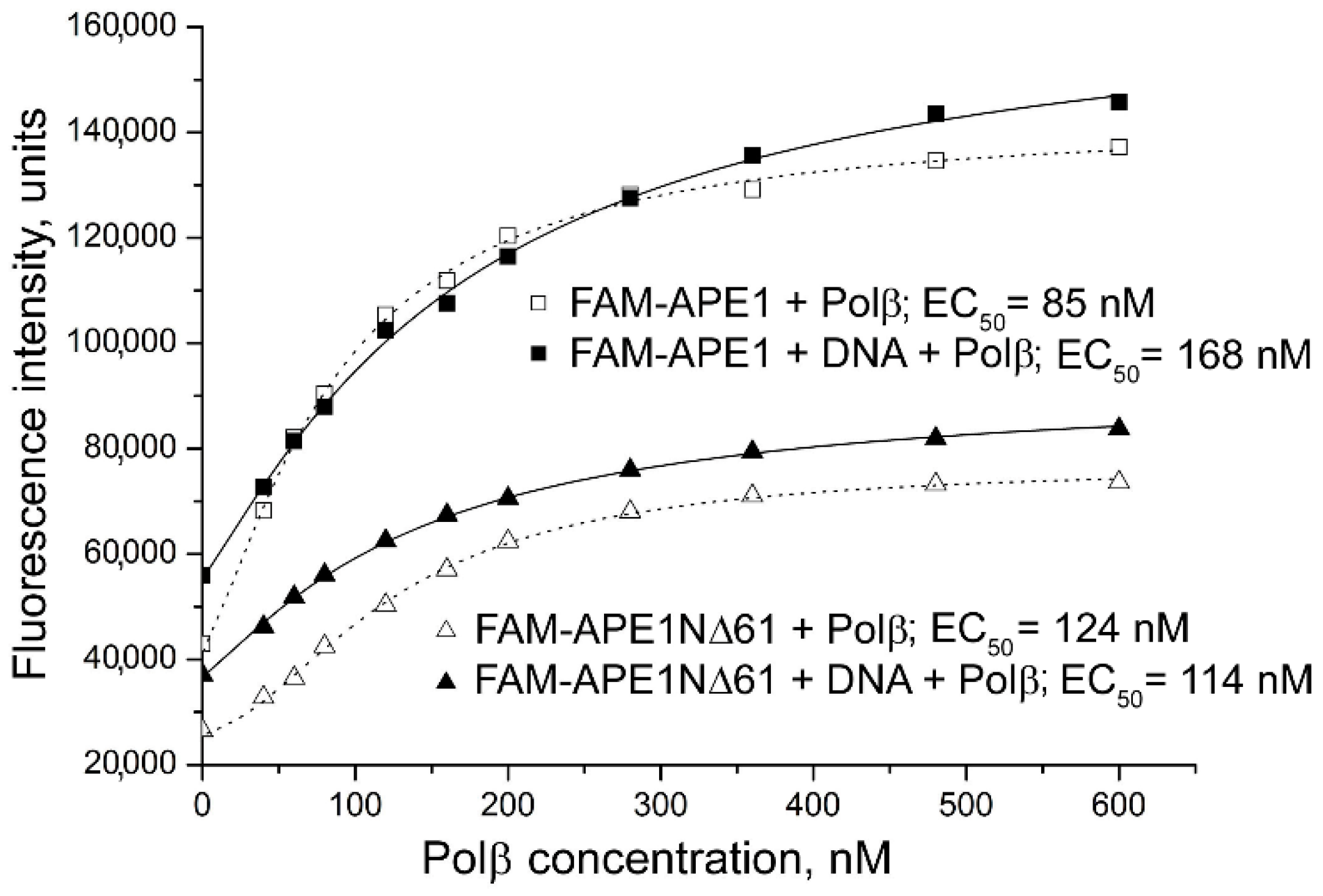
2.2. Involvement of N-Terminal Extension of APE in DNA-Dependent Modulation of Protein–Protein Interactions
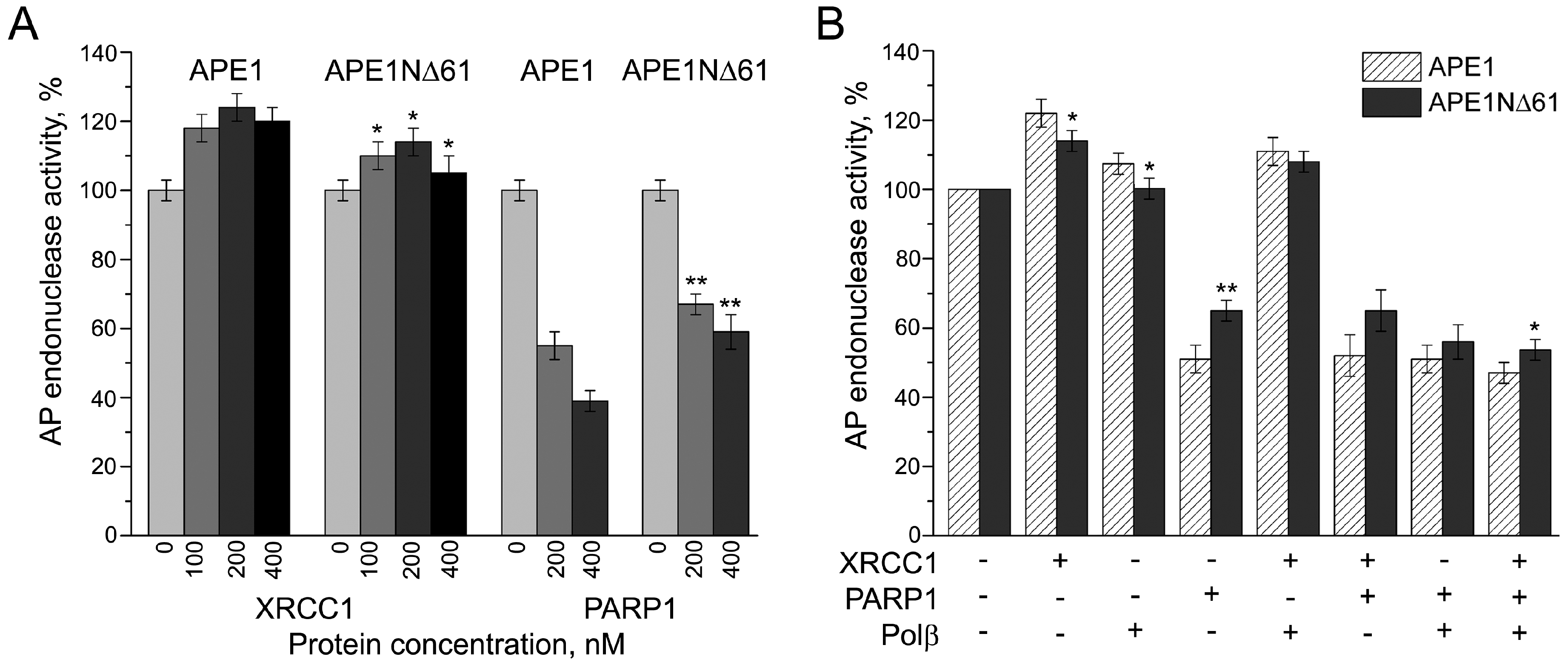
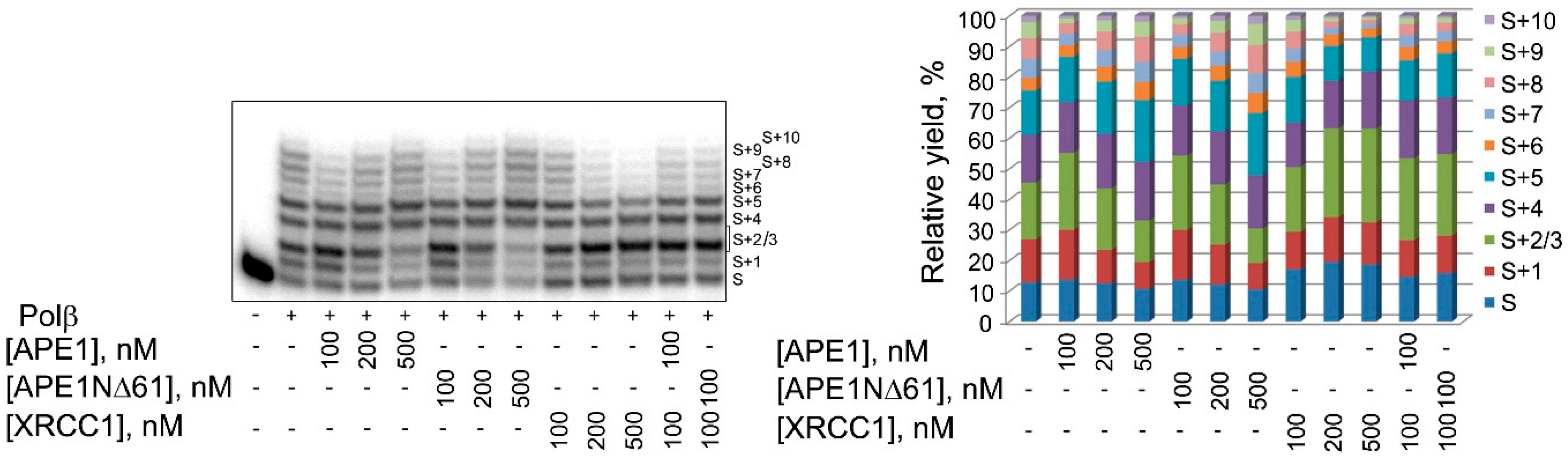
2.3. Influence of N-Terminal Truncation of APE1 on Functional Cooperation between BER Proteins
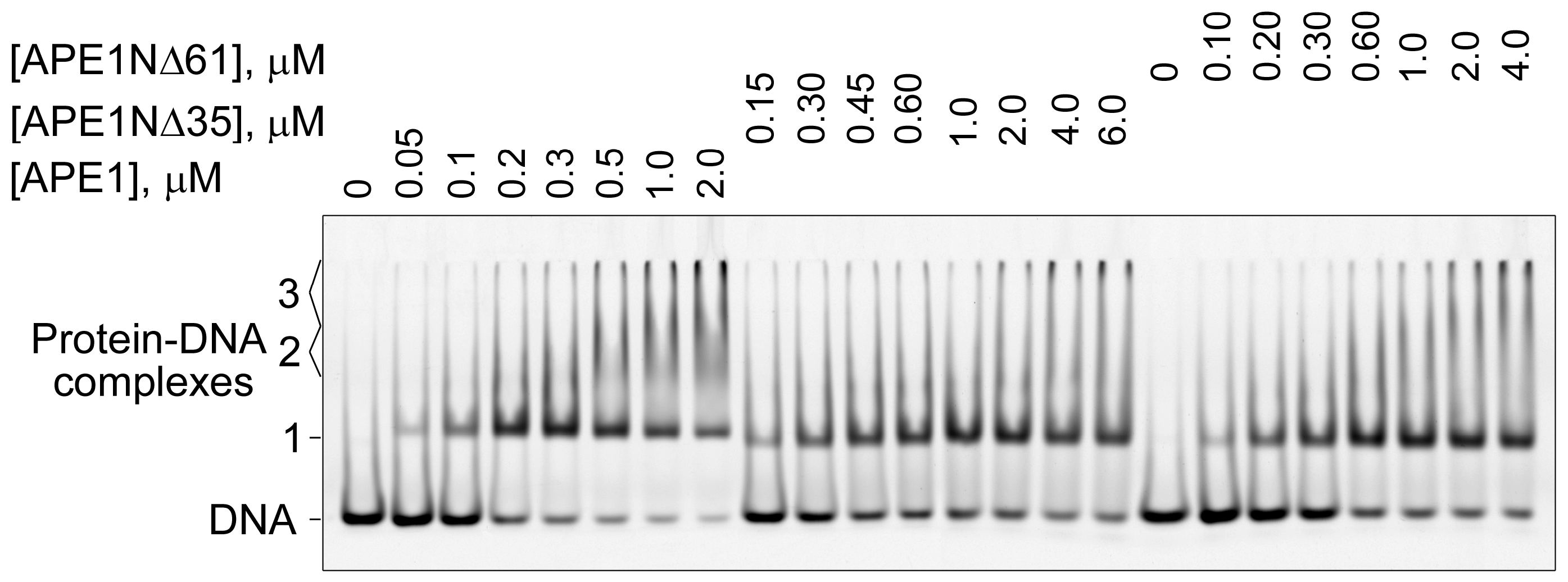
2.4. Contribution of N-Terminal Extension of APE1 to AP-DNA Binding
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Fluorescence Studies of Protein–Protein Interactions
4.3. AP Endonuclease Activity Assay
4.4. DNA Synthesis by Polβ
4.5. Electrophoretic-Mobility-Shift Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APE1 | apurinic/apyrimidinic endonuclease 1 |
| BER | base excision repair |
| AP site | apurinic/apyrimidinic site |
| SSB | single-strand break |
| Polβ | DNA polymerase β |
| XRCC1 | X-ray repair cross-complementing protein 1 |
| PARP1 | poly(ADP-ribose) polymerase 1 |
| APE1NΔ35/APE1NΔ61 | APE1 without 35/61 N-terminal amino acid residues |
| FAM | 5(6)-carboxyfluorescein |
| TMR | 5(6)-carboxytetramethylrhodamine |
| FRET | Fluorescence resonance energy transfer |
| AP-DNA/gap-DNA | DNA with a synthetic AP site/1-nucleotide gap |
| FAM-SE/TMR-SE | succinimidyl ester of 5(6)-carboxyfluorescein/5(6)-carboxytetramethylrhodamine |
References
- Li, M.; Wilson, D.M., 3rd. Human apurinic/apyrimidinic endonuclease 1. Antioxid. Redox. Signal. 2014, 20, 678–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniali, G.; Malfatti, M.C.; Tell, G. Unveiling the non-repair face of the Base Excision Repair pathway in RNA processing: A missing link between DNA repair and gene expression? DNA Repair 2017, 56, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitaker, A.M.; Freudenthal, B.D. APE1: A skilled nucleic acid surgeon. DNA Repair (Amst) 2018, 71, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Wilson, D.M., 3rd. Coordination of DNA single strand break repair. Free Radic. Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.R.; Flynn, T.S.; Freudenthal, B.D. Base excision repair of oxidative DNA damage: from mechanism to disease. Front. Biosci. (Landmark Ed) 2017, 22, 1493–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bont, R.; van Larebeke, N. Endogeneous DNA damage in humans: a review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [Green Version]
- Malfatti, M.C.; Balachander, S.; Antoniali, G.; Koh, K.D.; Saint-Pierre, C.; Gasparutto, D.; Chon, H.; Crouch, R.J.; Storici, F.; Tell, G. Abasic and oxidized ribonucleotides embedded in DNA are processed by human APE1 and not by RNase H2. Nucleic Acids Res. 2017, 45, 11193–11212. [Google Scholar] [CrossRef] [Green Version]
- Antoniali, G.; Serra, F.; Lirussi, L.; Tanaka, M.; D’Ambrosio, C.; Zhang, S.; Radovic, S.; Dalla, E.; Ciani, Y.; Scaloni, A.; et al. Mammalian APE1 controls miRNA processing and its interactome is linked to cancer RNA metabolism. Nat. Commun. 2017, 8, 797. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Dai, N.; Wang, D.; Zhong, Z. Distinct APE1 activities affect the regulation of VEGF transcription under hypoxic conditions. Comput. Struct. Biotechnol. J. 2019, 17, 324–332. [Google Scholar] [CrossRef]
- Frossi, B.; Antoniali, G.; Yu, K.; Akhtar, N.; Kaplan, M.H.; Kelley, M.R.; Tell, G.; Pucillo, C.E.M. Endonuclease and redox activities of human apurinic/apyrimidinic endonuclease 1 have distinctive and essential functions in IgA class switch recombination. J. Biol. Chem. 2019, 294, 5198–5207. [Google Scholar] [CrossRef]
- Limpose, K.L.; Corbett, A.H. Doetsch, P.W. BERing the burden of damage: pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair (Amst) 2017, 56, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Dhiman, M.; Tell, G.; Mantha, A.K. A review on protein-protein interaction network of APE1/Ref-1 and its associated biological functions. Cell Biochem. Funct. 2015, 33, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Moor, N.A.; Lavrik, O.I. Protein-protein interactions in DNA base excision repair. Biochemistry (Mosc) 2018, 83, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Malfatti, M.C.; Antoniali, G.; Codrich, M.; Burra, S.; Mangiapane, G.; Dalla, E.; Tell, G. New perspectives in cancer biology from a study of canonical and non-canonical functions of base excision repair proteins with a focus on early steps. Mutagenesis 2020, (in press). [Google Scholar] [CrossRef] [PubMed]
- Gorman, M.A.; Morera, S.; Rothwell, D.G.; de La Fortelle, E.; Mol, C.D.; Tainer, J.A.; Hickson, I.D.; Freemont, P.S. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997, 16, 6548–6558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, T.; Mitra, S. Deletion analysis of human AP-endonuclease: minimum sequence required for the endonuclease activity. Carcinogenesis 1998, 19, 525–527. [Google Scholar] [CrossRef]
- Yu, E.; Gaucher, S.P.; Hadi, M.Z. Probing conformational changes in Ape1 during the progression of base excision repair. Biochemistry 2010, 49, 3786–3796. [Google Scholar] [CrossRef]
- Fantini, D.; Vascotto, C.; Marasco, D.; D’Ambrosio, C.; Romanello, M.; Vitagliano, L.; Pedone, C.; Poletto, M.; Cesaratto, L.; Quadrifoglio, F.; et al. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010, 38, 8239–8256. [Google Scholar] [CrossRef] [Green Version]
- Lirussi, L.; Antoniali, G.; Vascotto, C.; D’Ambrosio, C.; Poletto, M.; Romanello, M.; Marasco, D.; Leone, M.; Quadrifoglio, F.; Bhakat, K.K.; et al. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol. Biol. Cell 2012, 23, 4079–4096. [Google Scholar] [CrossRef]
- Poletto, M.; Vascotto, C.; Scognamiglio, P.L.; Lirussi, L.; Marasco, D.; Tell, G. Role of the unstructured N-terminal domain of the hAPE1 (human apurinic/apyrimidinic endonuclease 1) in the modulation of its interaction with nucleic acids and NPM1 (nucleophosmin). Biochem. J. 2013, 452, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Roychoudhury, S.; Nath, S.; Song, H.; Hegde, M.L.; Bellot, L.J.; Mantha, A.K.; Sengupta, S.; Ray, S.; Natarajan, A.; Bhakat, K.K. Human apurinic/apyrimidinic endonuclease (APE1) is acetylated at DNA damage sites in chromatin, and acetylation modulates its DNA repair activity. Mol. Cell. Biol. 2017, 37, e00401-16. [Google Scholar] [CrossRef] [Green Version]
- Madlener, S.; Ströbel, T.; Vose, S.; Saydam, O.; Price, B.D.; Demple, B.; Saydam, N. Essential role for mammalian apurinic/apyrimidinic (AP) endonuclease Ape1/Ref-1 in telomere maintenance. Proc. Natl Acad. Sci. USA 2013, 110, 17844–17849. [Google Scholar] [CrossRef] [Green Version]
- Burra, S.; Marasco, D.; Malfatti, M.C.; Antoniali, G.; Virgilio, A.; Esposito, V.; Demple, B.; Galeone, A.; Tell, G. Human AP-endonuclease (Ape1) activity on telomeric G4 structures is modulated by acetylatable lysine residues in the N-terminal sequence. DNA Repair (Amst) 2019, 73, 129–143. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Sengupta, S.; Adeniyi, V.F.; Roychoudhury, S.; Nath, S.; Bellot, L.J.; Feng, D.; Mantha, A.K.; Sinha, M.; Qiu, S.; et al. Regulation of limited N-terminal proteolysis of APE1 in tumor via acetylation and its role in cell proliferation. Oncotarget 2016, 7, 22590–22604. [Google Scholar] [CrossRef] [Green Version]
- Scott, T.L.; Wicker, C.A.; Suganya, R.; Dhar, B.; Pittman, T.; Horbinski, C.; Izumi, T. Polyubiquitination of apurinic/apyrimidinic endonuclease 1 by Parkin. Mol. Carcinog. 2017, 56, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D’Ambrosio, C.; Scaloni, A.; et al. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol. Cell. Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef] [Green Version]
- Vidal, A.E.; Boiteux, S.; Hickson, I.D.; Radicella, J.P. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001, 20, 6530–6539. [Google Scholar] [CrossRef] [Green Version]
- Moor, N.A.; Vasil’eva, I.A.; Anarbaev, R.O.; Antson, A.A.; Lavrik, O.I. Quantitative characterization of protein-protein complexes involved in base excision DNA repair. Nucleic Acids Res. 2015, 43, 6009–6022. [Google Scholar] [CrossRef] [Green Version]
- Lalonde, S.; Ehrhardt, D.W.; Loqué, D.; Chen, J.; Rhee, S.Y.; Frommer, W.B. Molecular and cellular approaches for the detection of protein-protein interactions: latest techniques and current limitations. Plant J. 2008, 53, 610–635. [Google Scholar] [CrossRef]
- Wojtuszewski, K.; Harvey, J.J.; Han, M.K.; Knutson, J.R. Fluorescence detection of proximity. In Protein Interactions; Biophysical Approaches for the Study of Complex Reversible Systems; Schuck, P., Atassi, M.Z., Eds.; Springer: New York, USA, 2007; pp. 367–396. [Google Scholar]
- Liu, Y.; Prasad, R.; Beard, W.A.; Kedar, P.S.; Hou, E.W.; Shock, D.D.; Wilson, S.H. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase β. J. Biol. Chem. 2007, 282, 13532–13541. [Google Scholar] [CrossRef] [Green Version]
- D’Silva, I.; Pelletier, J.D.; Lagueux, D.; D’Amours, J.; Chaudhry, M.A.; Weinfeld, M.; Lees-Miller, S.P.; Poirier, G.G. Relative affinities of poly(ADP-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim. Biophys. Acta 1999, 1430, 119–126. [Google Scholar] [CrossRef]
- Mani, R.S.; Karimi-Busheri, F.; Fanta, M.; Caldecott, K.W.; Cass, C.E.; Weinfeld, M. Biophysical characterization of human XRCC1 and its binding to damaged and undamaged DNA. Biochemistry 2004, 43, 16505–16514. [Google Scholar] [CrossRef] [PubMed]
- Nazarkina, Z.K.; Khodyreva, S.N.; Marsin, S.; Lavrik, O.I.; Radicella, J.P. XRCC1 interactions with base excision repair DNA intermediates. DNA Repair (Amst) 2007, 6, 54–64. [Google Scholar] [CrossRef]
- Khodyreva, S.N.; Prasad, R.; Ilina, E.S.; Sukhanova, M.V.; Kutuzov, M.M.; Liu, Y.; Hou, E.W.; Wilson, S.H.; Lavrik, O.I. Apurinic/apyrimidinic (AP) site recognition by the 5’-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc. Natl Acad. Sci. USA 2010, 107, 22090–22095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilyestrom, W.; van der Woerd, N.M.; Clark, J.; Luger, K. Structural and biophysical studies of human PARP-1 in complex with damaged DNA. J. Mol. Biol. 2010, 395, 983–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, Y.; Nash, R.A.; Klungland, A.; Schär, P.; Barnes, D.E.; Lindahl, T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase β and the XRCC1 protein. EMBO J. 1996, 15, 6662–6670. [Google Scholar] [CrossRef]
- Bennett, R.A.; Wilson, D.M., 3rd; Wong, D.; Demple, B. Interaction of human apurinic endonuclease and DNA polymerase β in the base excision repair pathway. Proc. Natl Acad. Sci. USA 1997, 94, 7166–7169. [Google Scholar] [CrossRef] [Green Version]
- Masuda, Y.; Bennett, R.A.; Demple, B. Dynamics of the interaction of human apurinic endonuclease (Ape1) with its substrate and product. J. Biol. Chem. 1998, 273, 30352–30359. [Google Scholar] [CrossRef] [Green Version]
- Lavrik, O.I.; Prasad, R.; Sobol, R.W.; Horton, J.K.; Ackerman, E.J.; Wilson, S.H. Photoaffinity labeling of mouse fibroblast enzymes by a base excision repair intermediate. Evidence for the role of poly(ADP-ribose) polymerase-1 in DNA repair. J. Biol. Chem. 2001, 276, 25541–25548. [Google Scholar] [CrossRef] [Green Version]
- Sukhanova, M.V.; Khodyreva, S.N.; Lebedeva, N.A.; Prasad, R.; Wilson, S.H.; Lavrik, O.I. Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase β and poly(ADP-ribose) polymerase 1: interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 2005, 33, 1222–1229. [Google Scholar] [CrossRef] [Green Version]
- Dyrkheeva, N.S.; Khodyreva, S.N.; Lavrik, O.I. Interaction of APE1 and other repair proteins with DNA duplexes imitating intermediates of DNA repair and replication. Biochemistry (Mosc) 2008, 73, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Kladova, O.A.; Bazlekowa-Karaban, M.; Baconnais, S.; Piétrement, O.; Ishchenko, A.A.; Matkarimov, B.T.; Iakovlev, D.A.; Vasenko, A.; Fedorova, O.S.; Le Cam, E.; et al. The role of the N-terminal domain of human apurinic/apyrimidinic endonuclease 1, APE1, in DNA glycosylase stimulation. DNA Repair (Amst) 2018, 64, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Moor, N.; Lavrik, O. Coordination of DNA Base Excision Repair by Protein-Protein Interactions. In DNA Repair—An Update; Mognato, M., Ed.; IntechOpen Ltd.: London, UK, 2019; Chapter 2; pp. 1–22. [Google Scholar] [CrossRef] [Green Version]
- Vasil’eva, I.A.; Anarbaev, R.O.; Moor, N.A.; Lavrik, O.I. Dynamic light scattering study of base excision DNA repair proteins and their complexes. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 297–305. [Google Scholar] [CrossRef]
- London, R.E. The structural basis of XRCC1-mediated DNA repair. DNA Repair (Amst) 2015, 30, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Shall, S.; de Murcia, G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat. Res. 2000, 460, 1–15. [Google Scholar] [CrossRef]
- Dantzer, F.; de La Rubia, G.; Ménissier-De Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef]
- Cistulli, C.; Lavrik, O.I.; Prasad, R.; Hou, E.; Wilson, S.H. AP endonuclease and poly(ADP-ribose) polymerase-1 interact with the same base excision repair intermediate. DNA repair (Amst) 2004, 3, 581–591. [Google Scholar] [CrossRef]
- Kutuzov, M.M.; Ilina, E.S.; Sukhanova, M.V.; Pyshnaya, I.A.; Pyshnyi, D.V.; Lavrik, O.I.; Khodyreva, S.N. Interaction of poly(ADP-ribose) polymerase 1 with apurinic/apyrimidinic sites within clustered DNA damage. Biochemistry (Mosc) 2011, 76, 147–156. [Google Scholar] [CrossRef]
- Prasad, R.; Dyrkheeva, N.; Williams, J.; Wilson, S.H. Mammalian base excision repair: functional partnership between PARP-1 and APE1 in AP-site repair. PLoS ONE 2015, 10, e0124269. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Kong, M.; Gassman, N.R.; Freudenthal, B.D.; Prasad, R.; Zhen, S.; Watkins, S.C.; Wilson, S.H.; Van Houten, B. PARP1 changes from three-dimensional DNA damage searching to one-dimensional diffusion after auto-PARylation or in the presence of APE1. Nucleic Acids Res. 2017, 45, 12834–12847. [Google Scholar] [CrossRef]
- Moor, N.A.; Vasil’eva, I.A.; Kuznetsov, N.A.; Lavrik, O.I. Human apurinic/apyrimidinic endonuclease 1 is modified in vitro by poly(ADP-ribose) polymerase 1 under control of the structure of damaged DNA. Biochimie 2020, 168, 144–155. [Google Scholar] [CrossRef]
- Kurien, B.T.; Scofield, R.H. Western blotting: an introduction. Methods Mol. Biol. 2015, 1312, 17–30. [Google Scholar] [CrossRef]
- Mol, C.D.; Izumi, T.; Mitra, S.; Tainer, J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature 2000, 403, 451–456. [Google Scholar] [CrossRef]
- Hedge, M.L.; Hazra, T.K.; Mitra, S. Functions of disordered regions in mammalian early base excision repair proteins. Cell. Mol. Life Sci. 2010, 67, 3573–3587. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.; Dyson, H.J. Intrinsically disordered proteins in cellular signaling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Janoshazi, A.K.; Horton, J.K.; Zhao, M.L.; Prasad, R.; Scappini, E.L.; Tucker, C.J.; Wilson, S.H. Shining light on the response to repair intermediates in DNA of living cells. DNA Repair (Amst) 2020, 85, 102749. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Khodyreva, S.N.; Sukhanova, M.V.; Safronov, I.V.; Dezhurov, S.V.; Lavrik, O.I. 3’-5’ exonuclease activity of human apurinic/apyrimidinic endonuclease 1 towards DNAs containing dNMP and their modified analogs at the 3′ end of single strand DNA break. Biochemistry (Mosc) 2006, 71, 200–210. [Google Scholar] [CrossRef]
- Waters, T.R.; Gallinari, P.; Jiricny, J.; Swann, P.F. Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem. 1999, 274, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, M.R.; O’Brien, P.J. Defining the functional footprint for recognition and repair of deaminated DNA. Nucleic Acids Res. 2012, 40, 11638–11647. [Google Scholar] [CrossRef] [Green Version]
- Luncsford, P.J.; Manvilla, B.A.; Patterson, D.N.; Malik, S.S.; Jin, J.; Hwang, B.J.; Gunther, R.; Kalvakolanu, S.; Lipinski, L.J.; Yuan, W.; et al. Coordination of MYH DNA glycosylase and APE1 endonuclease activities via physical interactions. DNA Repair (Amst) 2013, 12, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Esadze, A.; Rodriguez, G.; Cravens, S.L.; Stivers, J.T. AP-Endonuclease 1 accelerates turnover of human 8-oxoguanine DNA glycosylase by preventing retrograde binding to the abasic-site product. Biochemistry 2017, 56, 1974–1986. [Google Scholar] [CrossRef]
- Popov, A.V.; Grin, I.R.; Dvornikova, A.P.; Matkarimov, B.T.; Groisman, R.; Saparbaev, M.; Zharkov, D.O. Reading targeted DNA damage in the active demethylation pathway: Role of accessory domains of eukaryotic AP endonucleases and thymine-DNA glycosylases. J. Mol. Biol. 2020, (in press). [Google Scholar] [CrossRef]
- Gagné, J.P.; Isabelle, M.; Lo, K.S.; Bourassa, S.; Hendzel, M.J.; Dawson, V.L.; Dawson, T.M.; Poirier, G.G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008, 36, 6959–6976. [Google Scholar] [CrossRef] [Green Version]
- Teloni, F.; Altmeyer, M. Readers of poly(ADP-ribose): designed to be fit for purpose. Nucleic Acids Res. 2016, 44, 993–1006. [Google Scholar] [CrossRef] [Green Version]
- Strauss, P.R.; Beard, W.A.; Patterson, T.A.; Wilson, S.H. Substrate binding by human apurinic/apyrimidinic endonuclease indicates a Briggs-Haldane mechanism. J. Biol. Chem. 1997, 272, 1302–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Widen, S.G.; Williams, K.R.; Kedar, P.; Karpel, R.L.; Wilson, S.H. Studies of the domain structure of mammalian DNA polymerase β. Identification of a discrete template binding domain. J. Biol. Chem. 1990, 265, 2124–2131. [Google Scholar] [PubMed]
- Daviet, S.; Couvé-Privat, S.; Gros, L.; Shinozuka, K.; Ide, H.; Saparbaev, M.; Ishchenko, A.A. Major oxidative products of cytosine are substrates for the nucleotide incision repair pathway. DNA Repair (Amst) 2007, 6, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, M.V.; Khodyreva, S.N.; Lavrik, O.I. Poly(ADP-ribose) polymerase-1 inhibits strand-displacement synthesis of DNA catalyzed by DNA polymerase β. Biochemistry (Mosc) 2004, 69, 558–568. [Google Scholar] [CrossRef]
- Belousova, E.A.; Vasil’eva, I.A.; Moor, N.A.; Zatsepin, T.S.; Oretskaya, T.S.; Lavrik, O.I. Clustered DNA lesions containing 5-formyluracil and AP site: repair via the BER system. PLoS One 2013, 8, e68576. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Labelled Protein a | Protein Partner | EC50 b, nM | Effect on Affinity c | E d |
|---|---|---|---|---|
| FAM-APE1 | Polβ | 84 ± 7 | 0.16 ± 0.02 | |
| FAM-APE1NΔ35 | Polβ | 97 ± 8 * | 1.2 | 0.16 ± 0.02 |
| FAM-APE1NΔ61 | Polβ | 120 ± 11 ** | 1.4 | 0.22 ± 0.02 * |
| FAM-APE1 | XRCC1 | 76 ± 8 | 0.14 ± 0.01 | |
| FAM-APE1NΔ35 | XRCC1 | 100 ± 10 ** | 1.3 | 0.11 ± 0.01 * |
| FAM-APE1NΔ61 | XRCC1 | 130 ± 12 *** | 1.7 | 0.09 ± 0.01 ** |
| FAM-APE1 | PARP1 | 45 ± 5 | 0.04 ± 0.01 | |
| FAM-APE1NΔ35 | PARP1 | 57 ± 5 ** | 1.3 | 0.13 ± 0.02 ** |
| FAM-APE1NΔ61 | PARP1 | 75 ± 7 *** | 1.7 | 0.10 ± 0.01 ** |
| Labelled Protein a | DNA a | Protein Partner | EC50 b, nM | Effect on Affinity c | Effect on FRET Efficiency d |
|---|---|---|---|---|---|
| FAM-APE1 | AP-DNA | Polβ | 92 ± 7 | 1.1 | +0.05 * |
| FAM-APE1 | AP-DNA inc | Polβ | 170 ± 13 *** | 2.0 | –0.06 ** |
| FAM-APE1 | gap-DNA | Polβ | 110 ± 8 * | 1.3 | –0.11 ** |
| FAM-APE1NΔ35 | AP-DNA | Polβ | 91 ± 7 | 0.94 | –0.08 ** |
| FAM-APE1NΔ35 | AP-DNA inc | Polβ | 110 ± 10 | 1.1 | –0.0 8** |
| FAM-APE1NΔ61 | AP-DNA | Polβ | 110 ± 10 | 0.92 | –0.08 ** |
| FAM-APE1NΔ61 | AP-DNA inc | Polβ | 130 ± 11 | 1.1 | –0.09 ** |
| FAM-APE1NΔ61 | gap-DNA | Polβ | 140 ± 13 | 1.2 | –0.07 * |
| FAM-APE1 | AP-DNA | XRCC1 | 78 ± 7 | 1.0 | –0.01 |
| FAM-APE1 | gap-DNA | XRCC1 | 51 ± 4 ** | 0.67 | +0.08 ** |
| FAM-APE1NΔ35 | AP-DNA | XRCC1 | 68 ± 6 ** | 0.68 | –0.03 * |
| FAM-APE1NΔ35 | gap-DNA | XRCC1 | 65 ± 6 ** | 0.65 | –0.03 * |
| FAM-APE1NΔ61 | AP-DNA | XRCC1 | 99 ± 8 * | 0.76 | –0.06 ** |
| FAM-APE1NΔ61 | gap-DNA | XRCC1 | 97 ± 8 * | 0.75 | –0.05 ** |
| FAM-APE1 | AP-DNA | PARP1 | 76 ± 6 ** | 1.7 | +0.05 ** |
| FAM-APE1 | gap-DNA | PARP1 | 54 ± 4 | 1.2 | +0.02 |
| FAM-APE1NΔ35 | AP-DNA | PARP1 | 49 ± 4 | 0.86 | –0.03 |
| FAM-APE1NΔ35 | gap-DNA | PARP1 | 48 ± 4 | 0.84 | +0.03 |
| FAM-APE1NΔ61 | AP-DNA | PARP1 | 55 ± 5 * | 0.73 | –0.02 |
| FAM-APE1NΔ61 | gap-DNA | PARP1 | 54 ± 4 * | 0.72 | +0.04 * |
| Protein | EC50 a, nM | Maximal Extent of Binding a |
|---|---|---|
| APE1 | 0.12 ± 0.02 | 0.96 ± 0.03 |
| APE1NΔ35 | 0.20 ± 0.03 | 0.77 ± 0.03 |
| APE1NΔ61 | 0.21 ± 0.03 | 0.84 ± 0.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moor, N.; Vasil’eva, I.; Lavrik, O. Functional Role of N-Terminal Extension of Human AP Endonuclease 1 In Coordination of Base Excision DNA Repair via Protein–Protein Interactions. Int. J. Mol. Sci. 2020, 21, 3122. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093122
Moor N, Vasil’eva I, Lavrik O. Functional Role of N-Terminal Extension of Human AP Endonuclease 1 In Coordination of Base Excision DNA Repair via Protein–Protein Interactions. International Journal of Molecular Sciences. 2020; 21(9):3122. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093122
Chicago/Turabian StyleMoor, Nina, Inna Vasil’eva, and Olga Lavrik. 2020. "Functional Role of N-Terminal Extension of Human AP Endonuclease 1 In Coordination of Base Excision DNA Repair via Protein–Protein Interactions" International Journal of Molecular Sciences 21, no. 9: 3122. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093122