Altered Ca2+ Homeostasis in Immune Cells during Aging: Role of Ion Channels

Center for Integrative Physiology and Molecular Medicine, Department of Biophysics, School of Medicine, Saarland University, 66421 Homburg, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(1), 110; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010110
Submission received: 11 December 2020
/
Revised: 21 December 2020
/
Accepted: 22 December 2020
/
Published: 24 December 2020
(This article belongs to the Special Issue STIMulating Ca2+ Homeostasis)
{kind=link}
Abstract
:Aging is an unstoppable process and begins shortly after birth. Each cell of the organism is affected by the irreversible process, not only with equal density but also at varying ages and with different speed. Therefore, aging can also be understood as an adaptation to a continually changing cellular environment. One of these very prominent changes in age affects Ca2+ signaling. Especially immune cells highly rely on Ca2+-dependent processes and a strictly regulated Ca2+ homeostasis. The intricate patterns of impaired immune cell function may represent a deficit or compensatory mechanisms. Besides, altered immune function through Ca2+ signaling can profoundly affect the development of age-related disease. This review attempts to summarize changes in Ca2+ signaling due to channels and receptors in T cells and beyond in the context of aging.
1. Introduction
Aging is often associated with a loss of function. It describes a cumulative phenomenon that contributes to morbidity and mortality in man due to the greater incidence of infection, autoimmune phenomena, ineffective vaccination and cancer in elderly individuals (reviewed in [1,2,3,4,5]). One may also look at aging to be a constant adaptation and remodeling to the various, and often continuous, stressors encountered during life to maintain the organism’s overall functionality. The adaptation of cells during aging to environmental changes may increase the susceptibility to diseases but often ensures survival.
Dysregulation and changes of ionic fluxes across membranes mediated by ion channels, transporters, and receptors probably form the basis for a modified cell function not only during aging but also in disease. Ion channels and transporters evolved various mechanisms through which the monovalent (K+, Na+, Cl−) and divalent (Mg2+, Ca2+, Zn2+) ions are gated in response to different cellular signals.
Considering T cells, calcium (Ca2+) is one of the critical ions in generation, coordination, and control of signals within and between cells. To fulfill these numerous and wide-ranging tasks, very strictly coordinated and regulated calcium homeostasis and its maintenance in the cell is required. In the last decades of research, an incline of functional and signaling defects in elderly T cells has accumulated (reviewed in [6,7,8]). Most of these events depend either on transient or sustained Ca2+-influx to keep the intracellular calcium concentration [Ca2+]i higher than basal levels for minutes to several hours. T cell function and maintenance are among the most remarkable and most pronounced changes occurring within an aging immune system.
Since the importance of Ca2+ for T cell function and in adaptive immunity has been already excessively and excellently reviewed by many groups [9,10,11,12,13], we focus more on the possibly altered channels and receptors during aging. These are expressed and play an essential role in T cells from human and mice. Although the signaling machinery in T cells is exceptionally complicated and many steps remain to be clarified, age-related changes in Ca2+ entry may be a critical cause of cell-mediated immune response decline with aging. This review reports findings on cellular mechanisms linked to Ca2+ homeostasis focusing on channels and their relevance in pathophysiological processes, mainly in T cells during aging.
2. Altered T Cell Function during Aging
One significant hallmark in immune system aging is the thymic involution with age, resulting in a steady decline of naïve T cells (TN) numbers [14,15,16,17] with restricted T cell receptor (TCR) repertoires [18,19,20], ending in disrupted T cell homeostasis. The lack of naïve T cells and increasing memory T cells (TM) accumulation contribute to a higher risk of severe infections in the elderly [21]. Interestingly, the naïve CD8+ T cell compartment is much more affected than the CD4+ T cells, including higher contraction rates [17,18,19]. However, a quantitative decrease of TN cells alone might not account for the functional differences in effector (TE) and TM cell responses. Different models are discussed in this context, such as distinctive phenotype between adult and elderly naïve CD8+ T cells with altered survival, developmental pathways, and responses to infection [22,23,24]. On the other hand, an increased adaptation to environmental changes during aging has been observed [25] consistent with loss of stem-like features lead to reduced plasticity [24,26] and accumulation of virtual-memory T cells without former antigen stimulation [27,28].
TCR antigen-binding triggers intracellular Ca2+ mobilization and is required for plethora of cellular processes and may account for reduced effector responses by aged T cells. Activation of TCR includes a firmly defined sequence of events, and numerous age-related deficits are described in T cell signaling pathways after its activation. Just recently, microRNA (miR-181a) expression emerged as a crucial regulator of controlling TCR activation thresholds in peripheral T cell response [29]. In naïve CD4+ T cells from elderly organisms, lower miR-181a expression leads to reduced extracellular regulated kinase (ERK) upon TCR activation [30], as well as in naïve CD8+ T cells [31]. The deletion of miR-181a in peripheral T cells in a mouse model causes defective viral response through impaired generation of CD8+ effector T cells [32]. Additionally, activation-induced upregulation of miR-21 shifts the transcriptome towards effector T cells and away from memory T cell differentiation [33].
Unfavorable alterations of T cells subpopulations result in a decreased CD4+/CD8+ ratio and the accumulation of senescent and terminally differentiated T cells (reviewed in [34,35]). The inversion of the CD4+/CD8+ ratio is associated with altered immune function, chronic viral infection, and chronic inflammation [36,37,38]. Besides the ratio, the CD4+ and CD8+ T cell subsets are affected differently by aging [39]. The aged naïve CD4+ T cells differentiate poorly to T-helper-cell-1 (Th1) and T-helper-cell-2 (Th2) effector subsets, but the ability to generate T-helper-cell-17 (Th17) is intact, reflected by their increased numbers during aging [40]. Moreover, the elderly have increased Th1/Th2 ratio [41], and data from murine studies supports a shift from a Th1-like to a Th2-like cytokine response [42]. Simultaneously, the subset of regulatory T cells (Tregs) increases compared to adult individuals [43]. The accumulation of functional Tregs contributes to the frequent reactivations of chronic infections often observed in aging. The aged-dependent decrease of Th17/Treg ratio after stimulation accompanying altered cytokine expression may contribute to the imbalance between pro-and anti-inflammatory immune responses [44].
Furthermore, in vitro stimulated T cells from humans and mice show altered cytokine secretion. However, the published results are ambiguous and inconsistent for many investigated cytokines, like interferon-gamma (IFN-γ) and interleukin-2 (IL-2) [40,41,45,46,47,48,49,50,51,52]. All the studies highlight the importance of used stimuli for cytokine induction and the resulting impact on immune responses. Additionally, one must consider T cell responsiveness’s altered kinetics with age as a possible cause impacting proliferation, upregulation of activation markers, and cytokine secretion [53]. Naïve CD4+ T cells from elderly mice secrete less than 50% IL-2 compared to adult cells, leading to decreased expression of CD25 (IL-2 receptor α), and show reduced proliferation and incomplete differentiation to effector cells [50,54]. The age-related reduction in IL-2 production by CD4+ T cells is not fully explained either by alterations of the actual structure of TCRs or by changes in the TCR-CD3 complex [55,56]. The defects in effector generation associated with aging are reversible by adding IL-2 but no other related gamma chain (γc)-receptor binding cytokines [50]. Already in 1985, the first experiments implicate a Ca2+ influx as an essential component for IL-2 function [57]. Nowadays, there is no doubt about the regulation of IL-2 and IL-2 receptor (IL2-R) mediated signaling through the nuclear factor of activated T cell (NFAT)/calcineurin pathway controlled by Ca2+ influx upon TCR and costimulatory signals [58,59].
The influence of aging is not only limited to T cell subtype distribution and cytokine production of CD4+ T cells but also the cytotoxicity of CD8+ T cells is changed. In a study by Fagnoni and colleagues, the CD3-mediated cytotoxicity of freshly isolated T cells from healthy aged donors against P815 target cells exhibited higher values than their younger counterparts [60]. This correlates with higher amounts of CD8+CD28− cells in elderly humans [60,61]. However, in the context of disease, the elderly human with COVID-19 show reduced overall CD8+ T cell numbers and granzyme A expression by CD8+ T cells. In effector memory (TEM) and TE cells, perforin’s expression decreases with age in those patients [62]. Furthermore, in vitro stimulated naϊve elderly CD4+ T cells exhibit impaired cytoskeleton signaling, LAT (linker of activated T cells) and ZAP-70 (Zeta-chain-associated protein kinase 70) recruitment, and CD3-zeta assembly with the cytoskeleton to the induction of NFAT [52,63]. Additionally, CD4+ T cells from aged TCR transgenic mice do not form immunological synapses (IS) with antigen-presenting cells (APC) as efficiently as in adult mice, with a reduction in the recruitment of signaling molecules in the elderly compared to adult CD4+ T cells [64,65].
Changes in Ca2+ influx in aged T cells are also reported [66,67]; however, the influence of aging after TCR activation and the underlying molecular players are still under investigation. Following T cell activation in mice, several groups reported a decline in the Ca2+ levels with age [68,69]. Comparing T cells from mice of any age, naïve T cells are much more likely than memory T cells to respond with an increase in [Ca2+]i in response to lectin, anti-CD3 plus anti-CD28, or Ca2+ ionophores [70,71]. These studies suggest that naïve and memory T cells differ fundamentally in their ability to increase [Ca2+]i following receptor-dependent or receptor-independent stimulation. Changes in basal Ca2+ levels reported by several studies are conflicting. The resting level of free Ca2+ is lower or unaffected in human aged T cells [72,73] but higher in T lymphocytes obtained from elderly mice [74].
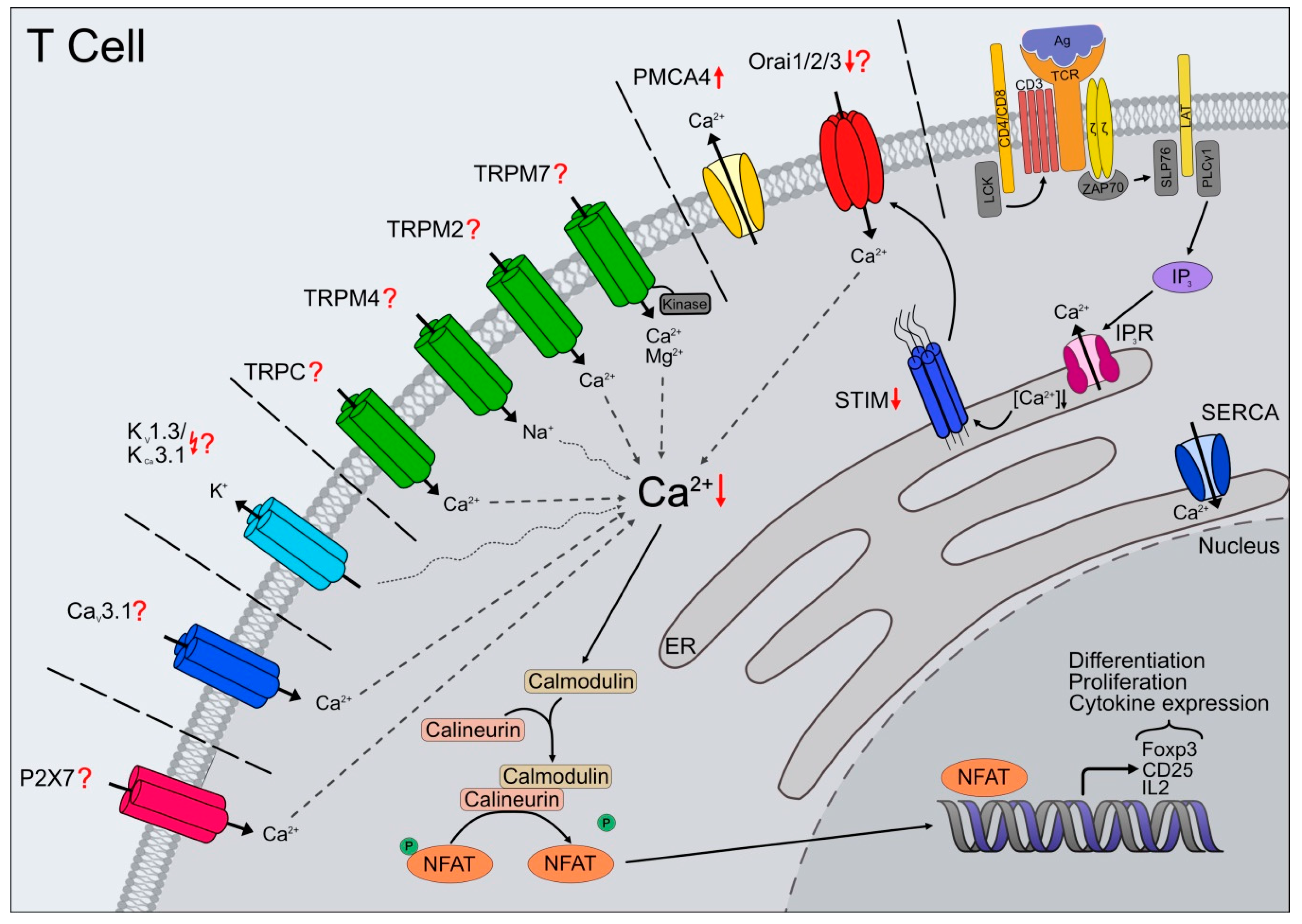
Many of the dysregulations described above are highly dependent or regulated by Ca2+ itself; however, the underlying molecular mechanisms are not well characterized and still under investigation. Altered Ca2+ fluctuations have already been associated with numerous age-related diseases, such as neurodegenerative [75], muscle-related diseases [76,77], autoimmune and inflammatory disorders [78,79]. Ca2+ responses are regulated negatively and positively by several mechanisms involving channels, pumps, and sensors (reviewed in [80]). Here we review the impact of possibly altered Ca2+-permeable channels expressed in T cells and their contribution to the altered processes observed during aging.
3. Orai/STIM
In many immunocytes, the main mechanism for Ca2+ entry is through SOCE (store-operated Ca2+ entry) [81] and involves the activation of CRAC (Ca2+-release activated Ca2+) channels. Genome-wide RNAi screens and linkage analysis in human patients with defects in SOCE identified two fundamental players of SOCE via ICRAC [82]: Stromal interaction molecules (STIM1), as the ER Ca2+ sensor [83,84] and the CRAC channel [85,86,87] itself. In addition to the identification and characterization of the Orai and STIM homologs [88,89,90], the research of the last 15 years has revealed numerous splice variants [91,92,93] that contribute to the diversity of the resulting Ca2+ signals.
SOCE pathway is important for the immunocytes and essential for numerous cellular processes, including sufficient T cell activation, development, differentiation, gene expression, the formation of the immunological synapse, and cytotoxicity (reviewed in [10,13,58,94,95,96]). For efficient development of an immune response, T cells require long-lasting Ca2+ influx through CRAC channels, and the formation of a stable IS with the antigen-presenting cell (APC) [97,98]. Orai1 and STIM1 translocate to IS accompanied by Ca2+ influx through CRAC channels [97,99,100]. Besides, mRNA expression for STIM1 and Orai homologs is upregulated. The generated distinctive Ca2+ patterns determined by the heterogeneous composition of channels and activators [101,102,103] allow not only their modulation but the transmission of extracellularly generated signals intracellularly.
The magnitude and duration of changes in [Ca2+]i are crucial determinants for T cell activation and other immune system responses. Prolonged elevations of [Ca2+]i are vital for activating transcription factors that initiate many changes in gene expression which drives T cell proliferation, cytokine, and chemokine production. The work in deficient mouse models gives an insight into the variety of processes mediated and determined by SOCE. Profound defects in key T cells cytokines such as IL-2, IL-4, IL-10, IFN-γ and TNF-α and apoptosis genes are found in CD4+ and CD8+ T cells from Orai1, STIM1, and STIM1/2-deficient mice [104,105,106]. Complete inhibition of SOCE in CD8+ T cells from STIM1/2-deficient mice impairs lytic granule exocytosis and elimination of tumor cells and virus-infected cells [107,108]. Additionally, CD8+ T cells and NK cells show Ca2+ dependent cytotoxicity with an optimum for cancer cell elimination at rather low free [Ca2+] concentrations. Downregulation of ORAI1 in cytotoxic T lymphocytes (CTLs) leads to decreased Ca2+ signals but increased efficiency to eliminate cancer cells [109]. It seems like delineation of the accurate STIM/Orai ratio could be a feature of the killing efficiency of CD8+ T cells by determining the Ca2+ killing optimum.
One of the T cells’ best studied Ca2+-dependent mechanism is the NFAT (nuclear factor of activated T cells)/calcineurin pathway [110]. The NFAT-driven gene expression is highly dependent on sustained Ca2+-influx. The activation of calmodulin-dependent enzyme calcineurin by the rise in [Ca2+]i levels leads to NFAT dephosphorylation followed by nucleus translocation. A decrease in [Ca2+]i levels leads to the export of NFAT from the nucleus [111]. Undoubtedly, the relevance of SOCE highlights the fact that lymphocytes with defective SOCE are unable to mount an immune response, and patients with such defects develop SCID. Studies of Orai1 in SCID patients have further confirmed that CRAC channels are the primary pathway for Ca2+ entry in naïve T cells. An Arg91Trp mutation in Orai1, as a pore-forming subunit of CRAC channels, is responsible for abolishing Ca2+ influx in T cells from these SCID patients [112]. Meanwhile, numerous other mutations in STIM and ORAI were identified, leading to distinctive phenotypes in patients (reviewed in [113]). Unexpectedly, immunodeficient patients with loss-of-function or null mutations in ORAI1 or STIM1 that abolish TCR-mediated Ca2+ influx in T cells have normal CD4+ and CD8+ T cell numbers with a normal TCR Vß repertoire [114,115]. These data indicate that CRAC channels do not play a significant role in the thymic development and selection of T cells. The functional defect is not limited to T cells and affects SOCE in B cells and fibroblasts [116]. Homozygous mice lacking STIM1, STIM2, or Orai1 are embryonic lethal or die soon after birth [104,105]. STIM1-deficient T cells completely lack SOCE, ICRAC, and Ca2+-dependent cytokine expression [105], but the STIM2-deficient naïve T cells show normal SOCE and cytokine production. T cells from Orai1-null mice also display an evident impairment in all three functions [104,105]. Moreover, Orai1/Orai2-deficient mice are protected from autoimmunity and alloimmunity in graft-versus-host disease. The deletion of Orai1/Orai2 in T cells abolishes SOCE leading to augmented T cell function and altered proliferation and cytokine production. Surprisingly, Orai2−/− T cells exhibit increased SOCE without improving T cell function in vivo and in vitro [117]. Additionally, Orai2 shapes the Ca2+ signaling profile in human Tregs after thapsigargin or TCR- induced SOCE. The enhanced Ca2+ signals, compared to the conventional CD4+ T cells, correlate with the lower expression of Orai2 in these cells [118].
Despite the substantial literature on SOCE associated with T cell function, the changes in Ca2+ homeostasis components and age-related changes in Ca2+ entry are less well understood. We recently linked the aging-related reduction in Ca2+ signals to reductions of the primary critical players in the Ca2+ signaling pathway [66]. The reduced expression of STIM and Orai mRNA and proteins leads to reduced Ca2+ entry. The upregulation of the plasma membrane Ca2+ ATPases 4 (PMCA4) contributes to faster extrusion in CD8+ T cells isolated from aged mice. Furthermore, these cells show a less efficient TCR-induced [Ca2+]i mobilization and increased insensitivity to Ca2+ fluctuations during cytotoxic activity [66].
4. TRP Channels
Next to STIM and Orai, other Ca2+ and ion channels, including TRP channels, are relevant for Ca2+ signaling. TRPV1 contributes to the TCR-induced Ca2+ entry in CD4+ T cells and is gated by phosphorylation depending on the lymphocyte-specific protein tyrosine kinase (LCK) [119]. Complete deletion of Trpv1 using a mouse model showed impaired TCR signaling resulting from reduced Ca2+ flux [119]. Furthermore, CD4+ T cells presented defects in T cell activation and cytokine production [119], also confirmed by using TRPV1 antagonists in T cells isolated from murine spleen [120]. However, additional electrophysiological data is missing to underline that TRPV1 is activated downstream of TCR. Besides, an inhibition of the TRPA1 channel can inhibit the TRPV1 activity, thereby reducing the Ca2+ influx. This inhibition is caused by a direct heteromerization of the two channels and such mutual modeling has also been described for other channels combination such as Orai1 and Orai2 [117] or TRPM7 and TRPM6 [121,122].
TRPC3, 5 and 6 are involved in T cell Ca2+ signaling. The Ca2+ influx via TRPC3 modulates cell proliferation [123,124]. TRPC5 seems to be important in mediating Treg-influenced inhibition of TE cells however the exact mechanism remains elusive [125]. The involvement of TRPC channels in T cells remains highly argumentative. More detailed and sophisticated studies (not only during aging) are necessary to address these issues.
TRPM2 is another channel expressed by different cell types of the peripheral immune system, including lymphocytes [126] and monocytes [127], which is involved in immune cells function. It is stimulated by oxidative stress and specifically activated by intracellular ADP-Ribose. Second messenger molecules like cyclic ADP-ribose (cADPR) and nicotinic acid adenine dinucleotide phosphate (NAADP) can activate and regulate the Ca2+ influx through TRPM2 channels in lymphocytes [128,129]. TCR engagement causes a sustained cADPR increase and an antagonist of cADPR inhibits T cell activation and proliferation in response to T cell stimulation [129]. However, it needs to be considered that this effect can also be explained by the effect of cADPR on RyRs. Trpm2−/− T cells exhibit reduced proliferation and proinflammatory cytokine secretion [130]. More evidence for the role of TRPM2 in inflammation can be found after induced inflammation by H2O2 or lipopolysaccharide (LPS). TRPM2 activation promotes immune responses through cytokines production like CXCL8, IL-6, IL-10 and TNF-alpha in monocytes [127,131]. The incubation of monocytes with LPS resulted in TRPM2 mRNA and protein upregulation and ADP-ribose-induced membrane currents [127]. By Trpm2-deficient mice, it was shown that TRPM2 minimizes excessive inflammation by dampening the inflammatory response through cellular depolarization and following reduction of ROS production in phagocytes [132]. The exposition to endotoxins demonstrated augmented inflammatory response and decreased survival compared to wild type mice. There is also good evidence that TRPM2 plays an essential role in ROS-coupled diseases since H2O2-mediated TRPM2 activation is a potential mechanism for pathogenic processes characterized by an increased oxidative microenvironment, including inflammation. Interestingly, its role in aging of immune cells has hardly been explored, although ROS plays a central role in aging theory (reviewed in [133]) and one could imagine TRPM2 physiological or pathophysiological role during age-associated inflammatory responses. The existing Trpm2-knock out mouse studies already provide initial information and coherences about this channel’s role in the development and course of the inflammatory processes because an exacerbating inflammation and age-related upregulation of pro-inflammatory cytokines were not observed in Trpm2-deficient mice at least in the brain [134]. However, more sophisticated studies are required to examine its role in immune cells in the aging context.
TRPM4 is involved in a diversity of physiological processes, including T cells [135], mast cells [136], and dendritic cells [137] activity. It has a profound impact on Ca2+ signaling because Na+ entry depolarizes the plasma membrane to reduce the driving force for calcium entry during SOCE. The Ca2+ induced TRPM4 activation serves as a negative feedback mechanism to prevent toxic Ca2+ overload and fine tunes T cell responses [135,138]. Small interfering RNA-mediated knockdown of Trpm4 amplified Ca2+ entry, NFAT translocation, and IL-2 production in mouse Th2 cells, but it had the opposite effect in Th1 cells [139]. The reasons for the differences are the diverse TRPM4 expression levels as well as different Ca2+ clearance dynamics in subtypes, leading to reduced TCR-mediated Ca2+ influx in Th2 cells [139] and the high sensitivity to FAS-dependent apoptosis in Th1 cells [140]. The deletion of Trpm4 gene impaired antigen- and stem cell factor- induced migration of bone marrow derived mast cells (BMMCs) [136] and chemokine-dependent migration of dendritic cells [137]. In a sepsis model, the ablation of Trpm4 gene decreased phagocytic function and pro-inflammatory cytokine production leading to increased mouse mortality [141]. In BMMC’s, Ca2+ influx via CRAC channels decreases critically after TRPM4 channels depolarize the membrane following adenosine- and FcεRI-stimulation. Accordingly, activated Trpm4−/− BMMCs have amplified degranulation and release excessive amounts of histamine, leukotrienes, and tumor necrosis factor [142]. Furthermore, TRPM4 channel activation is an efficient mechanism for limiting exaggerated antigen-induced mast cell activation that triggers inflammatory and allergic reactions [143].
The selective cation permeable channel TRPM7 with protein serine/threonine kinase activity [144,145,146] has been implicated in numerous physiological functions, including cell survival, proliferation, apoptosis as well as migration, and immune cell function (reviewed in [147,148,149,150,151,152,153]). TRPM7 is essential for T cell development since Trpm7 knock out mice have reduced numbers of T cells due to halting of thymocytes development at the double negative CD4−CD8− stage and resulted in altered chemokine and cytokine expression [154]. Moreover, the T cell specific Trpm7 deletion in vivo resulted in reduced expression of essential growth factors and progressive loss of medullary thymic epithelial cells [154], which regulate T cell development through their function as APC. The impact on proliferation efficiency depends mainly on the type of activation stimuli [155,156]. The channel itself seems to regulate T cell homeostasis by mediating Fas-depending T cell apoptosis through caspase activation [157]. TRPM7 can activate SOCE by phosphorylation of CRAC components leading to reduced SOCE in the absence of TRPM7 [158] and is implicated in receptor-induced Ca2+ release [159]. The positive regulation of SOCE requires the channel kinase activity but not the channel domain itself. The inactivation of TRPM7 kinase activity by introducing the K1646R mutation shows reduced SOCE [155] but normal T cell development except for a reduction in Th17 cell development [156]. The fact that the Treg cells were not affected in this context was very thrilling since both originate from the same precursor cells, and their differentiation requires the involvement of the TGF-β (transforming growth factor ß) [160,161,162,163]. Overall, Th17 cells up-regulate inflammation, while Treg cells have an immunosuppressive function [164,165]. Altered balance of Th17/Treg may play a critical role in the pathogenesis of autoimmune and chronic inflammatory diseases (reviewed in [166]). To ensure an effective immune response, the inflammatory response must be tightly regulated to avoid damage and destruction. However, a characteristic feature of aging and aging-related disease is “inflamm-aging”, associated with immune imbalance and cytokine dysregulation (reviewed in [167,168,169,170]). Several studies already reported a reciprocal connection between pro-inflammatory Th17 and anti-inflammatory Treg cells [162,171] during aging [44,172]. The balance between Th17/Treg and their generation and maintenance are influenced by many factors including TCR and cytokine signaling [166]. The study of Romagnani and colleagues [156] implicated a distinctive defect of small mothers against decapentaplegic family member 2 (SMAD2) signaling in T cells and highlight the role of TRPM7 kinase inhibition in immune homeostasis and in graft-versus-host disease. Although current studies are missing, the existing data provides an excellent foundation to study the involvement of TRPM7 not only in the context of inflammation but also in aging.
5. Potassium Channels
After TCR activation in immune cells, subsequent opening of calcium-activated and voltage gated K+ channels (KV1.3, KCa3.1) mediate K+ influx and hyperpolarization, providing an electrochemical gradient critical for sustained Ca2+ influx (reviewed in [173,174,175]). In T cells of human and mice several K+ channels have been reported and their expression depends on activation and differentiation status. Naïve human and mouse CD4+ and CD8+ T cells, as well as activated central memory T cells (TCM) predominantly express KV1.3 [176,177,178,179,180,181]. Furthermore, T cells from human and mice, upregulate the calcium-activated channel KCa3.1 following T cell activation to maximize Ca2+ influx and proliferation during the re-activation of TN and TCM [177,178,182]. Additionally, the sensitivity to selective blockers of KCa3.1 and KV1.3 differ in TN versus TM because of the different expression levels of these channels [176,177,178,180,183,184,185]. However, mouse TEM up-regulate KCa3.1 instead of KV1.3, like shown in humans and rats. Although in KCa3.1-deficient mice the CD4+ T cell differentiation was not affected but Ca2+ influx and cytokine production in Th1 and Th2 cells were impaired in contrast to Treg and Th17 cells [182]. The results from the KCa3.1−/− mice underlie the role of KCa3.1 function in the activation of CD4 subtypes [182].
Although the T cell homeostasis in humans and mice fundamentally differs [186], it is beyond question that these processes require stable and balanced calcium homeostasis [10]. A block of both KV1.3 and KCa3.1 abolishes Ca2+ oscillations, impacting T cell proliferation [187]. Overall, the pharmacological inhibition of K+ channels reduces Ca2+ influx and decreases cytokine expression profile [182,188,189]. The discovery of immunomodulatory actions [190] by inhibiting KV1.3 channels pave the way for intensive investigations on a therapeutic application in immune-mediated disorders [177,180,191]. Besides, the differentiation of CD8+ T cells into effector cells with cytotoxic ability requires KV1.3 channels. KV1.3 channels gather specifically at the IS between cytotoxic and target cells to modulate the killing process mediated by cytotoxic T lymphocytes [192,193].
Changes in the prevalence of distinct T cell subsets have already been studied extensively [17,62,66,194], however very little is known on functional alterations affecting activation and the underlying molecular mechanisms (not only) in aging. The influence of T cell activation by Ca2+ influx regulated by KV1.3 and IKCa1 potassium channels may alter T cell function during aging [195]. The use of specific inhibitors of KV1.3 and IKCa1, namely margatoxin (MGTX) and triarylmethane-34 (TRAM), reveals a different pattern of Ca2+ influx kinetics dependent on age and T cell subset. High Ca2+ influx observed in CD8+, and Th1 T cells decreased during aging. Surprisingly, the Ca2+ influx in Th2 is similar in all investigated age groups. MGTX inhibitory effect is even more pronounced in Th2 cells, whereas in Th1, the TRAM inhibition remains more potent. Ca2+ influx of CD8+ T cells is inhibited to a similar extent by both applied inhibitors in the two adult groups and does not affect in the elderly. KV1.3 and IKCa1 channel dysfunction, as essential regulators of Ca2+ influx kinetics, is associated with altered function and contribute to age-related changes of T cells [195]. Basically, any ion signaling dysregulation can have severe effects on immune function, leading to (age-related) diseases. During necrosis in the tumor microenvironment, exposure of T cells to high K+ concentrations inhibits TE cell function. The excessive extracellular potassium concentration ([K+]e) leads to increased [K+]i blocking the TCR/Akt/mTOR pathway via phosphatase [196]. The consequence is the inhibition of transcription of genes mediating T cells’ activation response to antigen presentation.
6. CaV Channels, Voltage Gated Channels
T lymphocytes express, among others, the ß3, ß4, and α1 subunit of voltage gated channels CaV1.1, 1.2, 1.3, 1.4, and CaV3.1 [197,198,199,200,201]. The increasing number of publications conducted using mice models provided useful insights of CaV1 channels and subunits in T cell biology and uncovered their role in the activation and survival of T cells [200,201,202,203]. Additionally, the plethora of newly discovered splice variants with altered gating characteristics [199] and partly complete insensitivity to membrane polarization [204] may play a critical role in shaping Cav-dependent Ca2+ signals [205]. Murine CD4+ and CD8+ T cells with a conventional CaV1.4-deficiency showed impaired Ca2+ influx and decreased ERK (extracellular-signal-regulated kinase) and NFAT activation response to TCR stimulation [201]. Furthermore, CaV1.4-deficiency is associated with increased apoptosis and a relative loss of naïve CD44lo T cells in vivo. Upon infection, the number of functional Ag-specific T cells is reduced, shifting towards TM cells phenotype with upregulated activation markers [201], and failed to mount an effective antigen-specific CD8+ T cell response. The lack of ß regulatory subunits in mice models resulted in compromised cytokine production in CD4+ T cells and decreased expression of the CaV1.1 pore-forming units [199] required for TCR-induced Ca2+ entry [198]. The lack of ß3 subunit in CD8 T cells leads to reduced cell numbers due to spontaneous apoptosis mediated by high expression of the Fas receptor [200]. Like the CD4+ T cells, the remaining CD8+ T cells showed activated memory character and defects in TCR-induced Ca2+ signaling and proliferation. Moreover, the lack of ß3 subunit in naïve CD8+ T cells resulted in compromised CaV1.4 protein expression, suggesting that CaV1.4 and β3 may form a Ca2+ channel complex [200]. CaV1.2 and CaV1.3 may play a role in Th2 cell activation since their deletion impaired TCR-induced Ca2+ influx and IL-4 production in vitro and prevented experimental asthma development [202,206]. Finally, the T-type channel CaV3.1-deficiency showed a protective role in EAE (Experimental Autoimmune Encephalomyelitis) mouse model due to reduced cytokine production of granulocyte macrophage colony-stimulating factor (GM-CSF), IL-17A, IL-17F, and IL-21 in Th1 and Th17 cells [197]. Although the overall published data implicate CaV channels’ involvement in Ag-receptor signaling, it is still a pending question how CaV channels work in T cells and in combination with other channels to shape a specific calcium signaling. Unfortunately, momentarily no data are available for CaV channels and function during aging in the immune system. The main reason might be the difficulty to separate the involvement of CaV in aged related defects in the interplay of channels, pumps, and receptors involved in the choreography of Ca2+ signaling in immune cells.
7. Purinergic Receptors
The members of the P2X receptor family are widely expressed among human and mice immune cells. Probably, the best-studied and characterized, not only in T cells, is the P2X7 receptor with an established role in inflammatory and immune responses [207,208]. At this point, it is worthy of mentioning that besides the P2X5 receptor, all other family members can facilitate extracellular adenosine triphosphate (ATP)-mediated Ca2+ entry [209,210].
Upon TCR activation, the increase in mitochondrial activity requires an increase in cytosolic Ca2+ concentration via CRAC channels to raise ATP secretion leading to autocrine activation via P2X receptors. In turn, P2X receptor activation causes a Ca+2 influx, IL-2 production, and proliferation by the activation of NFAT along with an increased expression of the P2RX7 gene [211,212]. ATP release via pannexin-1 hemichannels after TCR activation placed ATP as a mediator in an autocrine feedback loop intensifying T cell stimulation [213] and also helps to sustain P2 receptor signaling as well as NFAT activation [214]. A successful T cell activation requires forming of a stable IS with the antigen-presenting cell [215]. Pannexin-1 hemichannels, P2X1, and P2X4 receptors rapidly translocate to the IS after TCR stimulation facilitating ATP release and autocrine feedback mechanism, while P2X7 receptors remain uniformly distributed [216]. It implicates that P2X receptor subtypes may fulfill different functions in different steps during T cell activation. The colocalization with STIM1 and Orai1 enhances Ca2+ entry at the IS [216] which is necessary during weak TCR stimulation and supportive in antigen scanning or for the formation of Ca2+ microdomains.
Besides the autocrine signaling during T cell activation, paracrine effects have been observed. Extracellular ATP, as a danger signaling molecule during inflammation and injury, activates the innate immune system and mediates chronic pain through P2X7 receptors [217,218]. However, the latest work showed the unsuspected but critical involvement of P2X7 in generating resilient, long-lived central and tissue-resident memory CD8+ T cells (CD62L+) supporting adaptive immune system memory [212]. The induction of adenosine monophosphate (AMP)-activated protein kinase, metabolic reprogramming and mitochondrial maintenance promotes TCM’s metabolic fitness while TEM generation is much less affected [212]. On the other hand, P2X7 receptor inhibition supports CD4+ T cells’ differentiation into Tregs [219]. Furthermore, P2X7 and P2X4 receptors play an essential role in non-conventional γδ T cell differentiation and cytokine production mediated by amplification of TCR-mediated Ca2+ signaling [220,221]. Application of extracellular ATP on P2RX7-deficient T cells prevents shedding of CD62L (L-selectin) [222]. Furthermore, P2X7 receptor seems to be essential for ATP-induced shedding of CD23, CD27, and IL-6R mediated by metalloproteases and converts the membrane proteins into soluble effector proteins [222,223,224,225]. Additionally, ATP concentration seems to determine T cells’ fate, whether to keep them in a resting state, becomes activated, or undergo apoptosis [226]. In mature T cells, the P2X7 receptor is essential for the induction of apoptosis by ATP [227] and nicotinamide adenine dinucleotide (NAD) [228]. One another fascinating paracrine ATP-function is the influence of the P2X7 and X4 receptors on the migration or motility of T cells [229]. In lymph nodes, ATP-release from activated T cells reduces bystander T cells’ motility to support scanning of resident dendritic cells for better antigen recognition.
Although the field of purinergic receptors now contains quite a lot of substantial publications, the role of P2 receptors during immune system aging is still under investigation. Supportive data of a direct dysregulation on the receptors itself is missing. However, the evidence supports that alterations in the purinergic signaling pathways occur during aging. The survival of TE cells, their specific cytotoxic competence, activity, and necessary transition into TM cells is a critical step of the recall immune response strongly affected during aging. Additionally, there is evidence that changes in purinergic signaling pathways mediated by nucleotides influence inflammatory processes [230,231].
Two important enzymes in purinergic signaling are the ectoenzymes ectonucleoside triphosphate diphosphohydrolase-1 (NTPDase1, CD39) and ecto-5′-nucleotidase (CD73). They are expressed on endothelial and immune cells and play a central role in inflammation [232,233,234] and tumor immunity [235,236,237]. Severe P2 receptor-mediated stimulation of endothelium, lymphocytes, and monocytes might cause a pro-inflammatory environment [238]. The activation of the P2X7 receptor inhibits Tregs’ immunosuppressive potential and induces their conversion to Th17 effector cells in vivo during inflammation by increasing ATP levels via IL-6 [219,239,240]. The CD39/CD73 pathway counteracts through the degradation of excessive ATP levels into adenosine, leading to a more anti-inflammatory environment [241]. Interestingly Fang and colleagues identified CD39 as a cell surface marker for short-lived CD4+ effector T cells [242]. Furthermore, CD39 has been reported on exhausted CD8+ T cells [243] and CD8+ TM cells are more prone to express CD39 than CD4+ T cells [244]. In mice, they may function as an integral component of T cells’ suppressive machinery, as Tregs express CD39 and CD73 [245] and impact Th17 cell generation [246]. Increased induction of CD39 with age on human CD4+ T cells correlates with increased apoptosis after antigen encounter [242] and reduced generation of long-lived TM cells in vaccine response. In agreement with the in vitro observations, individuals with CD39 polymorphisms [247] show higher efficiency to vaccination [242]. Still, the follicular helper T (Tfh) cells and the survival as TM cells are compromised, leading to vaccination’s inefficacy. Overall the increased CD39 expression with age resulting in reduced ATP concentration and preventing signaling through P2X receptors will lead to higher apoptosis susceptibility and preferential generation of short-lived effector T cells [242].
Opposite to CD4+ T cells, isolated CD8+ T cells are overall relatively resistant to extracellular ATP [228,248,249]. Mellouk and colleague recently investigated the ATP-sensitivity of CD8+ T cells (TN, TCM/EM) isolated from secondary lymphoid organs during aging [250]. They identified a CD44hiCD45RBhi phenotype within the aged CD8+ T cell populations with total resistance to apoptosis induced by ATP in contrast to CD4+ T of the same age. Thus, their cytotoxic activity might be maintained even in inflammatory tissues where high ATP concentration is a common phenomenon [251], and the CD44hiCD45RBlo phenotype will probably undergo apoptosis. The level of P2X7 receptor expression is upregulated on TCM/EM CD44hiCD45RBlo cells compared to TN, but low on CD44hiCD45RBhi T cells and unaffected with aging. Furthermore, the P2X7RloCD44hiCD45RBhi phenotype with aging is entirely resistant to ATP-mediated channel formation and Ca2+ influx. The data suggest a minor role of the ATP/P2X7 receptor pathway in CD8+ T cell activation and differentiation during secondary immune responses [250].
8. Perspectives
Through increasing numbers of studies, it becomes more evident that ion signal transduction changes appear to have a strong influence on the development of age-associated diseases. Knowing the significant role of Ca2+ signaling for immune cells, a better understanding of ion channel and receptor biology is essential for the development of effective and targeted treatment strategies. Especially the ongoing SARS-CoV-2 pandemic reminds us how essential a functioning immune system is—the risk for severe illness with COVID-19 increases with age. The remodeled immune system of the elderly, with less naïve T cells, dysfunctional memory cells, and altered innate immune response, leads to greater susceptibility to infectious disease. Moreover, vaccinations do not always seem to provide sufficient immunity for the elderly, with less immunogenicity and effectiveness in the elderly than younger individuals [252]. Since immunological memory is the basis of vaccination, it is essential to understand the different T cell subsets’ changes during aging. T cells are extremely heterogeneous in terms of longevity, phenotype, distribution, and function, and the changes brought about by aging increase this complexity even further. Additionally, changes in the abundance and functionality of the Ca2+ and K+ channels may contribute to altered Ca2+ homeostasis in T cell subsets during aging (Figure 1). Therefore, more profound understanding of the dysregulation of ion channels contributing to the altered ion signaling transduction in immune cells with age is indispensable. More detailed and sophisticated studies are necessary to place channel and receptor dysfunction as a possible hallmark of aging.
Funding
We acknowledge support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) and Saarland University within the funding programme Open Access Publishing. This work was supported by the DFG by LI 1750/4-2 (to A.L.).
Acknowledgments
We are very thankful to Markus Hoth and Sandra Janku for helpful discussion and critical reading of the manuscript. We apologize to all colleagues whose papers are not cited because of space limitations or because we overlooked them.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grubeck-Loebenstein, B.; Berger, P.; Saurwein-Teissl, M.; Zisterer, K.; Wick, G. No immunity for the elderly. Nat. Med. 1998, 4, 870. [Google Scholar] [CrossRef]
- Weinberger, B.; Herndler-Brandstetter, D.; Schwanninger, A.; Weiskopf, D.; Grubeck-Loebenstein, B. Biology of immune responses to vaccines in elderly persons. Clin. Infect. Dis. 2008, 46, 1078–1084. [Google Scholar] [CrossRef]
- Nikolich-Zugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Goodwin, K.; Viboud, C.; Simonsen, L. Antibody response to influenza vaccination in the elderly: A quantitative review. Vaccine 2006, 24, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Weyand, C.M. Immune aging and autoimmunity. Cell Mol. Life Sci. 2012, 69, 1615–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.A. Effect of aging on T lymphocyte activation. Vaccine 2000, 18, 1654–1660. [Google Scholar] [CrossRef]
- Haynes, L.; Swain, S.L. Aged-related shifts in T cell homeostasis lead to intrinsic T cell defects. Semin. Immunol. 2012, 24, 350–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolich-Zugich, J.; Li, G.; Uhrlaub, J.L.; Renkema, K.R.; Smithey, M.J. Age-related changes in CD8 T cell homeostasis and immunity to infection. Semin. Immunol. 2012, 24, 356–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [Green Version]
- Trebak, M.; Kinet, J.P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef]
- Srikanth, S.; Woo, J.S.; Sun, Z.; Gwack, Y. Immunological Disorders: Regulation of Ca(2+) Signaling in T Lymphocytes. Adv. Exp. Med. Biol. 2017, 993, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Skolnik, E.Y.; Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 2012, 12, 532–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh-Hora, M.; Rao, A. Calcium signaling in lymphocytes. Curr. Opin. Immunol. 2008, 20, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, J.S.; Boursalian, T.E.; Turk, G.L.; Fink, P.J. Thymic output in aged mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8447–8452. [Google Scholar] [CrossRef] [Green Version]
- Rezzani, R.; Nardo, L.; Favero, G.; Peroni, M.; Rodella, L.F. Thymus and aging: Morphological, radiological, and functional overview. Age 2014, 36, 313–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, D.B. The effect of age on thymic function. Front. Immunol. 2013, 4, 316. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Yao, D.; Zeng, X.; Kasakovski, D.; Zhang, Y.; Chen, S.; Zha, X.; Li, Y.; Xu, L. Age related human T cell subset evolution and senescence. Immun. Ageing 2019, 16, 24. [Google Scholar] [CrossRef] [Green Version]
- Qi, Q.; Liu, Y.; Cheng, Y.; Glanville, J.; Zhang, D.; Lee, J.Y.; Olshen, R.A.; Weyand, C.M.; Boyd, S.D.; Goronzy, J.J. Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. USA 2014, 111, 13139–13144. [Google Scholar] [CrossRef] [Green Version]
- Egorov, E.S.; Kasatskaya, S.A.; Zubov, V.N.; Izraelson, M.; Nakonechnaya, T.O.; Staroverov, D.B.; Angius, A.; Cucca, F.; Mamedov, I.Z.; Rosati, E.; et al. The Changing Landscape of Naive T Cell Receptor Repertoire With Human Aging. Front. Immunol. 2018, 9, 1618. [Google Scholar] [CrossRef] [Green Version]
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z.; et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 2014, 192, 2689–2698. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.D.; Emerson, S.G.; Punt, J.; Goff, W.D. Decreased Naive T-cell Production Leading to Cytokine Storm as Cause of Increased COVID-19 Severity with Comorbidities. Aging Dis. 2020, 11, 742–745. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.L.; Patel, R.K.; Reynaldi, A.; Grenier, J.K.; Wang, J.; Watson, N.B.; Nzingha, K.; Yee Mon, K.J.; Peng, S.A.; Grimson, A.; et al. Developmental Origin Governs CD8(+) T Cell Fate Decisions during Infection. Cell 2018, 174, 117–130.e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynaldi, A.; Smith, N.L.; Schlub, T.E.; Tabilas, C.; Venturi, V.; Rudd, B.D.; Davenport, M.P. Fate mapping reveals the age structure of the peripheral T cell compartment. Proc. Natl. Acad. Sci. USA 2019, 116, 3974–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitz, D.M.; Zhang, D.W.; Hu, B.; Le Saux, S.; Yanes, R.E.; Ye, Z.; Buenrostro, J.D.; Weyand, C.M.; Greenleaf, W.J.; Goronzy, J.J. Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol. 2017, 2, eaag0192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rane, S.; Hogan, T.; Seddon, B.; Yates, A.J. Age is not just a number: Naive T cells increase their ability to persist in the circulation over time. PLoS Biol. 2018, 16, e2003949. [Google Scholar] [CrossRef]
- Hu, B.; Li, G.; Ye, Z.; Gustafson, C.E.; Tian, L.; Weyand, C.M.; Goronzy, J.J. Transcription factor networks in aged naive CD4 T cells bias lineage differentiation. Aging Cell 2019, 18, e12957. [Google Scholar] [CrossRef] [PubMed]
- Quinn, K.M.; Fox, A.; Harland, K.L.; Russ, B.E.; Li, J.; Nguyen, T.H.O.; Loh, L.; Olshanksy, M.; Naeem, H.; Tsyganov, K.; et al. Age-Related Decline in Primary CD8(+) T Cell Responses Is Associated with the Development of Senescence in Virtual Memory CD8(+) T Cells. Cell Rep. 2018, 23, 3512–3524. [Google Scholar] [CrossRef]
- White, J.T.; Cross, E.W.; Burchill, M.A.; Danhorn, T.; McCarter, M.D.; Rosen, H.R.; O’Connor, B.; Kedl, R.M. Virtual memory T cells develop and mediate bystander protective immunity in an IL-15-dependent manner. Nat. Commun. 2016, 7, 11291. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Yu, M.; Lee, W.W.; Tsang, M.; Krishnan, E.; Weyand, C.M.; Goronzy, J.J. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat. Med. 2012, 18, 1518–1524. [Google Scholar] [CrossRef]
- Ye, Z.; Li, G.; Kim, C.; Hu, B.; Jadhav, R.R.; Weyand, C.M.; Goronzy, J.J. Regulation of miR-181a expression in T cell aging. Nat. Commun. 2018, 9, 3060. [Google Scholar] [CrossRef]
- Gustafson, C.E.; Cavanagh, M.M.; Jin, J.; Weyand, C.M.; Goronzy, J.J. Functional pathways regulated by microRNA networks in CD8 T-cell aging. Aging Cell 2019, 18, e12879. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Jadhav, R.R.; Gustafson, C.E.; Smithey, M.J.; Hirsch, A.J.; Uhrlaub, J.L.; Hildebrand, W.H.; Nikolich-Zugich, J.; Weyand, C.M.; Goronzy, J.J. Defects in Antiviral T Cell Responses Inflicted by Aging-Associated miR-181a Deficiency. Cell Rep. 2019, 29, 2202–2216.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Hu, B.; Jadhav, R.R.; Jin, J.; Zhang, H.; Cavanagh, M.M.; Akondy, R.S.; Ahmed, R.; Weyand, C.M.; Goronzy, J.J. Activation of miR-21-Regulated Pathways in Immune Aging Selects against Signatures Characteristic of Memory T Cells. Cell Rep. 2018, 25, 2148–2162.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, Y.; Minato, N.; Hattori, M. The impact of senescence-associated T cells on immunosenescence and age-related disorders. Inflamm. Regen. 2018, 38, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goronzy, J.J.; Weyand, C.M. Understanding immunosenescence to improve responses to vaccines. Nat. Immunol. 2013, 14, 428–436. [Google Scholar] [CrossRef] [Green Version]
- Luz Correa, B.; Ornaghi, A.P.; Cerutti Muller, G.; Engroff, P.; Pestana Lopes, R.; Gomes da Silva Filho, I.; Bosch, J.A.; Bonorino, C.; Bauer, M.E. The inverted CD4:CD8 ratio is associated with cytomegalovirus, poor cognitive and functional states in older adults. Neuroimmunomodulation 2014, 21, 206–212. [Google Scholar] [CrossRef]
- McBride, J.A.; Striker, R. Imbalance in the game of T cells: What can the CD4/CD8 T-cell ratio tell us about HIV and health? PLoS Pathog. 2017, 13, e1006624. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Mehraj, V.; Vyboh, K.; Cao, W.; Li, T.; Routy, J.P. CD4:CD8 ratio as a frontier marker for clinical outcome, immune dysfunction and viral reservoir size in virologically suppressed HIV-positive patients. J. Int AIDS Soc. 2015, 18, 20052. [Google Scholar] [CrossRef]
- Czesnikiewicz-Guzik, M.; Lee, W.W.; Cui, D.; Hiruma, Y.; Lamar, D.L.; Yang, Z.Z.; Ouslander, J.G.; Weyand, C.M.; Goronzy, J.J. T cell subset-specific susceptibility to aging. Clin. Immunol. 2008, 127, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.C.; Liao, J.J.; Bonasera, S.; Longo, D.L.; Goetzl, E.J. Nuclear factor-kappaB-dependent reversal of aging-induced alterations in T cell cytokines. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 2142–2150. [Google Scholar] [CrossRef]
- Sakata-Kaneko, S.; Wakatsuki, Y.; Matsunaga, Y.; Usui, T.; Kita, T. Altered Th1/Th2 commitment in human CD4+ T cells with ageing. Clin. Exp. Immunol. 2000, 120, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Dayan, M.; Segal, R.; Globerson, A.; Habut, B.; Shearer, G.M.; Mozes, E. Effect of aging on cytokine production in normal and experimental systemic lupus erythematosus-afflicted mice. Exp. Gerontol. 2000, 35, 225–236. [Google Scholar] [CrossRef]
- Haynes, L.; Maue, A.C. Effects of aging on T cell function. Curr. Opin. Immunol. 2009, 21, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, V.; Rink, L.; Uciechowski, P. The Th17/Treg balance is disturbed during aging. Exp. Gerontol. 2013, 48, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Schindowski, K.; Frohlich, L.; Maurer, K.; Muller, W.E.; Eckert, A. Age-related impairment of human T lymphocytes’ activation: Specific differences between CD4(+) and CD8(+) subsets. Mech. Ageing Dev. 2002, 123, 375–390. [Google Scholar] [CrossRef] [Green Version]
- Gardner, E.M.; Murasko, D.M. Age-related changes in Type 1 and Type 2 cytokine production in humans. Biogerontology 2002, 3, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Zanni, F.; Vescovini, R.; Biasini, C.; Fagnoni, F.; Zanlari, L.; Telera, A.; Di Pede, P.; Passeri, G.; Pedrazzoni, M.; Passeri, M.; et al. Marked increase with age of type 1 cytokines within memory and effector/cytotoxic CD8+ T cells in humans: A contribution to understand the relationship between inflammation and immunosenescence. Exp. Gerontol. 2003, 38, 981–987. [Google Scholar] [CrossRef]
- Engwerda, C.R.; Fox, B.S.; Handwerger, B.S. Cytokine production by T lymphocytes from young and aged mice. J. Immunol. 1996, 156, 3621–3630. [Google Scholar]
- Ernst, D.N.; Weigle, W.O.; Noonan, D.J.; McQuitty, D.N.; Hobbs, M.V. The age-associated increase in IFN-gamma synthesis by mouse CD8+ T cells correlates with shifts in the frequencies of cell subsets defined by membrane CD44, CD45RB, 3G11, and MEL-14 expression. J. Immunol. 1993, 151, 575–587. [Google Scholar]
- Haynes, L.; Linton, P.J.; Eaton, S.M.; Tonkonogy, S.L.; Swain, S.L. Interleukin 2, but not other common gamma chain-binding cytokines, can reverse the defect in generation of CD4 effector T cells from naive T cells of aged mice. J. Exp. Med. 1999, 190, 1013–1024. [Google Scholar] [CrossRef]
- Adolfsson, O.; Huber, B.T.; Meydani, S.N. Vitamin E-enhanced IL-2 production in old mice: Naive but not memory T cells show increased cell division cycling and IL-2-producing capacity. J. Immunol. 2001, 167, 3809–3817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whisler, R.L.; Beiqing, L.; Chen, M. Age-related decreases in IL-2 production by human T cells are associated with impaired activation of nuclear transcriptional factors AP-1 and NF-AT. Cell. Immunol. 1996, 169, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Pieren, D.K.J.; Smits, N.A.M.; van de Garde, M.D.B.; Guichelaar, T. Response kinetics reveal novel features of ageing in murine T cells. Sci. Rep. 2019, 9, 5587. [Google Scholar] [CrossRef] [Green Version]
- Linton, P.J.; Haynes, L.; Klinman, N.R.; Swain, S.L. Antigen-independent changes in naive CD4 T cells with aging. J. Exp. Med. 1996, 184, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T., Jr.; Gagne, D.; Goulet, A.C.; Desgeorges, S.; Lacombe, G.; Arcand, M.; Dupuis, G. Age-related impairment of p56lck and ZAP-70 activities in human T lymphocytes activated through the TcR/CD3 complex. Exp. Gerontol. 1999, 34, 197–216. [Google Scholar] [CrossRef]
- Tamura, T.; Kunimatsu, T.; Yee, S.T.; Igarashi, O.; Utsuyama, M.; Tanaka, S.; Miyazaki, S.; Hirokawa, K.; Nariuchi, H. Molecular mechanism of the impairment in activation signal transduction in CD4(+) T cells from old mice. Int. Immunol. 2000, 12, 1205–1215. [Google Scholar] [CrossRef] [Green Version]
- Gearing, A.J.; Wadhwa, M.; Perris, A.D. Interleukin 2 stimulates T cell proliferation using a calcium flux. Immunol. Lett. 1985, 10, 297–302. [Google Scholar] [CrossRef]
- Hogan, P.G. Calcium-NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 2017, 63, 66–69. [Google Scholar] [CrossRef] [Green Version]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar] [CrossRef]
- Fagnoni, F.F.; Vescovini, R.; Mazzola, M.; Bologna, G.; Nigro, E.; Lavagetto, G.; Franceschi, C.; Passeri, M.; Sansoni, P. Expansion of cytotoxic CD8+ CD28- T cells in healthy ageing people, including centenarians. Immunology 1996, 88, 501–507. [Google Scholar] [CrossRef]
- Herndler-Brandstetter, D.; Landgraf, K.; Tzankov, A.; Jenewein, B.; Brunauer, R.; Laschober, G.T.; Parson, W.; Kloss, F.; Gassner, R.; Lepperdinger, G.; et al. The impact of aging on memory T cell phenotype and function in the human bone marrow. J. Leukoc. Biol. 2012, 91, 197–205. [Google Scholar] [CrossRef]
- Westmeier, J.; Paniskaki, K.; Karakose, Z.; Werner, T.; Sutter, K.; Dolff, S.; Overbeck, M.; Limmer, A.; Liu, J.; Zheng, X.; et al. Impaired Cytotoxic CD8(+) T Cell Response in Elderly COVID-19 Patients. mBio 2020, 11. [Google Scholar] [CrossRef]
- Miller, R.A.; Berger, S.B.; Burke, D.T.; Galecki, A.; Garcia, G.G.; Harper, J.M.; Sadighi Akha, A.A. T cells in aging mice: Genetic, developmental, and biochemical analyses. Immunol. Rev. 2005, 205, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G.G.; Miller, R.A. Single-cell analyses reveal two defects in peptide-specific activation of naive T cells from aged mice. J. Immunol. 2001, 166, 3151–3157. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G.G.; Miller, R.A. Age-related defects in CD4+ T cell activation reversed by glycoprotein endopeptidase. Eur. J. Immunol. 2003, 33, 3464–3472. [Google Scholar] [CrossRef]
- Angenendt, A.; Steiner, R.; Knorck, A.; Schwar, G.; Konrad, M.; Krause, E.; Lis, A. Orai, STIM, and PMCA contribute to reduced calcium signal generation in CD8(+) T cells of elderly mice. Aging 2020, 12, 3266. [Google Scholar] [CrossRef]
- Hartmann, H.; Eckert, A.; Forstl, H.; Muller, W.E. Similar age-related changes of free intracellular calcium in lymphocytes and central neurons: Effects of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1994, 243, 218–223. [Google Scholar] [CrossRef]
- Miller, R.A.; Jacobson, B.; Weil, G.; Simons, E.R. Diminished calcium influx in lectin-stimulated T cells from old mice. J. Cell Physiol. 1987, 132, 337–342. [Google Scholar] [CrossRef]
- Grossmann, A.; Maggio-Price, L.; Jinneman, J.C.; Rabinovitch, P.S. Influence of aging on intracellular free calcium and proliferation of mouse T-cell subsets from various lymphoid organs. Cell Immunol. 1991, 135, 118–131. [Google Scholar] [CrossRef]
- Dobber, R.; Tielemans, M.; de Weerd, H.; Nagelkerken, L. Mel14+ CD4+ T cells from aged mice display functional and phenotypic characteristics of memory cells. Int. Immunol. 1994, 6, 1227–1234. [Google Scholar] [CrossRef]
- Rajasekar, R.; Augustin, A. Antigen-dependent selection of T cells that are able to efficiently regulate free cytoplasmic Ca2+ levels. J. Immunol. 1994, 153, 1037–1045. [Google Scholar]
- Fulop, T., Jr.; Seres, I. Age-related changes in signal transduction. Implications for neuronal transmission and potential for drug intervention. Drugs Aging 1994, 5, 366–390. [Google Scholar] [CrossRef]
- Gupta, S. Membrane signal transduction in T cells in aging humans. Ann. N. Y. Acad. Sci. 1989, 568, 277–282. [Google Scholar] [CrossRef]
- Proust, J.J.; Filburn, C.R.; Harrison, S.A.; Buchholz, M.A.; Nordin, A.A. Age-related defect in signal transduction during lectin activation of murine T lymphocytes. J. Immunol. 1987, 139, 1472–1478. [Google Scholar]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef]
- Walter, M.C.; Rossius, M.; Zitzelsberger, M.; Vorgerd, M.; Muller-Felber, W.; Ertl-Wagner, B.; Zhang, Y.; Brinkmeier, H.; Senderek, J.; Schoser, B. 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation. Neuromuscul. Disord. 2015, 25, 577–584. [Google Scholar] [CrossRef]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [Green Version]
- Schuhmann, M.K.; Stegner, D.; Berna-Erro, A.; Bittner, S.; Braun, A.; Kleinschnitz, C.; Stoll, G.; Wiendl, H.; Meuth, S.G.; Nieswandt, B. Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J. Immunol. 2010, 184, 1536–1542. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, U.; Shaw, P.J.; Kozhaya, L.; Subramanian, R.; Gaida, K.; Unutmaz, D.; McBride, H.J.; Feske, S. Selective ORAI1 Inhibition Ameliorates Autoimmune Central Nervous System Inflammation by Suppressing Effector but Not Regulatory T Cell Function. J. Immunol. 2016, 196, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Bose, T.; Cieslar-Pobuda, A.; Wiechec, E. Role of ion channels in regulating Ca(2)(+) homeostasis during the interplay between immune and cancer cells. Cell Death Dis. 2015, 6, e1648. [Google Scholar] [CrossRef] [Green Version]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [Green Version]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Putney, J.W., Jr. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J. Biol. Chem. 2007, 282, 17548–17556. [Google Scholar] [CrossRef] [Green Version]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. 2007, 17, 794–800. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.T.; Manji, S.S.; Parker, N.J.; Hancock, M.S.; Van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R.; et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: Coding for a novel class of transmembrane proteins. Biochem. J. 2001, 357, 673–685. [Google Scholar] [CrossRef]
- Miederer, A.M.; Alansary, D.; Schwar, G.; Lee, P.H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 2015, 6, 6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjo, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669. [Google Scholar] [CrossRef] [Green Version]
- Bergmeier, W.; Weidinger, C.; Zee, I.; Feske, S. Emerging roles of store-operated Ca(2)(+) entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels 2013, 7, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, E.C.; Qu, B.; Hoth, M. Calcium, cancer and killing: The role of calcium in killing cancer cells by cytotoxic T lymphocytes and natural killer cells. Biochim. Biophys. Acta 2013, 1833, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Lioudyno, M.I.; Kozak, J.A.; Penna, A.; Safrina, O.; Zhang, S.L.; Sen, D.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc. Natl. Acad. Sci. USA 2008, 105, 2011–2016. [Google Scholar] [CrossRef] [Green Version]
- Quintana, A.; Schwindling, C.; Wenning, A.S.; Becherer, U.; Rettig, J.; Schwarz, E.C.; Hoth, M. T cell activation requires mitochondrial translocation to the immunological synapse. Proc. Natl. Acad. Sci. USA 2007, 104, 14418–14423. [Google Scholar] [CrossRef] [Green Version]
- Quintana, A.; Pasche, M.; Junker, C.; Al-Ansary, D.; Rieger, H.; Kummerow, C.; Nunez, L.; Villalobos, C.; Meraner, P.; Becherer, U.; et al. Calcium microdomains at the immunological synapse: How ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. Embo J. 2011, 30, 3895–3912. [Google Scholar] [CrossRef] [Green Version]
- Barr, V.A.; Bernot, K.M.; Srikanth, S.; Gwack, Y.; Balagopalan, L.; Regan, C.K.; Helman, D.J.; Sommers, C.L.; Oh-Hora, M.; Rao, A.; et al. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: Puncta and distal caps. Mol. Biol. Cell 2008, 19, 2802–2817. [Google Scholar] [CrossRef]
- Yen, M.; Lewis, R.S. Numbers count: How STIM and Orai stoichiometry affect store-operated calcium entry. Cell Calcium 2019, 79, 35–43. [Google Scholar] [CrossRef]
- Fahrner, M.; Schindl, R.; Romanin, C. Studies of Structure-Function and Subunit Composition of Orai/STIM Channel. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2018; pp. 25–50. [Google Scholar] [CrossRef]
- Lewis, R.S. Store-Operated Calcium Channels: From Function to Structure and Back Again. Cold Spring Harb Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Srikanth, S.; Oh-Hora, M.; Hogan, P.G.; Lamperti, E.D.; Yamashita, M.; Gelinas, C.; Neems, D.S.; Sasaki, Y.; Feske, S.; et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol. Cell Biol. 2008, 28, 5209–5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh-Hora, M.; Yamashita, M.; Hogan, P.G.; Sharma, S.; Lamperti, E.; Chung, W.; Prakriya, M.; Feske, S.; Rao, A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 2008, 9, 432–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.D.; Srikanth, S.; Yee, M.K.; Mock, D.C.; Lawson, G.W.; Gwack, Y. ORAI1 deficiency impairs activated T cell death and enhances T cell survival. J. Immunol. 2011, 187, 3620–3630. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, C.; Shaw, P.J.; Feske, S. STIM1 and STIM2-mediated Ca(2+) influx regulates antitumour immunity by CD8(+) T cells. EMBO Mol. Med. 2013, 5, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Weidinger, C.; Vaeth, M.; Luethy, K.; Kaech, S.M.; Feske, S. CD4(+) and CD8(+) T cell-dependent antiviral immunity requires STIM1 and STIM2. J. Clin. Investig. 2014, 124, 4549–4563. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Friedmann, K.S.; Lyrmann, H.; Zhou, Y.; Schoppmeyer, R.; Knorck, A.; Mang, S.; Hoxha, C.; Angenendt, A.; Backes, C.S.; et al. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. J. Physiol. 2018, 596, 2681–2698. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Oh-hora, M.; Rao, A. The calcium/NFAT pathway: Role in development and function of regulatory T cells. Microbes Infect. 2009, 11, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarl, C.A.; Picard, C.; Khalil, S.; Kawasaki, T.; Rother, J.; Papolos, A.; Kutok, J.; Hivroz, C.; Ledeist, F.; Plogmann, K.; et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. 2009, 124, 1311–1318.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, S.; Rensing-Ehl, A.; Speckmann, C.; Bengsch, B.; Schmitt-Graeff, A.; Bondzio, I.; Maul-Pavicic, A.; Bass, T.; Vraetz, T.; Strahm, B.; et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J. Immunol. 2012, 188, 1523–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S. ORAI1 and STIM1 deficiency in human and mice: Roles of store-operated Ca2+ entry in the immune system and beyond. Immunol. Rev. 2009, 231, 189–209. [Google Scholar] [CrossRef]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Karoly Jani, P.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [Green Version]
- Kircher, S.; Merino-Wong, M.; Niemeyer, B.A.; Alansary, D. Profiling calcium signals of in vitro polarized human effector CD4(+) T cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 932–943. [Google Scholar] [CrossRef]
- Bertin, S.; Aoki-Nonaka, Y.; de Jong, P.R.; Nohara, L.L.; Xu, H.; Stanwood, S.R.; Srikanth, S.; Lee, J.; To, K.; Abramson, L.; et al. The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4(+) T cells. Nat. Immunol. 2014, 15, 1055–1063. [Google Scholar] [CrossRef]
- Majhi, R.K.; Sahoo, S.S.; Yadav, M.; Pratheek, B.M.; Chattopadhyay, S.; Goswami, C. Functional expression of TRPV channels in T cells and their implications in immune regulation. FEBS J. 2015, 282, 2661–2681. [Google Scholar] [CrossRef]
- Ferioli, S.; Zierler, S.; Zaisserer, J.; Schredelseker, J.; Gudermann, T.; Chubanov, V. TRPM6 and TRPM7 differentially contribute to the relief of heteromeric TRPM6/7 channels from inhibition by cytosolic Mg(2+) and Mg.ATP. Sci. Rep. 2017, 7, 8806. [Google Scholar] [CrossRef]
- Li, M.; Jiang, J.; Yue, L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philipp, S.; Strauss, B.; Hirnet, D.; Wissenbach, U.; Mery, L.; Flockerzi, V.; Hoth, M. TRPC3 mediates T-cell receptor-dependent calcium entry in human T-lymphocytes. J. Biol. Chem. 2003, 278, 26629–26638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenning, A.S.; Neblung, K.; Strauss, B.; Wolfs, M.J.; Sappok, A.; Hoth, M.; Schwarz, E.C. TRP expression pattern and the functional importance of TRPC3 in primary human T-cells. Biochim. Biophys. Acta 2011, 1813, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lu, Z.H.; Gabius, H.J.; Rohowsky-Kochan, C.; Ledeen, R.W.; Wu, G. Cross-linking of GM1 ganglioside by galectin-1 mediates regulatory T cell activity involving TRPC5 channel activation: Possible role in suppressing experimental autoimmune encephalomyelitis. J. Immunol. 2009, 182, 4036–4045. [Google Scholar] [CrossRef] [Green Version]
- Syed Mortadza, S.A.; Wang, L.; Li, D.; Jiang, L.H. TRPM2 Channel-Mediated ROS-Sensitive Ca(2+) Signaling Mechanisms in Immune Cells. Front. Immunol. 2015, 6, 407. [Google Scholar] [CrossRef] [Green Version]
- Wehrhahn, J.; Kraft, R.; Harteneck, C.; Hauschildt, S. Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J. Immunol. 2010, 184, 2386–2393. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Kolisek, M.; Bagley, L.A.; Fleig, A.; Penner, R. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 962–964. [Google Scholar] [CrossRef] [Green Version]
- Guse, A.H.; da Silva, C.P.; Berg, I.; Skapenko, A.L.; Weber, K.; Heyer, P.; Hohenegger, M.; Ashamu, G.A.; Schulze-Koops, H.; Potter, B.V.; et al. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature 1999, 398, 70–73. [Google Scholar] [CrossRef]
- Melzer, N.; Hicking, G.; Gobel, K.; Wiendl, H. TRPM2 cation channels modulate T cell effector functions and contribute to autoimmune CNS inflammation. PLoS ONE 2012, 7, e47617. [Google Scholar] [CrossRef]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef]
- Di, A.; Gao, X.P.; Qian, F.; Kawamura, T.; Han, J.; Hecquet, C.; Ye, R.D.; Vogel, S.M.; Malik, A.B. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 2011, 13, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakae, M.; Miyanohara, J.; Morishima, M.; Nagayasu, K.; Mori, Y.; Shirakawa, H.; Kaneko, S. Pathophysiological Role of TRPM2 in Age-Related Cognitive Impairment in Mice. Neuroscience 2019, 408, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Launay, P.; Cheng, H.; Srivatsan, S.; Penner, R.; Fleig, A.; Kinet, J.P. TRPM4 regulates calcium oscillations after T cell activation. Science 2004, 306, 1374–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Owsianik, G.; Freichel, M.; Flockerzi, V.; Nilius, B.; Vennekens, R. TRPM4 regulates migration of mast cells in mice. Cell Calcium 2009, 45, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Barbet, G.; Demion, M.; Moura, I.C.; Serafini, N.; Leger, T.; Vrtovsnik, F.; Monteiro, R.C.; Guinamard, R.; Kinet, J.P.; Launay, P. The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat. Immunol. 2008, 9, 1148–1156. [Google Scholar] [CrossRef] [Green Version]
- Launay, P.; Fleig, A.; Perraud, A.L.; Scharenberg, A.M.; Penner, R.; Kinet, J.P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.S.; Hildner, K.; Murphy, K.M.; Allen, P.M. Trpm4 differentially regulates Th1 and Th2 function by altering calcium signaling and NFAT localization. J. Immunol. 2010, 185, 2836–2846. [Google Scholar] [CrossRef]
- Zhang, X.; Brunner, T.; Carter, L.; Dutton, R.W.; Rogers, P.; Bradley, L.; Sato, T.; Reed, J.C.; Green, D.; Swain, S.L. Unequal death in T helper cell (Th)1 and Th2 effectors: Th1, but not Th2, effectors undergo rapid Fas/FasL-mediated apoptosis. J. Exp. Med. 1997, 185, 1837–1849. [Google Scholar] [CrossRef]
- Serafini, N.; Dahdah, A.; Barbet, G.; Demion, M.; Attout, T.; Gautier, G.; Arcos-Fajardo, M.; Souchet, H.; Jouvin, M.H.; Vrtovsnik, F.; et al. The TRPM4 channel controls monocyte and macrophage, but not neutrophil, function for survival in sepsis. J. Immunol. 2012, 189, 3689–3699. [Google Scholar] [CrossRef]
- Vennekens, R.; Olausson, J.; Meissner, M.; Bloch, W.; Mathar, I.; Philipp, S.E.; Schmitz, F.; Weissgerber, P.; Nilius, B.; Flockerzi, V.; et al. Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat. Immunol. 2007, 8, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Rixecker, T.; Mathar, I.; Medert, R.; Mannebach, S.; Pfeifer, A.; Lipp, P.; Tsvilovskyy, V.; Freichel, M. TRPM4-mediated control of FcepsilonRI-evoked Ca(2+) elevation comprises enhanced plasmalemmal trafficking of TRPM4 channels in connective tissue type mast cells. Sci. Rep. 2016, 6, 32981. [Google Scholar] [CrossRef] [Green Version]
- Nadler, M.J.; Hermosura, M.C.; Inabe, K.; Perraud, A.L.; Zhu, Q.; Stokes, A.J.; Kurosaki, T.; Kinet, J.P.; Penner, R.; Scharenberg, A.M.; et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 2001, 411, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Runnels, L.W.; Yue, L.; Clapham, D.E. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 2001, 291, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, C.; Perraud, A.L.; Johnson, C.O.; Inabe, K.; Smith, M.K.; Penner, R.; Kurosaki, T.; Fleig, A.; Scharenberg, A.M. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 2003, 114, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Chubanov, V.; Mittermeier, L.; Gudermann, T. Role of kinase-coupled TRP channels in mineral homeostasis. Pharm. Ther. 2018, 184, 159–176. [Google Scholar] [CrossRef]
- Abumaria, N.; Li, W.; Liu, Y. TRPM7 functions in non-neuronal and neuronal systems: Perspectives on its role in the adult brain. Behav. Brain Res. 2018, 340, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Nadolni, W.; Zierler, S. The Channel-Kinase TRPM7 as Novel Regulator of Immune System Homeostasis. Cells 2018, 7, 109. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.S. Role of TRPM7 in cerebral ischaemia and hypoxia. J. Physiol. 2017, 595, 3077–3083. [Google Scholar] [CrossRef] [Green Version]
- Komiya, Y.; Runnels, L.W. TRPM channels and magnesium in early embryonic development. Int. J. Dev. Biol. 2015, 59, 281–288. [Google Scholar] [CrossRef]
- Fleig, A.; Chubanov, V. Trpm7. Handb. Exp. Pharmacol. 2014, 222, 521–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, C.; Brandao, K.; Perraud, A.L. The channel-kinase TRPM7, revealing the untold story of Mg(2+) in cellular signaling. Magnes Res. 2014, 27, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Desai, B.N.; Navarro, B.; Donovan, A.; Andrews, N.C.; Clapham, D.E. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 2008, 322, 756–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beesetty, P.; Wieczerzak, K.B.; Gibson, J.N.; Kaitsuka, T.; Luu, C.T.; Matsushita, M.; Kozak, J.A. Inactivation of TRPM7 kinase in mice results in enlarged spleens, reduced T-cell proliferation and diminished store-operated calcium entry. Sci. Rep. 2018, 8, 3023. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, A.; Vettore, V.; Rezzonico-Jost, T.; Hampe, S.; Rottoli, E.; Nadolni, W.; Perotti, M.; Meier, M.A.; Hermanns, C.; Geiger, S.; et al. TRPM7 kinase activity is essential for T cell colonization and alloreactivity in the gut. Nat. Commun. 2017, 8, 1917. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.N.; Krapivinsky, G.; Navarro, B.; Krapivinsky, L.; Carter, B.C.; Febvay, S.; Delling, M.; Penumaka, A.; Ramsey, I.S.; Manasian, Y.; et al. Cleavage of TRPM7 releases the kinase domain from the ion channel and regulates its participation in Fas-induced apoptosis. Dev. Cell 2012, 22, 1149–1162. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, M.; Kilch, T.; Horgen, F.D.; Fleig, A.; Penner, R. The TRPM7 channel kinase regulates store-operated calcium entry. J. Physiol. 2017, 595, 3165–3180. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Lis, A.; Schmitz, C.; Penner, R.; Fleig, A. The TRPM7 kinase limits receptor-induced calcium release by regulating heterotrimeric G-proteins. Cell Mol. Life Sci. 2018, 75, 3069–3078. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Zheng, S.G.; Gray, J.D.; Ohtsuka, K.; Yamagiwa, S.; Horwitz, D.A. Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25- precursors. J. Immunol. 2002, 169, 4183–4189. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Kitani, A.; Fuss, I.; Strober, W. Cutting edge: Regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J. Immunol. 2007, 178, 6725–6729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- van der Geest, K.S.; Abdulahad, W.H.; Tete, S.M.; Lorencetti, P.G.; Horst, G.; Bos, N.A.; Kroesen, B.J.; Brouwer, E.; Boots, A.M. Aging disturbs the balance between effector and regulatory CD4+ T cells. Exp. Gerontol. 2014, 60, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Chandy, K.G. Potassium channels, memory T cells, and multiple sclerosis. Neuroscientist 2005, 11, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Panyi, G. Biophysical and pharmacological aspects of K+ channels in T lymphocytes. Eur. Biophys. J. 2005, 34, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Beeton, C.; Chandy, K.G. Potassium channels as therapeutic targets for autoimmune disorders. Curr. Opin. Drug Discov. Devel. 2003, 6, 640–647. [Google Scholar] [PubMed]
- Beeton, C.; Pennington, M.W.; Wulff, H.; Singh, S.; Nugent, D.; Crossley, G.; Khaytin, I.; Calabresi, P.A.; Chen, C.Y.; Gutman, G.A.; et al. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol. Pharmacol. 2005, 67, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Wulff, H.; Standifer, N.E.; Azam, P.; Mullen, K.M.; Pennington, M.W.; Kolski-Andreaco, A.; Wei, E.; Grino, A.; Counts, D.R.; et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 17414–17419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanshani, S.; Wulff, H.; Miller, M.J.; Rohm, H.; Neben, A.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 2000, 275, 37137–37149. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.H.; Fleischmann, B.K.; Hondowicz, B.; Maier, C.C.; Turka, L.A.; Yui, K.; Kotlikoff, M.I.; Wells, A.D.; Freedman, B.D. Modulation of Kv channel expression and function by TCR and costimulatory signals during peripheral CD4(+) lymphocyte differentiation. J. Exp. Med. 2002, 196, 897–909. [Google Scholar] [CrossRef] [Green Version]
- Wulff, H.; Calabresi, P.A.; Allie, R.; Yun, S.; Pennington, M.; Beeton, C.; Chandy, K.G. The voltage-gated Kv1.3 K(+) channel in effector memory T cells as new target for MS. J. Clin. Investig. 2003, 111, 1703–1713. [Google Scholar] [CrossRef] [Green Version]
- Fanger, C.M.; Neben, A.L.; Cahalan, M.D. Differential Ca2+ influx, KCa channel activity, and Ca2+ clearance distinguish Th1 and Th2 lymphocytes. J. Immunol. 2000, 164, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Srivastava, S.; Zhdanova, O.; Ding, Y.; Li, Z.; Wulff, H.; Lafaille, M.; Skolnik, E.Y. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proc. Natl. Acad. Sci. USA 2010, 107, 1541–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, M.W.; Beeton, C.; Galea, C.A.; Smith, B.J.; Chi, V.; Monaghan, K.P.; Garcia, A.; Rangaraju, S.; Giuffrida, A.; Plank, D.; et al. Engineering a stable and selective peptide blocker of the Kv1.3 channel in T lymphocytes. Mol. Pharmacol. 2009, 75, 762–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalman, K.; Pennington, M.W.; Lanigan, M.D.; Nguyen, A.; Rauer, H.; Mahnir, V.; Paschetto, K.; Kem, W.R.; Grissmer, S.; Gutman, G.A.; et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 1998, 273, 32697–32707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulff, H.; Miller, M.J.; Hansel, W.; Grissmer, S.; Cahalan, M.D.; Chandy, K.G. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc. Natl. Acad. Sci. USA 2000, 97, 8151–8156. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheugen, J.A.; Le Deist, F.; Devignot, V.; Korn, H. Enhancement of calcium signaling and proliferation responses in activated human T lymphocytes. Inhibitory effects of K+ channel block by charybdotoxin depend on the T cell activation state. Cell Calcium 1997, 21, 1–17. [Google Scholar] [CrossRef]
- Hu, L.; Pennington, M.; Jiang, Q.; Whartenby, K.A.; Calabresi, P.A. Characterization of the functional properties of the voltage-gated potassium channel Kv1.3 in human CD4+ T lymphocytes. J. Immunol. 2007, 179, 4563–4570. [Google Scholar] [CrossRef] [Green Version]
- Koch Hansen, L.; Sevelsted-Moller, L.; Rabjerg, M.; Larsen, D.; Hansen, T.P.; Klinge, L.; Wulff, H.; Knudsen, T.; Kjeldsen, J.; Kohler, R. Expression of T-cell KV1.3 potassium channel correlates with pro-inflammatory cytokines and disease activity in ulcerative colitis. J. Crohns Colitis 2014, 8, 1378–1391. [Google Scholar] [CrossRef]
- Chandy, K.G.; Wulff, H.; Beeton, C.; Pennington, M.; Gutman, G.A.; Cahalan, M.D. K+ channels as targets for specific immunomodulation. Trends Pharmacol. Sci. 2004, 25, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Matheu, M.P.; Beeton, C.; Garcia, A.; Chi, V.; Rangaraju, S.; Safrina, O.; Monaghan, K.; Uemura, M.I.; Li, D.; Pal, S.; et al. Imaging of effector memory T cells during a delayed-type hypersensitivity reaction and suppression by Kv1.3 channel block. Immunity 2008, 29, 602–614. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Wang, T.; Gocke, A.R.; Nath, A.; Zhang, H.; Margolick, J.B.; Whartenby, K.A.; Calabresi, P.A. Blockade of Kv1.3 potassium channels inhibits differentiation and granzyme B secretion of human CD8+ T effector memory lymphocytes. PLoS ONE 2013, 8, e54267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panyi, G.; Vamosi, G.; Bacso, Z.; Bagdany, M.; Bodnar, A.; Varga, Z.; Gaspar, R.; Matyus, L.; Damjanovich, S. Kv1.3 potassium channels are localized in the immunological synapse formed between cytotoxic and target cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1285–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; McElhaney, J.E. Age-related changes in memory and effector T cells responding to influenza A/H3N2 and pandemic A/H1N1 strains in humans. Vaccine 2011, 29, 2169–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollar, S.; Berta, L.; Vasarhelyi, Z.E.; Balog, A.; Vasarhelyi, B.; Rigo, J., Jr.; Toldi, G. Impact of aging on calcium influx and potassium channel characteristics of T lymphocytes. Oncotarget 2015, 6, 13750–13756. [Google Scholar] [CrossRef] [Green Version]
- Eil, R.; Vodnala, S.K.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Pan, J.H.; Palmer, D.C.; Gros, A.; Yamamoto, T.N.; Patel, S.J.; et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016, 537, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, X.; Xue, L.; Xing, J.; Jouvin, M.H.; Putney, J.W.; Anderson, M.P.; Trebak, M.; Kinet, J.P. Low-Voltage-Activated CaV3.1 Calcium Channels Shape T Helper Cell Cytokine Profiles. Immunity 2016, 44, 782–794. [Google Scholar] [CrossRef] [Green Version]
- Matza, D.; Badou, A.; Klemic, K.G.; Stein, J.; Govindarajulu, U.; Nadler, M.J.; Kinet, J.P.; Peled, A.; Shapira, O.M.; Kaczmarek, L.K.; et al. T Cell Receptor Mediated Calcium Entry Requires Alternatively Spliced Cav1.1 Channels. PLoS ONE 2016, 11, e0147379. [Google Scholar] [CrossRef] [Green Version]
- Badou, A.; Jha, M.K.; Matza, D.; Mehal, W.Z.; Freichel, M.; Flockerzi, V.; Flavell, R.A. Critical role for the beta regulatory subunits of Cav channels in T lymphocyte function. Proc. Natl. Acad. Sci. USA 2006, 103, 15529–15534. [Google Scholar] [CrossRef] [Green Version]
- Jha, M.K.; Badou, A.; Meissner, M.; McRory, J.E.; Freichel, M.; Flockerzi, V.; Flavell, R.A. Defective survival of naive CD8+ T lymphocytes in the absence of the beta3 regulatory subunit of voltage-gated calcium channels. Nat. Immunol. 2009, 10, 1275–1282. [Google Scholar] [CrossRef] [Green Version]
- Omilusik, K.; Priatel, J.J.; Chen, X.; Wang, Y.T.; Xu, H.; Choi, K.B.; Gopaul, R.; McIntyre-Smith, A.; Teh, H.S.; Tan, R.; et al. The Ca(v)1.4 calcium channel is a critical regulator of T cell receptor signaling and naive T cell homeostasis. Immunity 2011, 35, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Robert, V.; Triffaux, E.; Paulet, P.E.; Guery, J.C.; Pelletier, L.; Savignac, M. Protein kinase C-dependent activation of CaV1.2 channels selectively controls human TH2-lymphocyte functions. J. Allergy Clin. Immunol. 2014, 133, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Kotturi, M.F.; Carlow, D.A.; Lee, J.C.; Ziltener, H.J.; Jefferies, W.A. Identification and functional characterization of voltage-dependent calcium channels in T lymphocytes. J. Biol. Chem. 2003, 278, 46949–46960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, L.; Gordon, J.; Grafton, G. Non-voltage-gated L-type Ca2+ channels in human T cells: Pharmacology and molecular characterization of the major alpha pore-forming and auxiliary beta-subunits. J. Biol. Chem. 2004, 279, 19566–19573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, G.M.; Yu, D.; Wang, J.; Soong, T.W. Alternative splicing at C terminus of Ca(V)1.4 calcium channel modulates calcium-dependent inactivation, activation potential, and current density. J. Biol. Chem. 2012, 287, 832–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral, M.D.; Paulet, P.E.; Robert, V.; Gomes, B.; Renoud, M.L.; Savignac, M.; Leclerc, C.; Moreau, M.; Lair, D.; Langelot, M.; et al. Knocking down Cav1 calcium channels implicated in Th2 cell activation prevents experimental asthma. Am. J. Respir. Crit. Care Med. 2010, 181, 1310–1317. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Chiozzi, P.; Ferrari, D.; Falzoni, S.; Sanz, J.M.; Morelli, A.; Torboli, M.; Bolognesi, G.; Baricordi, O.R. Nucleotide receptors: An emerging family of regulatory molecules in blood cells. Blood 2001, 97, 587–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, R.A. Molecular physiology of P2X receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef] [PubMed]
- Yip, L.; Woehrle, T.; Corriden, R.; Hirsh, M.; Chen, Y.; Inoue, Y.; Ferrari, V.; Insel, P.A.; Junger, W.G. Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Borges da Silva, H.; Beura, L.K.; Wang, H.; Hanse, E.A.; Gore, R.; Scott, M.C.; Walsh, D.A.; Block, K.E.; Fonseca, R.; Yan, Y.; et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature 2018, 559, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Schenk, U.; Westendorf, A.M.; Radaelli, E.; Casati, A.; Ferro, M.; Fumagalli, M.; Verderio, C.; Buer, J.; Scanziani, E.; Grassi, F. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci. Signal. 2008, 1, ra6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Onnis, A.; Baldari, C.T. Orchestration of Immunological Synapse Assembly by Vesicular Trafficking. Front. Cell Dev. Biol. 2019, 7, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woehrle, T.; Yip, L.; Elkhal, A.; Sumi, Y.; Chen, Y.; Yao, Y.; Insel, P.A.; Junger, W.G. Pannexin-1 hemichannel-mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood 2010, 116, 3475–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef]
- Chessell, I.P.; Hatcher, J.P.; Bountra, C.; Michel, A.D.; Hughes, J.P.; Green, P.; Egerton, J.; Murfin, M.; Richardson, J.; Peck, W.L.; et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain 2005, 114, 386–396. [Google Scholar] [CrossRef]
- Schenk, U.; Frascoli, M.; Proietti, M.; Geffers, R.; Traggiai, E.; Buer, J.; Ricordi, C.; Westendorf, A.M.; Grassi, F. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci. Signal. 2011, 4, ra12. [Google Scholar] [CrossRef]
- Manohar, M.; Hirsh, M.I.; Chen, Y.; Woehrle, T.; Karande, A.A.; Junger, W.G. ATP release and autocrine signaling through P2X4 receptors regulate gammadelta T cell activation. J. Leukoc. Biol. 2012, 92, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Frascoli, M.; Marcandalli, J.; Schenk, U.; Grassi, F. Purinergic P2X7 receptor drives T cell lineage choice and shapes peripheral gammadelta cells. J. Immunol. 2012, 189, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Gu, B.; Bendall, L.J.; Wiley, J.S. Adenosine triphosphate-induced shedding of CD23 and L-selectin (CD62L) from lymphocytes is mediated by the same receptor but different metalloproteases. Blood 1998, 92, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Janner, N.; Chalaris, A.; Moss, M.L.; Floss, D.M.; Meyer, D.; Koch-Nolte, F.; Rose-John, S.; Scheller, J. Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. J. Biol. Chem. 2011, 286, 14804–14811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, H.; Na, H.Y.; Chong, K.H.; Kim, T.J. P2X7 receptor-dependent ATP-induced shedding of CD27 in mouse lymphocytes. Immunol. Lett. 2006, 102, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Sluyter, R.; Wiley, J.S. P2X7 receptor activation induces CD62L shedding from human CD4+ and CD8+ T cells. Inflamm. Cell Signal. 2014, 1, 44–49. [Google Scholar] [CrossRef]
- Trabanelli, S.; Ocadlikova, D.; Gulinelli, S.; Curti, A.; Salvestrini, V.; Vieira, R.P.; Idzko, M.; Di Virgilio, F.; Ferrari, D.; Lemoli, R.M. Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J. Immunol. 2012, 189, 1303–1310. [Google Scholar] [CrossRef] [Green Version]
- Lepine, S.; Le Stunff, H.; Lakatos, B.; Sulpice, J.C.; Giraud, F. ATP-induced apoptosis of thymocytes is mediated by activation of P2 X 7 receptor and involves de novo ceramide synthesis and mitochondria. Biochim. Biophys. Acta 2006, 1761, 73–82. [Google Scholar] [CrossRef]
- Seman, M.; Adriouch, S.; Scheuplein, F.; Krebs, C.; Freese, D.; Glowacki, G.; Deterre, P.; Haag, F.; Koch-Nolte, F. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity 2003, 19, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.M.; Ploia, C.; Anselmi, F.; Sarukhan, A.; Viola, A. Adenosine triphosphate acts as a paracrine signaling molecule to reduce the motility of T cells. EMBO J. 2014, 33, 1354–1364. [Google Scholar] [CrossRef] [Green Version]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhao, Y.; Liu, Y. The role of nucleotides and purinergic signaling in apoptotic cell clearance-implications for chronic inflammatory diseases. Front. Immunol. 2014, 5, 656. [Google Scholar] [CrossRef] [Green Version]
- Theatre, E.; Frederix, K.; Guilmain, W.; Delierneux, C.; Lecut, C.; Bettendorff, L.; Bours, V.; Oury, C. Overexpression of CD39 in mouse airways promotes bacteria-induced inflammation. J. Immunol. 2012, 189, 1966–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Ni, X.; Pan, X.; Lu, H.; Lu, Y.; Zhao, J.; Guo Zheng, S.; Hippen, K.L.; Wang, X.; Lu, L. Human CD39(hi) regulatory T cells present stronger stability and function under inflammatory conditions. Cell Mol. Immunol. 2017, 14, 521–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolova, M.; Carriere, M.; Jenabian, M.A.; Limou, S.; Younas, M.; Kok, A.; Hue, S.; Seddiki, N.; Hulin, A.; Delaneau, O.; et al. CD39/adenosine pathway is involved in AIDS progression. PLoS Pathog. 2011, 7, e1002110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi, A.; Battaglia, F.; Kalli, F.; Ferrera, F.; Conteduca, G.; Tardito, S.; Stringara, S.; Ivaldi, F.; Negrini, S.; Borgonovo, G.; et al. CD39 is highly involved in mediating the suppression activity of tumor-infiltrating CD8+ T regulatory lymphocytes. Cancer Immunol. Immunother. 2013, 62, 851–862. [Google Scholar] [CrossRef]
- Aliagas, E.; Vidal, A.; Texido, L.; Ponce, J.; Condom, E.; Martin-Satue, M. High expression of ecto-nucleotidases CD39 and CD73 in human endometrial tumors. Mediat. Inflamm. 2014, 2014, 509027. [Google Scholar] [CrossRef] [Green Version]
- Perrot, I.; Michaud, H.A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Dejou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425.e9. [Google Scholar] [CrossRef] [Green Version]
- la Sala, A.; Ferrari, D.; Di Virgilio, F.; Idzko, M.; Norgauer, J.; Girolomoni, G. Alerting and tuning the immune response by extracellular nucleotides. J. Leukoc. Biol. 2003, 73, 339–343. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.; Jia, X.; Zhang, X.; Xia, Y.; Wang, Y.; Fu, L.; Xiao, C.; Geng, D. Increased expression of P2X7 receptor in peripheral blood mononuclear cells correlates with clinical severity and serum levels of Th17-related cytokines in patients with myasthenia gravis. Clin. Neurol. Neurosurg. 2017, 157, 88–94. [Google Scholar] [CrossRef]
- Atarashi, K.; Nishimura, J.; Shima, T.; Umesaki, Y.; Yamamoto, M.; Onoue, M.; Yagita, H.; Ishii, N.; Evans, R.; Honda, K.; et al. ATP drives lamina propria T(H)17 cell differentiation. Nature 2008, 455, 808–812. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Yu, M.; Cavanagh, M.M.; Hutter Saunders, J.; Qi, Q.; Ye, Z.; Le Saux, S.; Sultan, W.; Turgano, E.; Dekker, C.L.; et al. Expression of CD39 on Activated T Cells Impairs their Survival in Older Individuals. Cell Rep. 2016, 14, 1218–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.K.; Godec, J.; Wolski, D.; Adland, E.; Yates, K.; Pauken, K.E.; Cosgrove, C.; Ledderose, C.; Junger, W.G.; Robson, S.C.; et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015, 11, e1005177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, A.; Moss, A.; Rothweiler, S.; Serena Longhi, M.; Wu, Y.; Junger, W.G.; Robson, S.C. NADH oxidase-dependent CD39 expression by CD8(+) T cells modulates interferon gamma responses via generation of adenosine. Nat. Commun. 2015, 6, 8819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, D.; Flores-Santibanez, F.; Neira, J.; Osorio-Barrios, F.; Tejon, G.; Nunez, S.; Hidalgo, Y.; Fuenzalida, M.J.; Meza, D.; Ureta, G.; et al. Purinergic Signaling as a Regulator of Th17 Cell Plasticity. PLoS ONE 2016, 11, e0157889. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Kunzli, B.M.; Yi, A.R.; Sevigny, J.; Berberat, P.O.; Enjyoji, K.; Csizmadia, E.; Friess, H.; Robson, S.C. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16788–16793. [Google Scholar] [CrossRef] [Green Version]
- Aswad, F.; Kawamura, H.; Dennert, G. High sensitivity of CD4+CD25+ regulatory T cells to extracellular metabolites nicotinamide adenine dinucleotide and ATP: A role for P2X7 receptors. J. Immunol. 2005, 175, 3075–3083. [Google Scholar] [CrossRef] [Green Version]
- Aswad, F.; Dennert, G. P2X7 receptor expression levels determine lethal effects of a purine based danger signal in T lymphocytes. Cell. Immunol. 2006, 243, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Mellouk, A.; Bobe, P. CD8(+), but not CD4(+) effector/memory T cells, express the CD44(high)CD45RB(high) phenotype with aging, which displays reduced expression levels of P2X7 receptor and ATP-induced cellular responses. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 3225–3236. [Google Scholar] [CrossRef]
- Cauwels, A.; Rogge, E.; Vandendriessche, B.; Shiva, S.; Brouckaert, P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014, 5, e1102. [Google Scholar] [CrossRef] [Green Version]
- Osterholm, M.T.; Kelley, N.S.; Sommer, A.; Belongia, E.A. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 36–44. [Google Scholar] [CrossRef]
Figure 1.
Altered Ca2+ signaling in T cells during aging—impact of ion channel expression and function. Calcium signaling in T cells, mediated by several types of channels, pumps, and receptors, is part of a crucial activation pathway, leading to proliferation, differentiation, and various effector functions. Antigen recognition through T cell receptors (TCRs) initiates the phosphorylation of multiple adaptor proteins. Activation of PLCγ1, followed by inositol triphosphate (IP3) production, induces Ca2+ release from ER calcium stores by binding to its receptor (IP3R). The decrease of [Ca2+]ER activates STIM, which translocates to the plasma membrane and causes store-operated calcium entry (SOCE) through direct interactions with Orai channels. The increased cytosolic Ca2+ concentration leads to the activation of calcineurin, dephosphorylation and nuclear translocation of NFAT resulting in expression of IL2, CD25, Foxp3, and further components essential for T cell function. Other ion channels including (non-selective) TRPC and TRPM2/7, CaV channels (e.g., CaV3.1), and purinergic ionotropic receptors (e.g., P2X7) mediate Ca2+ influx during T cell activation. Additionally, ion channels like potassium channels (e.g., KV1.3, KCa3.1) or TRPM4 indirectly regulate Ca2+ influx through de- or hyperpolarization of the plasma membrane. Besides alterations in TCR activation, also Ca2+ signaling changes during aging. STIM1 and STIM2 expression levels are reduced (↓). Orai channel expression is still under investigation but Orai2 mRNA expression is decreased (↓). In contrast, PMCA4 expression increases with age (↑) leading to higher Ca2+ extrusion. Inhibition of KV1.3 or KCa3.1 causes age- and subtype- dependent differences in Ca2+ influx patterns (↯). Possible changes in expression or function of other ion channels during aging are largely unknown (?).
Figure 1.
Altered Ca2+ signaling in T cells during aging—impact of ion channel expression and function. Calcium signaling in T cells, mediated by several types of channels, pumps, and receptors, is part of a crucial activation pathway, leading to proliferation, differentiation, and various effector functions. Antigen recognition through T cell receptors (TCRs) initiates the phosphorylation of multiple adaptor proteins. Activation of PLCγ1, followed by inositol triphosphate (IP3) production, induces Ca2+ release from ER calcium stores by binding to its receptor (IP3R). The decrease of [Ca2+]ER activates STIM, which translocates to the plasma membrane and causes store-operated calcium entry (SOCE) through direct interactions with Orai channels. The increased cytosolic Ca2+ concentration leads to the activation of calcineurin, dephosphorylation and nuclear translocation of NFAT resulting in expression of IL2, CD25, Foxp3, and further components essential for T cell function. Other ion channels including (non-selective) TRPC and TRPM2/7, CaV channels (e.g., CaV3.1), and purinergic ionotropic receptors (e.g., P2X7) mediate Ca2+ influx during T cell activation. Additionally, ion channels like potassium channels (e.g., KV1.3, KCa3.1) or TRPM4 indirectly regulate Ca2+ influx through de- or hyperpolarization of the plasma membrane. Besides alterations in TCR activation, also Ca2+ signaling changes during aging. STIM1 and STIM2 expression levels are reduced (↓). Orai channel expression is still under investigation but Orai2 mRNA expression is decreased (↓). In contrast, PMCA4 expression increases with age (↑) leading to higher Ca2+ extrusion. Inhibition of KV1.3 or KCa3.1 causes age- and subtype- dependent differences in Ca2+ influx patterns (↯). Possible changes in expression or function of other ion channels during aging are largely unknown (?).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zöphel, D.; Hof, C.; Lis, A. Altered Ca2+ Homeostasis in Immune Cells during Aging: Role of Ion Channels. Int. J. Mol. Sci. 2021, 22, 110. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010110
AMA Style
Zöphel D, Hof C, Lis A. Altered Ca2+ Homeostasis in Immune Cells during Aging: Role of Ion Channels. International Journal of Molecular Sciences. 2021; 22(1):110. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010110
Chicago/Turabian StyleZöphel, Dorina, Chantal Hof, and Annette Lis. 2021. "Altered Ca2+ Homeostasis in Immune Cells during Aging: Role of Ion Channels" International Journal of Molecular Sciences 22, no. 1: 110. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010110
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.