Fake It ‘Till You Make It—The Pursuit of Suitable Membrane Mimetics for Membrane Protein Biophysics


1
Wallenberg Centre for Molecular and Translational Medicine, University of Gothenburg, 405 30 Göteborg, Sweden
2
Department of Chemistry and Molecular Biology, University of Gothenburg, 405 30 Göteborg, Sweden
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(1), 50; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010050
Submission received: 9 December 2020
/
Revised: 17 December 2020
/
Accepted: 19 December 2020
/
Published: 23 December 2020
(This article belongs to the Special Issue Membrane Proteins: Structure, Function and Motion)
Abstract
:Membrane proteins evolved to reside in the hydrophobic lipid bilayers of cellular membranes. Therefore, membrane proteins bridge the different aqueous compartments separated by the membrane, and furthermore, dynamically interact with their surrounding lipid environment. The latter not only stabilizes membrane proteins, but directly impacts their folding, structure and function. In order to be characterized with biophysical and structural biological methods, membrane proteins are typically extracted and subsequently purified from their native lipid environment. This approach requires that lipid membranes are replaced by suitable surrogates, which ideally closely mimic the native bilayer, in order to maintain the membrane proteins structural and functional integrity. In this review, we survey the currently available membrane mimetic environments ranging from detergent micelles to bicelles, nanodiscs, lipidic-cubic phase (LCP), liposomes, and polymersomes. We discuss their respective advantages and disadvantages as well as their suitability for downstream biophysical and structural characterization. Finally, we take a look at ongoing methodological developments, which aim for direct in-situ characterization of membrane proteins within native membranes instead of relying on membrane mimetics.
1. Introduction
The overwhelming majority of scientific articles on membrane proteins introduces this class of proteins by mentioning their contribution of roughly 30% to organisms’ genomes, thus, highlighting their importance and resulting self-explanatory relevance. In the interest of avoiding a repetition of what has been written countless times before, let us instead spend a moment of consideration on the endangered species of the polar bear. The polar bear (Ursus maritimus) is perfectly adapted to life on the annual sea ice of the arctic circle, a very specific habitat, which it roams as wide-ranging predator hunting seals [1]. When polar bears are relocated from their natural environment to live in captivity in zoological gardens, they have a high tendency to develop abnormal repetitive behavior, such as stereotypical pacing and head nodding [2], and have severely increased infant mortality rates [3]. Similar alterations of behavior and infant mortality in captivity have been reported for several different species, whereupon the degree of these alterations is directly correlated to the extent of the environmental difference, but rarely as pronounced as in polar bears [3]. Much like this admittedly farfetched example, membrane proteins evolved to reside in the very specific amphipathic lipid bilayer environment of biological membranes and their removal from this environment often results in pronounced structural and functional ramifications [4,5,6]. It is, thus, one of the great challenges of membrane protein biophysics to characterize membrane proteins, while maintaining the specific nature of their lipid bilayer environment to be able study this class of proteins in a biologically meaningful context. The continuously progressing efforts to recreate this environment, in order to facilitate the biophysical characterization of membrane proteins are the subject of this review.
Lipid bilayers form the physical permeability barriers, which segregate cells and cellular compartments. Driven by the hydrophobic effect, amphiphilic lipid molecules self-assemble to form lamellar bilayers, with their hydrophobic moieties facing the core of the bilayer and their hydrophilic head groups facing the surrounding aqueous environment [7]. Cellular membranes are formed from a large variety of chemically very diverse lipids [8], ranging from hundreds of different lipid species in “simple” prokaryotic organisms like Escherichia coli to thousands in more complex eukaryotic organisms [9,10]. The diverse physiochemical properties of different cellular membranes are shaped by their lipid composition [9]. Biological membranes obtain their functionality only through the presence of specialized integral membrane proteins, which transmit molecules, energy and stimuli across these physical barriers. To fulfil these crucial functions membrane proteins depend on the properties of the surrounding membrane environment [11,12,13]. Factors, such as the lipid composition and bilayer asymmetry, membrane curvature, tension as well as the fluidity of the bilayer directly impact the structural and functional integrity of membrane proteins [13,14,15].
Unfortunately, in their native cellular form, membrane proteins immanently defy the requirements of biophysical experiments, which demand protein samples of high purity and high concentration, often in form of a solution. In addition to being insoluble, individual membrane protein species occur in rather low densities in cellular membranes. To bridge this gap and make membrane proteins experimentally accessible a multitude of membrane mimetics have been developed over the last decades. As implied by their name, membrane mimetics attempt to imitate the environment of lipid bilayers. In their most fundamental forms, as detergent micelles, this means simply to recreate the hydrophobic core environment of a lipid bilayer [16]. However, more complex forms such as bicelles and nanodiscs try to incorporate a certain number of lipid molecules (Figure 1). Moreover, purified membrane proteins can be reconstituted into bilayers of synthetic lipids or lipid extracts, attempting to closely resemble the original lipid bilayer a membrane protein was purified from.
Naturally, all these methods have their pros and cons and not all of them are compatible with different biophysical methods. For example, solution NMR (Nuclear magnetic resonance) spectroscopy, single-particle cryo-EM (electron microscopy), and X-ra/neutron solution scattering methods demand membrane proteins in a solubilized form, typically in the form of micelles, bicelles or nanodiscs. Solid-state-NMR, negative stain EM, and AFM (atomic force microscopy) are suitable for larger membrane assemblies such as proteoliposomes, whereas X-Ray crystallography requires samples in a three-dimensional (3D) crystalline form. The hydrophobic contacts required to maintain the structural integrity of membrane proteins within these crystals can either be satisfied by detergents or in case of lipidic cubic phases (LCP) by lipid molecules. In this review, we survey the currently available membrane mimetic systems, weigh their advantages, as well as their disadvantages, evaluate how they impact the structural and functional states of membrane proteins and assess their suitability for various biophysical methods.
2. Detergent Micelles
The archetype of membrane mimetics are detergents, which are routinely used to solubilize membrane proteins. Detergents are amphipathic molecules, which self-assemble to form micelles in aqueous solutions. Based on their molecular structure and properties, namely the charge of their hydrophilic head groups, detergent molecules can be subdivided into ionic, non-ionic, and zwitterionic detergents. Thereby, ionic detergents such as sodium dodecyl sulfate (SDS) are considered “harsh”, due to their (differently pronounced) denaturing effect on membrane proteins, ranging from minor structural alterations [17] to loss of function and complete unfolding [18]. Notably, some membrane proteins remain rather unaffected by ionic detergents. In particular, bacterial outer membrane proteins have shown to be very resistant to SDS denaturing, due to extensive hydrogen bonding networks stabilizing the transmembrane β-barrels of these proteins. In fact, the resulting altered migration behavior in SDS page is frequently used as an indicator of the folding state of outer membrane proteins [19,20].
For the solubilization of membrane proteins in a functional form more widely used are “mild” non-ionic detergents, such as Octyl-L-D-glucoside (OG) and Dodecyl-L-D-maltoside (DDM), which tend to retain the structural integrity of solubilized proteins and leave inter- and intra-molecular protein-protein interactions intact. The latter is particularly important for the solubilization of multimeric membrane protein complexes and especially DDM has proven quite useful for the purification of intact complexes [21,22,23,24]. The third group are zwitterionic detergents, such as Lauryldimethylamine-N-oxide (LDAO), the hydrophilic head groups of which have a positive, as well as a negative charge. While, the overall electrically neutral, zwitterionic detergents form an intermediate between ionic and non-ionic detergents, with a stronger solubilizing potential than non-ionic detergents and a less pronounced deactivating effect than ionic detergents [25].
The solubilization of biological membranes usually occurs via two stages with increasing detergent concentration (Figure 2). Initially, at low detergent concentrations, the detergent molecules insert into the lipid bilayer, resulting in destabilization and fragmentation. At high concentration, typically exceeding the detergents critical micellar concentration (CMC), the lipid bilayer is dissolved, resulting in binary lipid-detergent or protein-detergent, as well as ternary lipid-protein-detergent mixed micelles [26,27]. Thereby different detergents vary in their capacity to solubilize different cellular membranes. For example detergents like Triton X-100 and Sarkosyl have been shown to selectively solubilize inner membranes of Gram-negative bacteria and mitochondria, while leaving their outer membranes largely unaffected [28,29]. Lipid microdomains (sometimes referred to as lipid rafts), which typically contain cholesterol and saturated sphingo- and glycerophospholipids in a liquid ordered phase, are resistant to mild detergents, such as Triton X-100, and thus, remain as detergent-resistant membranes after solubilization [30,31]. Likewise, the amount of endogenous lipid molecules bound to membrane proteins varies greatly depending on the detergent used to solubilize a membrane [32]. In this context, mass spectrometry and in particular tandem mass spectrometry (MS/MS) is a powerful method to characterize not only the binding of lipids to membrane proteins, but also their effects on membrane protein oligomerization [33,34]. The choice of detergent depends ultimately on the planned downstream biophysical characterization and especially detergents with a low CMC, despite effectively solubilizing most membranes, can be difficult to remove and are, thus, of limited suitability for methods requiring detergent removal [35].
The purification of a membrane protein from a cellular membrane typically begins with the isolation and solubilization of the membrane of interest [36]. Due to the differences in the way detergents interact with cellular membranes, their efficient solubilization involves screening for a suitable detergent [37]. Finding a detergent that also stably maintains a membrane protein and is suitable for downstream applications can require substantial screening work [38,39]. Once solubilized, the purification of membrane proteins follows similar principles as the routine purification of soluble proteins, relying on chromatographic methods including affinity, gel filtration, and ion exchange chromatography [36]. An alternative route to obtaining solubilized membrane proteins in a pure and folded state is refolding into detergent micelles. To this end, membrane proteins are transferred from a fully unfolded state in concentrated solutions of chaotropic salts into a detergent containing refolding buffer to adopt a folded state within the detergent micelles [40,41]. Interestingly, it was shown that in a similar way the cellular chaperone machinery can be exploited and refolding into micelles results in exactly the same folded protein state, regardless whether folding was initiated from a chaperone or chaotropic reagents [42]. Combined with recombinant protein expression in inclusion bodies, this method can expedite protein purification and can prove particularly useful when large amounts of a protein are required. However, despite routinely used, membrane proteins do not necessarily fold into a native structure and refolding can result in non-native multimers and structural intermediates [43,44].
Using NMR spectroscopy membrane proteins can be directly characterized in a detergent solubilized form, without the need for additional downstream modifications [45]. Particularly useful when analyzing membrane proteins with NMR spectroscopy is the use of deuterated detergents, which eliminate interfering proton signals originating from the detergent [46]. While, a handful of membrane protein structures have been determined using NMR spectroscopy [47,48,49,50], the real strength of the method lies in its ability to probe dynamic processes [51]. In the micellar state NMR spectroscopy determines the residue-specific dynamics, can probe interactions with ligands and detect conformational changes in solution [45,52]. Moreover, unlike other structural techniques, NMR spectroscopy can yield detailed information on highly dynamic and unstructured regions of membrane proteins, such as loops [53,54]. Therefore, NMR spectroscopy could capture the subtle differences imposed on the structural conformation of membrane proteins by different detergents [54]. In this context, it should be stressed that, despite often resulting in high-resolution structural information, detergent stabilized states of membrane proteins, especially in Dodecylphosphorylcholine (DPC), might often be non-functional states [55].
Actuated by the recent resolution revolution [56], single-particle cryo-EM emerged as another powerful technique to characterize membrane proteins in detergent micelles. Unlike NMR spectroscopy, which excels at characterizing small proteins, single-particle cryo-EM is best suited for large proteins and protein complexes [57]. Several membrane protein structures stabilized in detergent micelles could be solved by cryo-EM, including bacterial β-barrel assembly machinery (BAM) complex, mitochondrial TOM core complex, and the spinach cytochrome b6f complex [58,59,60]. However, despite efforts to streamline the preparation of membrane proteins for cryo-EM, the methodology is far from being routine work [61,62]. Detergent concentrations typically used in preparations of membrane proteins tend to interfere with the controlled formation of thin vitrified ice, often resulting in reduced image contrast [63]. Therefore, the preparation of membrane proteins for cryo-EM requires thorough removal of excess detergent [64]. Moreover, the surface to volume ratio of a sub-micrometer thin water film on an EM grid is much higher than in conventional liquid droplets and it is not yet fully clear how the consequentially altered fluid dynamics and the air-water interface impact solubilized macromolecules [61,65].
While, detergents arguably form a less than ideal environment for many membrane proteins their usage remains in most cases unavoidable. With very few exceptions, the solubilization of membrane proteins from the membranes of an expression host is commonly accomplished through the use of detergents [66,67]. The majority of membrane mimetics require the reconstitution of a membrane protein, which naturally requires the prior solubilization and purification of said membrane protein. Detergent micelles are therefore almost always the starting point for additional downstream applications involving more complex membrane mimetics.
3. 3D Crystals and Lipidic Cubic Phase
Despite a growing toolbox of alternative membrane mimetics, to date the majority of membrane protein structures have been solved by X-ray crystallography, utilizing 3D crystals of membrane proteins. The crystallization of detergent-stabilized solubilized membrane proteins follows similar methods and principles as the crystallization of soluble proteins based on vapor diffusion, microdialysis, and batch crystallization [68,69]. Crystals are, thereby, formed from a protein solution, supersaturated with precipitating agents such as salts or polyethylene glycol (PEG), which drive the aggregation of protein-detergent-complexes into ordered crystals [68]. Within these type II crystals the hydrophobic surfaces of membrane proteins remain satisfied through the co-crystallized detergent micelles, while the crystal lattice is preferentially formed by polar protein-protein interactions [70,71]. Successful crystallization often depends on the choice and nature of the detergent and high detergent concentrations or the use of detergents with a large micelle size can impair crystal formation [69].
Following the first high-resolution crystal structure of a membrane protein, the photosynthetic reaction center from Rhodopseudomonas viridis [72], a great variety of membrane protein structures could be solved using X-ray crystallography. These include groundbreaking structures such as bacterial potassium channel KcsA [73] and lactose permease LacY [74], which resulted in unprecedented insight into the molecular details of these proteins in particular and membrane proteins in general. Nevertheless, it should be noted that solving crystal structures of membrane proteins often only is possible due to substantial molecular engineering. This includes the introduction of mutations, which arrest proteins in a certain conformation [75]—an effect, which can also be achieved through the binding of ligands or antibody fragments [76,77]—deletions of parts of the proteins to eliminate unfavorable crystal contacts [78], or chimeric modifications of proteins [79]. In addition, ambiguities can inhere in crystal structures, imposed by the crystallization conditions, together with uncertainties in the identity and position of atoms and molecules within crystal structures [80]. Even similar sample preparations can lead to altered structures, as for example evidenced in the translocator protein (TSPO) that yielded two different α-helical bundles depending if the structure was determined in milder DM or DDM [81,82], compared to the harsher zwitterionic DPC [83] and it remains unclear whether the different structures represent simply alternative states of the protein. However, the structures, obtained in the milder detergents, are in better agreement with known functional mutations and the ligand binding site shows a larger degree of conservation [81].
A valuable alternative to detergent-micelle mediated 3D crystallization of membrane proteins is crystallization in the lipidic cubic phase (LCP) [84]. LCP takes advantage of the propensity of monounsaturated monoacylglycerols, such as monoolein, to form a bicontinuous cubic mesophase, a single lipid bilayer organized into a three-dimensional bilayer structure containing an aqueous channel system [85]. The cubic phase is formed spontaneously when the lipid is mixed with solubilized or dispersed protein solutions, while crystal formation is driven by the addition of a precipitant [86]. In addition to monoolein, LCP can contain various accessory lipids, which can either remain from co-purification with a membrane protein or can be specifically added during the crystallization process [87]. Through the stabilization in a lipid bilayer, LCP is thought to provide a more natural environment for membrane proteins and LCP has been successfully used, particularly with membrane proteins which have small polar surfaces, such as the seven-helix-bundles of rhodopsins [88,89,90] and G-protein-coupled receptors (GPCRs) [91,92].
One limitation of LCP crystallization is its propensity to result in microcrystals [85]. However, microcrystals grown in LCP are ideally suited to be studied using serial femtosecond crystallography (SFX) [93]. The latter utilizes ultrashort pulses generated by an X-ray free-electron laser (XFEL) [94] to sequentially collect data from a continuous stream of microcrystals [95]. By the short duration of highly intense X-ray pulses the crystals are vaporized before radiation damage can occur [94], thus facilitating the time-resolved characterization of dynamic structural transitions in crystallized proteins [96]. These studies have provided remarkable insight into the activation of different photoreceptors, by visualizing the so-called protein quake [97,98,99]. In addition to SFX, another method that has been show to provide high-resolution structural data of microcrystalline membrane proteins is so-called microcrystal electron diffraction (Micro ED) [100,101], which was recently applied to study the tetrameric sodium channel NaK in DDM based crystals [102] as well as the human adenosine A2A receptor in LCP [103].
4. Bicelles and Nanodiscs
The first attempt to incorporate a substantial amount of lipids into solubilized membrane protein systems were bicelles. Bicelles (bilayered micelles; Figure 1) are formed by phospholipids, traditionally dimyristoyl-phosphatidylcholine (DMPC), in a planar discoidal bilayer assembly, which are surrounded by a scaffold of either a detergent, such as CHAPS, or short-chained lipids, such as dihexanoyl-phosphatidylcholine (DHPC) [104]. Thereby, bicelles can be designed to adopt a variety of shapes, ranging from small bilayer discs over wormlike structures to large perforated lamellar assemblies, dictated by the lipid-to-detergent, or longchain-to-shortchain lipid ratio, respectively [105,106]. In order to reconstitute membrane proteins into bicelles either a detergent-stabilized membrane protein is integrated into preformed bicelles or the bicelles can be formed through the addition of detergents to proteoliposomes that were assembled beforehand [106]. Through the incorporation of lipid molecules, bicelles have been shown to outperform micelles in their ability to maintain membrane proteins in a functional state [107,108]. However, it has also been shown that different lipid compositions of bicelles significantly influence membrane protein dynamics [109]. Moreover, molecular dynamics (MD) simulations indicate increased peptide solvation of transmembrane segments within small bicelles compared to larger bilayer systems [110].
While, originally developed as a membrane system for solid state NMR spectroscopy [111], bicelles found a broad audience in biophysics [106,112]. Due to the increased lipid content, bicelles are larger than most purely detergent-based micellar systems. Yet, their molecular tumbling permits detailed characterization of reconstituted membrane proteins by solution NMR spectroscopy [113,114]. By stabilizing the transmembrane region of the HIV-1 envelope spike (Env) in DMPC/DHPC bicelles the atomic resolution structure of the trimeric assembly could be determined [115,116,117], which remained elusive in the previous cryo-EM structure, possibly due to the detrimental influence of the DDM/sodium deoxycholate micelles [118]. Likewise, a direct comparison of the dimeric transmembrane domain of Glycophorin A in DPC micelles and DMPC/DHPC bicelles, respectively, showed reduced conformational fluctuation and enhanced stability of the transmembrane α-helixes in the bicellar lipid environment [119].
Bicelles have also been implemented as an alternative crystallization method trying to combine the incorporation of lipids, as used in LCP, with the facility of detergent based crystallization [120]. Bicelle crystallization exploits the temperature-dependent ability of lipid/amphiphile mixtures to exchange between different three-dimensional structural arrangements [121]. When this concept was initially introduced with studies of bacteriorhodopsin, the protein was found to be embedded in bicelles as a stable monomer instead of its usual trimeric assembly [120]. Bicelle-based 3D crystals have since these early studies successfully been used with multiple membrane proteins, including the human G-protein-coupled receptor (GPCR) β2 adrenergic receptor (β2AR) [122], and the eukaryotic mitochondrial voltage-dependent anion channel (VDAC) [123].
Evolving from bicelles, the recent years have seen great development in the field of nanodiscs. Collectively, the term nanodiscs refers to the lipid bilayer particles similar to discoidal bicelles, which are surrounded by a scaffolding molecule (Figure 1). Yet, the size-range in which scaffolded nanodiscs can be prepared is somewhat limited, compared to bicelles. Following the first account of a lipid nanodisc surrounded by membrane scaffold protein (MSP) [124], several other types of scaffolds have been described, including saposin proteins (salipro) [125], as well as copolymer-scaffolded nanodiscs utilizing styrene-maleic acid (SMA) and diisobutylene/maleic acid (DIBMA) [126,127]. The formation of nanodiscs follows similar principles as the formation of bicelles, starting from a mixture of detergent-solubilized lipid, detergent-solubilized protein, and MSP or saposin, respectively, and is driven by subsequent detergent removal [125,128,129].
Unlike protein-based nanodiscs, which require the reconstitution of membrane proteins from a detergent-solubilized micellar state, co-polymers have shown certain detergent-like properties. When mixed with membrane preparations, these polymers can extract “native nanodiscs” containing membrane proteins together with a fraction of the lipid molecules surrounding the proteins [130,131]. While, nanodiscs certainly have the ability to preserve the local lipid composition around a membrane protein, they cannot maintain membrane asymmetry [132]. The latter is due to the dynamic nature of the different nanodisc systems. On the one hand equilibration between both leaflets can occur when lipid molecules flip around the edges of the nanodisc and on the other through diffusional as well as collisional transfer. Polymer-scaffolded nanodiscs have been shown to readily exchange proteins, lipids, and polymer components at much higher rates compared to MSP-scaffolded nanodiscs or unilamellar lipid vesicles [133,134]. In contrast, MSP-scaffolded nanodiscs have been found to exhibit internal lipid dynamics, which are comparable to lipids in liposomes [135].
Nanodiscs have been widely used with a wide range of biophysical methods, including NMR spectroscopy [136,137,138], electron microscopy [139,140] and atomic force microscopy [141]. Therefore, the use of nanodiscs has proven successful even with complex systems. For example, individually MSP-nanodisc-stabilized inner and outer membrane components of bacterial tripartite efflux pumps MexAB–OprM and AcrAB–TolC could be recombined to form stable intact complexes bearing two separate nanodiscs [142]. The open pore state of bacterial Tc toxin complex TcdA1, which could not be sufficiently stabilized in Tween-20 detergent micelles or liposomes [143,144], could be resolved in high detail embedded in nanodiscs [145], whereas the corresponding crystal structure could only reveal a closed pre-pore state [143]. Likewise, when characterized in nanodiscs, aforementioned HIV envelope protein Env showed substantial differences in its arrangement and orientation to the membrane surface, compared to micellar and bicellar environments [146].
Direct comparisons of membrane proteins in micellar, bicellar and nanodisc systems revealed distinct differences between these membrane mimetics. For example, the bacterial outer membrane protein OmpX has been shown to form a stable β-barrel in all three environments, however, the dynamics and molecular motion differed substantially between the mimetics [147,148]. Similarly, the comparison of outer membrane protein BamA in LDAO micelles, DMPC/DHPC bicelles, and MSP-bounded DMPC nanodiscs suggested altered dynamics between the three environments [149]. Likewise, the α-helical tetrameric potassium channel KcsA has been shown to have reduced stability in nanodiscs, indicating altered dynamic properties, compared to DDM micelles [150]. In contrast, the CC-chemokine receptor 5 (CCR5), a GPCR which is inherently instable in detergents, such as DDM, could be stabilized in nanodiscs for prolonged NMR studies [151]. The human anion channel VDAC showed nearly identical folds in micelles, bicelles, and nanodiscs, however, unlike the first two, nanodiscs incorporated not only monomeric VDAC, but multimeric states, similar to the ones observed in native membranes [152].
5. Liposomes (and Polymersomes)
Native lipid bilayers contain a diverse blend of membrane proteins, interweaved with additional components, such as lipoproteins and carbohydrates in the form of glycoproteins and glycolipids [7]. In most native membranes any membrane protein of interest is vastly outnumbered by these additional components, making it inaccessible to biophysical investigations. Few exceptions, such as the purple membranes from Halobacteria [153,154] or disc membranes from vertebral rod cells [155], in fact allow proteins to be studied in native membrane isolates, which are covered in the next section. For the vast majority of membrane proteins, the alternative lies in the bottom-up reconstruction of an artificial lipid membrane. To this end, a membrane protein of interest is solubilized and isolated from the membrane of a suitable expression system using detergents and, naturally, the same principles and limitations of detergent micelles apply as covered above [156,157]. Following purification, in a subsequent step, membrane proteins are reconstituted into an artificial lipid bilayer. Reconstitution is generally achieved via one of two alternative routes (Figure 3). In one method the self-assembly of a lipid bilayer is driven by detergent removal from a ternary mixture of micelles containing the protein and separately detergent-solubilized lipids [158]. Alternatively, the solubilized membrane protein can be inserted into preformed liposomes [159]. In both cases assembly and membrane insertion are driven by the removal of the detergent and can be achieved by several means, all aiming at reducing the detergent concentration (far) below the CMC. Thereby, the method of detergent removal can have a strong influence on the resulting proteoliposomes. Dialysis can result in homogeneous vesicles, but is time-consuming and limited to detergents with high CMC. More rapid methods, such as size exclusion chromatography, rapid dilution, and the use hydrophobic adsorbents often result in inhomogeneous protein distribution and in-complete detergent removal [35].
Self-assembled lipid bilayers allow better control over the lipid-to-protein ratio and thus can result in a very dense protein packing within the bilayer, culminating in a 2D crystalline assembly [160,161]. However, due to the rotational freedom during assembly, membranes formed this way typically contain membrane proteins in a non-native up-down configuration, with either half of the proteins inserted in the bilayer in opposite directions (Figure 3). While random orientation can occur as well during insertion into preformed liposomes, asymmetry between the two solvent accessible poles of a membrane protein, for example in membrane proteins containing a soluble domain, can bias insertion with the soluble domain facing outwards (Figure 3) [159,162,163]. In fact, by fusing a soluble domain to one side of a membrane protein, this phenomenon can be utilized to drive unidirectional insertion of the protein into liposomes [164].
Naturally, a reconstituted artificial lipid bilayer cannot fully reassemble the complexity of a native membrane and features like membrane asymmetry or local variations in the lipid composition, sometimes referred to as lipid rafts, are impossible to mimic. Nevertheless, since artificial bilayers allow precise control over the lipid composition they are ideal proxies for detailed studies on how different lipid composition of a membrane affect membrane proteins. The utilization of reconstituted lipid bilayers for example allowed detailed characterization of lipid-induced topological switches in proteins of the major facilitator family [165,166], which could be confirmed in vivo [167]. Similarly, liposomes of varying lipid composition are a valuable tool to understanding membrane protein folding at a molecular level. Therefore, not only the folding process from chemically denatured states is of interest [168,169], but much focus has been laid on chaperone-dependent protein folding [170,171].
Until challenged by the advent of single particle cryo-EM, two-dimensional (2D) crystalline membrane protein assemblies were the system of choice for structure determination by EM [172,173]. Unlike X-Ray crystallography, which requires 3D crystals that are sometimes difficult to obtain from membrane proteins, electron crystallography is ideally suited for the 2D crystals of membrane proteins. Electron crystallography revealed the first high resolution structures of Bacteriorhodopsin [174], Aquaporin [175], and light-harvesting complex [176]. A rather recent development is the use of protein-containing liposomes in cryo-electron tomography combined with subtomogram averaging for the high-resolution reconstruction of membrane proteins [177,178,179]. Moreover, liposomes have been increasingly used to investigate membrane proteins using solid-state NMR spectroscopy. Recent advances in fast and ultra-fast (>100 kHZ) magic angle spinning (MAS) solid-state NMR spectroscopy based on 1H-detection resulted in enhanced sensitivity and resolution comparable to solution NMR, required to facilitate high resolution NMR studies of membrane proteins [180,181]. Solid-state NMR spectroscopy has, for example, been used to determine structural the details and conformational rearrangements of α-helical transmembrane proteins such as KcsA reconstituted in lipid bilayers [182,183], as well as bacterial β-barrel outer membrane proteins such as OmpA and OmpG [184,185]. Importantly, all of these structures revealed nuanced differences, such as structural alterations and altered dynamics compared to previously reported structures.
Another powerful tool for characterizing the membrane proteins reconstituted in lipid bilayers is atomic force microscopy (AFM), due to its ability to be operated in liquid environments at physiological temperatures [186,187]. Adsorbed to atomically flat surfaces, such as muscovite mica or highly oriented pyrolytic graphite (HOPG), AFM allows imaging of the membrane topographies of solid-supported planar lipid bilayers at molecular resolution [188,189,190]. AFM could reveal ligand-induced conformational changes of membrane proteins [191], details on the electrostatics of their accessible surfaces [192,193] and their dynamic behavior in lipid bilayers [194,195]. Moreover, AFM allows the direct physical manipulation of individual membrane proteins, facilitating detailed studies of their behavior under force [196,197,198], their folding behavior [199,200,201], as well as their interaction with tethered ligands [202].
In addition to being used for the structural characterization of membrane proteins, lipid bilayers allow the functional examination of membrane proteins, in particular molecular transport phenomena, which cannot be probed in solubilized states. Therefore, electrophysiological measurements, either based on fused liposomes or utilizing black lipid membranes, allow detailed characterization of diffusion through transmembrane pores as well as probing their electrochemical properties [203,204,205,206]. Similarly, molecular transport is routinely studied in intact liposomes based on osmotic swelling. The latter is particularly powerful when combined with fluorogenic reactions conducted inside the liposomes lumen in stopped-flow experiments [207,208].
The ability of artificial lipid bilayers to maintain membrane proteins in a functional state is showcased by their use as a bottom-up platform for the assembly of nanocells in synthetic biology, whereby membrane proteins are embedded in liposomes to create functional systems with potential applications as nano-sized reaction compartments, drug delivery vehicles and novel therapeutics [209,210,211]. In this context, a biomimetic alternative to lipid bilayers are membranes formed from amphiphilic block copolymers [212,213,214]. While the physical properties of these are very different from lipids, their bilayer forming abilities are driven by the same principles, and polymersomes can effectively maintain membrane proteins in a functional form [215,216]. In fact, in mixed polymer-lipid bilayers under certain conditions membrane proteins have been shown to preferentially reside within the polymer rather than the lipid fraction, depending on their relative fluidity [217,218].
6. Native Membranes
Whereas so far no mimetic entirely met the physiological requirements of membrane proteins, native membranes, in the majority of cases, cannot meet the experimental requirements of the modern biophysicist. Native membranes are notoriously difficult to handle. Typically, they contain a particular protein of interest only in small quantities over a much larger background of other membrane proteins. Nevertheless, native membrane preparations from E. coli, also known as Kabackosomes, were used to characterize molecular transport through membrane proteins long before the first membrane protein structures were uncovered [219,220]. Moreover, some specialized cellular membranes natively contain high densities of distinct membrane proteins, which are sufficiently pure to permit biophysical studies directly in the unaltered native membranes. The best example are the purple membranes from archaea such as Halobacterium salinarum and Halobacterium halobium, which naturally contain bacteriorhodopsin in a 2D crystalline form [154]. In fact, the first structural models of bacteriorhodopsin were determined using native purple membranes isolated from H. halobium [221]. While later preparations employing increased 2D crystals, obtained through detergent treatment of purple membranes, resulted in models with increased resolution [174,222], direct comparison of detergent treated crystals to native membranes showed that lipid boundaries separating individual trimers within the native membranes were removed [223]. A similar approach, based on partial de-lipidation was used to observe crystalline assemblies of porins in situ, in bacterial outer membrane sacculi [224].
While these early electron crystallographic studies required 2D crystalline assemblies of membrane proteins, advancements in cryo-electron tomography nowadays allow direct in situ analysis of membrane proteins in non-crystalline native membranes [225,226]. Although, data on integral membrane proteins so far remains sparse, the method has already proven its potential. Aided by membrane targeting nanoparticles, active preprotein-carrying TOM–TIM23 supercomplexes could be localized and subsequently visualized in situ in yeast mitochondrial membranes [227]. Intriguing examples showcasing the potential of cryo-electron tomography are the in situ structures of envelope spanning bacterial secretion systems, such as the structure of an intact primordial type III secretion system, which could be determined in Chlamydia trachomatis elementary bodies [228]. Other examples are the in situ structures of AcrAB–TolC efflux pump in intact E. coli cells [229], the type IV secretion system in intact Legionella pneumophila cells [230], as well as the membrane complex of a type VI secretion system in E. coli [231]. A very promising approach to study membrane proteins in situ is the enrichment of secreted extracellular vesicles with specific proteins, which could be exploited to study integral type I membrane proteins from C. elegans as well as Herpes simplex virus [232].
In addition to electron microscopy, solid-state NMR spectroscopy has been successfully employed to study membrane proteins embedded in native membranes. In this context, native Escherichia coli inner membranes were used as a proxy to characterize Anabaena sensory rhodopsin (ASR), which was found to form hexagonal packing in DMPC/DMPA liposomes but a square lattice assembly in the E. coli membrane [233]. Furthermore, solid-state NMR spectroscopy was used to characterize bacterial BAM complexes in native outer membranes, suggesting increased structural disorder in the native environment [234]. Recently, a combination of cryo-electron tomography and of 1H-detected solid-state NMR spectroscopy was employed to reveal different conformations of the bacterial membrane protein YidC in native membranes [235].
One exceptional method to study native membranes is AFM, which does not depend on the preparation of highly homogeneous samples [236,237]. The ability of AFM to imaging native membranes at molecular resolution was initially demonstrated with native purple membranes. Following initial topographies, which allowed discrimination of individual proteins [238], AFM was optimized to reveal the submolecular details of single bacteriorhodopsin molecules in great detail [239]. Using AFM, two-dimensional arrays of aquaporin 0 (AQP0) could be observed in native lens core membranes, surrounded by densely packed gap junction channels, and AQP0 array formation could be followed using time-lapse AFM [240]. Similarly, AFM could reveal native supramolecular assembly of VDAC in dense and sparse domains in native yeast mitochondrial outer membranes [241] as well as the closely packed, paracrystalline dimeric arrangement of rhodopsin arrays in native mouse disc membranes [242]. More recently, AFM was used to distinguish small and large protruding proteins in dimeric photosystem II oxygen-evolving complexes within native spinach grana membranes [243]. In addition to high-resolution imaging, AFM-based single-molecule force spectroscopy (SMFS) was used to probe the force response of individual membrane proteins in native membranes [196]. In this context, protein-enriched outer membrane vesicles from Escherichia coli were recently employed to reveal subtle differences in the dynamics of outer membrane proteins between native membranes and reconstituted lipid bilayers [244,245].
7. Conclusions and Future Perspectives
The last several decades have seen an enormous increase in efforts to optimize membrane mimetics to facilitate the structural and functional characterization of membrane proteins using biophysical methods leading to a constantly growing toolbox of diverse options (Table 1). Multiple membrane proteins could be characterized in several different mimetics, allowing the direct comparison and revealing the influence of the different mimetics on the structure and dynamics of these membrane proteins. Whereas for some membrane proteins these different tools are all in good agreement, for others vast differences have been observed, imposed by the respective mimetics used. For some membrane proteins, only a small set of mimetics could stabilize their folded state sufficiently to permit biophysical characterization, whereas other mimetics let to destabilization, impairing detailed characterization. Only in very few cases could membrane proteins be characterized in their native bilayer environment, some of which only revealed subtle, possibly negligible differences to mimetic systems, whereas others were substantially impacted by the mimetic environment.
Unlike the case of the polar bear, which can in fact be observed in its natural environment, as well as in captivity, thus, allowing a direct comparison between the two environments, for the majority of membrane proteins observations embedded in the native lipid environment are until now inexistent. Despite decades of studying membrane proteins with exceptional effort and the development of a plethora of groundbreaking sophisticated methods, we have only caught the first glimpses providing snapshots of their structural details within artificial environments. For many membrane proteins, these studies yielded priceless insight into their molecular architecture, which are supported by a multitude of functional investigations in vitro, as well as in vivo, yet, their behavior under native cellular conditions remains elusive. Clearly, to close this gap much more research on membrane proteins embedded in their native environment is necessary in the future. The recent trend towards in situ membrane protein biophysics will certainly help to illuminate this blind spot and provide in-depth insight into the details underlying membrane protein function.
Author Contributions
Conceptualization: J.T. and B.M.B.; writing—original draft preparation: J.T.; writing—review and editing: J.T. and B.M.B.; visualization: J.T. All authors have read and agreed to the published version of the manuscript.
Funding
J.T. was supported by an EMBO Long-Term Fellowship (ALTF 413-2018). B.M.B. gratefully acknowledges funding from the Swedish Research Council (Vetenskapsrådet Starting Grant 2016-04721) and the Knut och Alice Wallenberg Foundation through a Wallenberg Academy Fellowship (2016.0163) as well as through the Wallenberg Centre for Molecular and Translational Medicine, University of Gothenburg, Sweden.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AFM | Atomic force microscopy |
| AQP0 | Aquaporin 0 |
| ASR | Anabaena sensory rhodopsin |
| β2AR | β2 adrenergic receptor |
| BAM | β-barrel assembly machinery |
| CCR5 | CC-chemokine receptor 5 |
| CMC | Critical micellar concentration |
| DM | Decyl-L-D-maltoside |
| DHPC | Dihexanoyl-phosphatidylcholine |
| DIBMA | Diisobutylene/maleic acid |
| DMPC | Dimyristoyl-phosphatidylcholine |
| DDM | Dodecyl-L-D-maltoside |
| DPC | Dodecylphosphorylcholine |
| EM | Electron microscopy |
| ET | Electron tomography |
| GPCRs | G-protein-coupled receptors |
| HOPG | Highly oriented pyrolytic graphite |
| LDAO | Lauryldimethylamine-N-oxide |
| LCP | Lipidic cubic phases |
| MAS | Magic angle spinning |
| MS | Mass spectrometry |
| MSP | Membrane scaffold protein |
| MicroED | Microcrystal electron diffraction |
| MD | Molecular dynamics |
| NMR | Nuclear magnetic resonance |
| OG | Octyl-L-D-glucoside |
| PEG | Polyethylene glycol |
| SDS | Sodium dodecyl sulfate |
| SFX | Serial femtosecond crystallography |
| SMA | Styrene-maleic acid |
| SMFS | Single-molecule force spectroscopy |
| Salipro | Saposin lipo-protein |
| XFEL | X-ray free-electron laser |
| VDAC | Voltage-dependent anion channel |
References
- Stirling, I.; Derocher, A.E. Possible impacts of climatic warming on polar bears. Arctic 1993, 46, 240–245. [Google Scholar] [CrossRef]
- Cremers, P.W.F.H.; Geutjes, S.L. The cause of stereotypic behaviour in a male polar bear (Ursus maritimus). Proc. Meas. Behav. 2012, 2012, 28–31. [Google Scholar]
- Clubb, R.; Mason, G. Captivity effects on wide-ranging carnivores. Nature 2003, 425, 473–474. [Google Scholar] [CrossRef] [PubMed]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta Biomembr. 2004, 1666, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Chorev, D.S.; Robinson, C.V. The importance of the membrane for biophysical measurements. Nat. Chem. Biol. 2020, 16, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-X.; Cross, T.A. Influences of membrane mimetic environments on membrane protein structures. Annu. Rev. Biophys. 2013, 42, 361–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, S.J.; Nicolson, G.L. The fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Dowhan, W.; Bogdanov, M. Functional roles of lipids in membranes. In Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier Science: Amsterdam, The Netherlands, 2002; pp. 1–35. ISBN 9780444634382. [Google Scholar]
- Shevchenko, A.; Simons, K. Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef]
- Andersen, O.S.; Koeppe, R.E., II. Bilayer thickness and membrane protein function: An energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 107–130. [Google Scholar] [CrossRef] [Green Version]
- Phillips, R.; Ursell, T.; Wiggins, P.; Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature 2009, 459, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coskun, Ü.; Simons, K. Cell membranes: The lipid perspective. Structure 2011, 19, 1543–1548. [Google Scholar] [CrossRef] [Green Version]
- Oates, J.; Watts, A. Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr. Opin. Struct. Biol. 2011, 21, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Van Klompenburg, W.; Nilsson, I.; Von Heijne, G.; De Kruijff, B. Anionic phospholipids are determinants of membrane protein topology. EMBO J. 1997, 16, 4261–4266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garavito, R.M.; Ferguson-Miller, S. Detergents as tools in membrane biochemistry. J. Biol. Chem. 2001, 276, 32403–32406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamani, V.; Hegde, B.G.; Langen, R.; Lanyi, J.K. Secondary and tertiary structure of bacteriorhodopsin in the SDS denatured state. Biochemistry 2012, 51, 1051–1060. [Google Scholar] [CrossRef]
- Lau, F.W.; Bowie, J.U. A method for assessing the stability of a membrane protein. Biochemistry 1997, 36, 5884–5892. [Google Scholar] [CrossRef]
- Noinaj, N.; Kuszak, A.J.; Buchanan, S.K. Heat modifiability of outer membrane proteins from gram-negative bacteria. Methods Mol. Biol. 2015, 1329, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Kleinschmidt, J.H.; Wiener, M.C.; Tamm, L.K. Outer membrane protein A of E. coli folds into detergent micelles, but not in the presence of monomeric detergent. Protein Sci. 1999, 8, 2065–2071. [Google Scholar] [CrossRef] [Green Version]
- Sachelaru, I.; Winter, L.; Knyazev, D.G.; Zimmermann, M.; Vogt, A.; Kuttner, R.; Ollinger, N.; Siligan, C.; Pohl, P.; Koch, H.G. YidC and SecYEG form a heterotetrameric protein translocation channel. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Sverzhinsky, A.; Chung, J.W.; Deme, J.C.; Fabre, L.; Levey, K.T.; Plesa, M.; Carter, D.M.; Lypaczewski, P.; Coulton, J.W. Membrane protein complex ExbB4-ExbD1-TonB1 from Escherichia coli demonstrates conformational plasticity. J. Bacteriol. 2015, 197, 1873–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Li, H.; Dong, H.; Zeng, Y.; Zhang, Z.; Paterson, N.G.; Stansfeld, P.J.; Wang, Z.; Zhang, Y.; Wang, W.; et al. Structural basis of outer membrane protein insertion by the BAM complex. Nature 2016, 531, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, W.C.Y.; Rubinstein, J.L. Structure of intact Thermus thermophilus V-ATPase by cryo-EM reveals organization of the membrane-bound VO motor. Proc. Natl. Acad. Sci. USA 2010, 107, 1367–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luche, S.; Santoni, V.; Rabilloud, T. Evaluation of nonionic and zwitterionic detergents as membrane protein solubilizers in two-dimensional electrophoresis. Proteomics 2003, 3, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almgren, M. Mixed micelles and other structures in the solubilization of bilayer lipid membranes by surfactants. Biochim. Biophys. Acta Biomembr. 2000, 1508, 146–163. [Google Scholar] [CrossRef] [Green Version]
- Le Maire, M.; Champeil, P.; Møller, J.V. Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta Biomembr. 2000, 1508, 86–111. [Google Scholar] [CrossRef] [Green Version]
- Schnaitman, C. Solubilization of the cytoplasmic membrane of Escherichia coli by Triton X-100. J. Bacteriol. 1971, 108, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Filip, C.; Fletcher, G.; Wulff, J.L.; Earhart, C.F. Solubilization of the cytoplasmic membrane of Escherichia coli by the ionic detergent sodium-lauryl sarcosinate. J. Bacteriol. 1973, 115, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Schuck, S.; Honsho, M.; Ekroos, K.; Shevchenko, A.; Simons, K. Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. USA 2003, 100, 5795–5800. [Google Scholar] [CrossRef] [Green Version]
- London, E.; Brown, D.A. Insolubility of lipids in Triton X-100: Physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts). Biochim. Biophys. Acta Biomembr. 2000, 1508, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Ilgü, H.; Jeckelmann, J.M.; Gachet, M.S.; Boggavarapu, R.; Ucurum, Z.; Gertsch, J.; Fotiadis, D. Variation of the detergent-binding capacity and phospholipid content of membrane proteins when purified in different detergents. Biophys. J. 2014, 106, 1660–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.; Donlan, J.A.C.; Hopper, J.T.S.; Uzdavinys, P.; Landreh, M.; Struwe, W.B.; Drew, D.; Baldwin, A.J.; Stansfeld, P.J.; Robinson, C.V. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 2017, 541, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Rimon, A.; Mondal, R.; Friedler, A.; Padan, E. Cardiolipin is an optimal phospholipid for the assembly, stability, and proper functionality of the dimeric form of NhaA Na+/H+ antiporter. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, J.; Levy, D.; Mosser, G.; Lambert, O. Detergent removal by non-polar polystyrene beads. Eur. Biophys. J. 1998, 27, 305–319. [Google Scholar] [CrossRef]
- Lin, S.H.; Guidotti, G. Chapter 35 Purification of Membrane Proteins, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2009; Volume 463. [Google Scholar]
- Lantez, V.; Nikolaidis, I.; Rechenmann, M.; Vernet, T.; Noirclerc-Savoye, M. Rapid automated detergent screening for the solubilization and purification of membrane proteins and complexes. Eng. Life Sci. 2015, 15, 39–50. [Google Scholar] [CrossRef]
- Kotov, V.; Bartels, K.; Veith, K.; Josts, I.; Subhramanyam, U.K.T.; Günther, C.; Labahn, J.; Marlovits, T.C.; Moraes, I.; Tidow, H.; et al. High-throughput stability screening for detergent-solubilized membrane proteins. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Champeil, P.; Orlowski, S.; Babin, S.; Lund, S.; le Maire, M.; Møller, J.; Lenoir, G.; Montigny, C. A robust method to screen detergents for membrane protein stabilization, revisited. Anal. Biochem. 2016, 511, 31–35. [Google Scholar] [CrossRef]
- Buchanan, S.K. β-barrel proteins from bacterial outer membranes: Structure, function and refolding. Curr. Opin. Struct. Biol. 1999, 9, 455–461. [Google Scholar] [CrossRef]
- Fairman, J.W.; Noinaj, N.; Buchanan, S.K. The structural biology of β-barrel membrane proteins: A summary of recent reports. Curr. Opin. Struct. Biol. 2011, 21, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Burmann, B.M.; Hiller, S. Solution NMR studies of membrane-protein-chaperone complexes. Chimia 2012, 66, 759–763. [Google Scholar] [CrossRef] [Green Version]
- Visudtiphole, V.; Thomas, M.B.; Chalton, D.A.; Lakey, J.H. Refolding of Escherichia coli outer membrane protein F in detergent creates LPS-free trimers and asymmetric dimers. Biochem. J. 2005, 392, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Andersen, K.K.; Vad, B.S.; Otzen, D.E. OmpA can form folded and unfolded oligomers. Biochim. Biophys. Acta Proteins Proteomics 2013, 1834, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Tamm, L.K. NMR as a tool to investigate the structure, dynamics and function of membrane proteins. Nat. Struct. Mol. Biol. 2016, 23, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiruma-Shimizu, K.; Shimizu, H.; Thompson, G.S.; Kalverda, A.P.; Patching, S.G. Deuterated detergents for structural and functional studies of membrane proteins: Properties, chemical synthesis and applications. Mol. Membr. Biol. 2015, 32, 139–155. [Google Scholar] [CrossRef] [Green Version]
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 2008, 321, 1206–1210. [Google Scholar] [CrossRef] [Green Version]
- Gautier, A.; Mott, H.R.; Bostock, M.J.; Kirkpatrick, J.P.; Nietlispach, D. Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat. Struct. Mol. Biol. 2010, 17, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Cierpicki, T.; Jimenez, R.H.F.; Lukasik, S.M.; Ellena, J.F.; Cafiso, D.S.; Kadokura, H.; Beckwith, J.; Bushweller, J.H. NMR solution structure of the integral membrane enzyme DsbB: Functional insights into DsbB-catalyzed disulfide bond formation. Mol. Cell 2008, 31, 896–908. [Google Scholar] [CrossRef] [Green Version]
- Arora, A.; Abildgaard, F.; Bushweller, J.H.; Tamm, L.K. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat. Struct. Biol. 2001, 8, 334–338. [Google Scholar] [CrossRef]
- Hiller, S.; Wagner, G. The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins. Curr. Opin. Struct. Biol. 2009, 19, 396–401. [Google Scholar] [CrossRef] [Green Version]
- Opella, S.J.; Marassi, F.M. Applications of NMR to membrane proteins. Arch. Biochem. Biophys. 2017, 628, 92–101. [Google Scholar] [CrossRef]
- Kucharska, I.; Seelheim, P.; Edrington, T.; Liang, B.; Tamm, L.K. OprG harnesses the dynamics of its extracellular loops to transport small amino acids across the outer membrane of Pseudomonas aeruginosa. Structure 2015, 23, 2234–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renault, M.; Saurel, O.; Czaplicki, J.; Demange, P.; Gervais, V.; Löhr, F.; Réat, V.; Piotto, M.; Milon, A. Solution state NMR structure and dynamics of KpOmpA, a 210 residue transmembrane domain possessing a high potential for immunological applications. J. Mol. Biol. 2009, 385, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Kurauskas, V.; Hessel, A.; Ma, P.; Lunetti, P.; Weinhäupl, K.; Imbert, L.; Brutscher, B.; King, M.S.; Sounier, R.; Dolce, V.; et al. How detergent impacts membrane proteins: Atomic-level views of mitochondrial carriers in dodecylphosphocholine. J. Phys. Chem. Lett. 2018, 9, 933–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühlbrandt, W. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y. Single-particle Cryo-EM at crystallographic resolution. Cell 2015, 161, 450–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadanza, M.G.; Higgins, A.J.; Schiffrin, B.; Calabrese, A.N.; Brockwell, D.J.; Ashcroft, A.E.; Radford, S.E.; Ranson, N.A. Lateral opening in the intact β-barrel assembly machinery captured by cryo-EM. Nat. Commun. 2016, 7, 12865. [Google Scholar] [CrossRef]
- Bausewein, T.; Mills, D.J.; Langer, J.D.; Nitschke, B.; Nussberger, S.; Kühlbrandt, W. Cryo-EM Structure of the TOM Core Complex from Neurospora crassa. Cell 2017, 170, 693–700. [Google Scholar] [CrossRef]
- Malone, L.A.; Qian, P.; Mayneord, G.E.; Hitchcock, A.; Farmer, D.A.; Thompson, R.F.; Swainsbury, D.J.K.; Ranson, N.A.; Hunter, C.N.; Johnson, M.P. Cryo-EM structure of the spinach cytochrome b 6 f complex at 3.6 Å resolution. Nature 2019, 575, 535–539. [Google Scholar] [CrossRef]
- Sgro, G.G.; Costa, T.R.D. Cryo-EM grid preparation of membrane protein samples for single particle analysis. Front. Mol. Biosci. 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Carvalho, V.; Pronk, J.W.; Engel, A.H. Characterization of membrane proteins using cryo-electron microscopy. Curr. Protoc. Protein Sci. 2018, 94, 1–30. [Google Scholar] [CrossRef]
- Schmidt-Krey, I.; Rubinstein, J.L. Electron cryomicroscopy of membrane proteins: Specimen preparation for two-dimensional crystals and single particles. Micron 2011, 42, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauer, F.; Gerle, C.; Fischer, N.; Oshima, A.; Shinzawa-Itoh, K.; Shimada, S.; Yokoyama, K.; Fujiyoshi, Y.; Stark, H. GraDeR: Membrane protein complex preparation for single-particle cryo-EM. Structure 2015, 23, 1769–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaeser, R.M.; Han, B.-G. Opinion: Hazards faced by macromolecules when confined to thin aqueous films. Biophys. Rep. 2017, 3, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Jagow, G.; Link, T.A.; Schägger, H. Purification Strategies for Membrane Proteins. In A Practical Guide to Membrane Protein Purification; Separation, Detection, and Characterization of Biological Macromolecules; Von Jagow, G., Schägger, H., Eds.; Academic Press: San Diego, CA, USA, 1994; Volume 2, pp. 3–21. ISBN 978-0-08-057172-0. [Google Scholar]
- Schägger, H. Chromatographic Techniques and Basic Operations in Membrane Protein Purification. In A Practical Guide to Membrane Protein Purification; Separation, Detection, and Characterization of Biological Macromolecules; Von Jagow, G., Schägger, H., Eds.; Academic Press: San Diego, CA, USA, 1994; Volume 2, pp. 23–57. ISBN 978-0-08-057172-0. [Google Scholar]
- Wiener, M.C. A pedestrian guide to membrane protein crystallization. Methods 2004, 34, 364–372. [Google Scholar] [CrossRef]
- Birch, J.; Axford, D.; Foadi, J.; Meyer, A.; Eckhardt, A.; Thielmann, Y.; Moraes, I. The fine art of integral membrane protein crystallisation. Methods 2018, 147, 150–162. [Google Scholar] [CrossRef]
- Michel, H. Crystallization of membrane proteins. Trends Biochem. Sci. 1983, 8, 56–59. [Google Scholar] [CrossRef]
- Ostermeier, C.; Michel, H. Crystallization of membrane proteins. Curr. Opin. Struct. Biol. 1997, 7, 697–701. [Google Scholar] [CrossRef]
- Deisenhofer, J.; Epp, O.; Miki, K.; Huber, R.; Michel, H. Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3Å resolution. Nature 1985, 318, 618–624. [Google Scholar] [CrossRef]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Abramson, J.; Smirnova, I.; Kasho, V.; Verner, G.; Kaback, H.R.; Iwata, S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003, 301, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Vaidehi, N.; Grisshammer, R.; Tate, C.G. How can mutations thermostabilize G-protein-coupled receptors? Trends Pharmacol. Sci. 2016, 37, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manglik, A.; Kobilka, B.K.; Steyaert, J. Nanobodies to study G protein-coupled receptor structure and function. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 19–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How ligands illuminate GPCR molecular pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbaum, D.M.; Cherezov, V.; Hanson, M.A.; Rasmussen, S.G.F.; Foon, S.T.; Kobilka, T.S.; Choi, H.J.; Yao, X.J.; Weis, W.I.; Stevens, R.C.; et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 2007, 318, 1266–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsen, T.S.; Matt, R.; Weis, W.I.; Kobilka, B.K. Modified T4 lysozyme fusion proteins facilitate G protein-coupled receptor crystallogenesis. Structure 2014, 22, 1657–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.M.; Teague, S.J.; Kleywegt, G.J. Application and limitations of x-ray crystallographic data in structure-based ligand and drug design. Angew. Chem. Int. Ed. 2003, 42, 2718–2736. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kalathur, R.C.; Liu, Q.; Kloss, B.; Bruni, R.; Ginter, C.; Kloppmann, E.; Rost, B.; Hendrickson, W.A. Structure and activity of tryptophan-rich TSPO proteins. Science 2015, 347, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Liu, J.; Zheng, Y.; Garavito, R.M.; Ferguson-Miller, S. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015, 347, 555–558. [Google Scholar] [CrossRef] [Green Version]
- Jaremko, Ł.; Jaremko, M.; Giller, K.; Becker, S.; Zweckstetter, M. Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 2014, 343, 1363–1366. [Google Scholar] [CrossRef] [Green Version]
- Landau, E.M.; Rosenbusch, J.P. Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 14532–14535. [Google Scholar] [CrossRef] [Green Version]
- Caffrey, M. Crystallizing membrane proteins for structure determination: Use of lipidic mesophases. Annu. Rev. Biophys. 2009, 38, 29–51. [Google Scholar] [CrossRef]
- Caffrey, M. Membrane protein crystallization. J. Struct. Biol. 2003, 142, 108–132. [Google Scholar] [CrossRef]
- Cherezov, V.; Clogston, J.; Misquitta, Y.; Abdel-Gawad, W.; Caffrey, M. Membrane protein crystallization in meso: Lipid type-tailoring of the cubic phase. Biophys. J. 2002, 83, 3393–3407. [Google Scholar] [CrossRef] [Green Version]
- Pebay-Peyroula, E.; Rummel, G.; Rosenbusch, J.P.; Landau, E.M. X-ray structure of bacteriorhodopsin at 2.5 Ångstroms from microcrystals grown in lipidic cubic phases. Science 1997, 277, 1676–1681. [Google Scholar] [CrossRef] [Green Version]
- Kolbe, M.; Besir, H.; Essen, L.O.; Oesterhelt, D. Structure of the light-driven chloride pump halorhodopsin at 1.8 Å Resolution. Science 2000, 288, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Royant, A.; Nollert, P.; Edman, K.; Neutze, R.; Landau, E.M.; Pebay-Peyroula, E.; Navarro, J. X-ray structure of sensory rhodopsin II at 2.1-Å resolution. Proc. Natl. Acad. Sci. USA 2001, 98, 10131–10136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.F.; Foon, S.T.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, E.Y.T.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Weierstall, U.; James, D.; Wang, C.; White, T.A.; Wang, D.; Liu, W.; Spence, J.C.H.; Bruce Doak, R.; Nelson, G.; Fromme, P.; et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Neutze, R.; Wouts, R.; Van Der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 752–757. [Google Scholar] [CrossRef]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; Deponte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–78. [Google Scholar] [CrossRef]
- Hajdu, J.; Neutze, R.; Sjögren, T.; Edman, K.; Szöke, A.; Wilmouth, R.C.; Wilmot, C.M. Analyzing protein functions in four dimensions. Nat. Struct. Biol. 2000, 7, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Arnlund, D.; Johansson, L.C.; Wickstrand, C.; Barty, A.; Williams, G.J.; Malmerberg, E.; Davidsson, J.; Milathianaki, D.; DePonte, D.P.; Shoeman, R.L.; et al. Visualizing a protein quake with time-resolved X-ray scattering at a free-electron laser. Nat. Methods 2014, 11, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.; Tanaka, R.; et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 2016, 354, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Dods, R.; Båth, P.; Morozov, D.; Gagnér, V.A.; Arnlund, D.; Luk, H.L.; Kübel, J.; Maj, M.; Vallejos, A.; Wickstrand, C.; et al. Ultrafast structural changes within a photosynthetic reaction centre. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Nannenga, B.L.; Iadanza, M.G.; Gonen, T. Three-dimensional electron crystallography of protein microcrystals. eLife 2013, 2, 1–17. [Google Scholar] [CrossRef]
- Nannenga, B.L.; Shi, D.; Leslie, A.G.W.; Gonen, T. High-resolution structure determination by continuous-rotation data collection in MicroED. Nat. Methods 2014, 11, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Gonen, T. MicroED structure of the NaK ion channel reveals a Na+ partition process into the selectivity filter. Commun. Biol. 2018, 1, 1–6. [Google Scholar] [CrossRef]
- Zhu, L.; Bu, G.; Jing, L.; Shi, D.; Lee, M.Y.; Gonen, T.; Liu, W.; Nannenga, B.L. Structure determination from lipidic cubic phase embedded microcrystals by MicroED. Structure 2020, 28, 1149–1159. [Google Scholar] [CrossRef]
- Sanders, C.R.; Prosser, R.S. Bicelles: A model membrane system for all seasons? Structure 1998, 6, 1227–1234. [Google Scholar] [CrossRef] [Green Version]
- Harroun, T.A.; Koslowsky, M.; Nieh, M.P.; De Lannoy, C.F.; Raghunathan, V.A.; Katsaras, J. Comprehensive examination of mesophases formed by DMPC and DHPC mixtures. Langmuir 2005, 21, 5356–5361. [Google Scholar] [CrossRef] [PubMed]
- Dürr, U.H.N.; Gildenberg, M.; Ramamoorthy, A. The magic of bicelles lights up membrane protein structure. Chem. Rev. 2012, 112, 6054–6074. [Google Scholar] [CrossRef] [PubMed]
- Sanders, C.R.; Landis, G.C. Reconstitution of membrane proteins into lipid-rich bilayered mixed micelles for NMR studies. Biochemistry 1995, 34, 4030–4040. [Google Scholar] [CrossRef] [PubMed]
- Fanucci, G.; Lee, J.; Cafiso, D. Membrane mimetic environments alter the conformation of the outer membrane protein BtuB. J. Am. Chem. Soc. 2003, 125, 13932–13933. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.A.; Henzler-Wildman, K.A. Reconstitution of integral membrane proteins into isotropic bicelles with improved sample stability and expanded lipid composition profile. Biochim. Biophys. Acta Biomembr. 2012, 1818, 814–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, M.; Kraft, J.F.; Vosegaard, T.; Thøgersen, L.; Schiøtt, B. Bicelles and other membrane mimics: Comparison of structure, properties, and dynamics from MD simulations. J. Phys. Chem. B 2015, 119, 15831–15843. [Google Scholar] [CrossRef]
- Sanders, C.R.; Hare, B.J.; Howard, K.P.; Prestegard, J.H. Magnetically-oriented phospholipid micelles as a tool for the study of membrane-associated molecules. Prog. Nucl. Magn. Reson. Spectrosc. 1994, 26, 421–444. [Google Scholar] [CrossRef]
- Diller, A.; Loudet, C.; Aussenac, F.; Raffard, G.; Fournier, S.; Laguerre, M.; Grélard, A.; Opella, S.J.; Marassi, F.M.; Dufourc, E.J. Bicelles: A natural “molecular goniometer” for structural, dynamical and topological studies of molecules in membranes. Biochimie 2009, 91, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Prosser, R.S.; Evanics, F.; Kitevski, J.L.; Al-Abdul-Wahid, M.S. Current applications of bicelles in NMR studies of membrane-associated amphiphiles and proteins. Biochemistry 2006, 45, 8453–8465. [Google Scholar] [CrossRef]
- Piai, A.; Fu, Q.; Dev, J.; Chou, J.J. Optimal bicelle size q for solution NMR studies of the protein transmembrane partition. Chem. A Eur. J. 2017, 23, 1361–1367. [Google Scholar] [CrossRef] [Green Version]
- Dev, J.; Park, D.; Fu, Q.; Chen, J.; Ha, H.J.; Ghantous, F.; Herrmann, T.; Chang, W.; Liu, Z.; Frey, G.; et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science 2016, 353, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Shaik, M.M.; Cai, Y.; Ghantous, F.; Piai, A.; Peng, H.; Rits-Volloch, S.; Liu, Z.; Harrison, S.C.; Seaman, M.S.; et al. Structure of the membrane proximal external region of HIV-1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2018, 115, E8892–E8899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiliveri, S.C.; Louis, J.M.; Ghirlando, R.; Baber, J.L.; Bax, A. Tilted, Uninterrupted, monomeric HIV-1 gp41 transmembrane helix from residual dipolar couplings. J. Am. Chem. Soc. 2018, 140, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ozorowski, G.; Ward, A.B. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 2016, 351, 1043–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mineev, K.S.; Bocharov, E.V.; Volynsky, P.E.; Goncharuk, M.V.; Tkach, E.N.; Ermolyuk, Y.S.; Schulga, A.A.; Chupin, V.V.; Maslennikov, I.V.; Efremov, R.G.; et al. Dimeric structure of the transmembrane domain of Glycophorin A in lipidic and detergent environments. Acta Nat. 2011, 3, 90–98. [Google Scholar] [CrossRef]
- Faham, S.; Bowie, J.U. Bicelle crystallization: A new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. J. Mol. Biol. 2002, 316, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ujwal, R.; Bowie, J.U. Crystallizing membrane proteins using lipidic bicelles. Methods 2011, 55, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.G.F.; Choi, H.J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.P.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Colletier, J.P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The crystal structure of mouse VDAC1 at 2.3 Å resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef] [Green Version]
- Bayburt, T.H.; Sligar, S.G. Self-assembly of single integral membrane proteins into soluble nanoscale phospholipid bilayers. Protein Sci. 2003, 12, 2476–2481. [Google Scholar] [CrossRef] [Green Version]
- Frauenfeld, J.; Löving, R.; Armache, J.P.; Sonnen, A.F.P.; Guettou, F.; Moberg, P.; Zhu, L.; Jegerschöld, C.; Flayhan, A.; Briggs, J.A.G.; et al. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat. Methods 2016, 13, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.J.; Finka, R.; Smith, C.; Lin, Y.-P.P.; Dafforn, T.; Overduin, M. Membrane proteins solubilized intact in lipid containing nanoparticles bounded by styrene maleic acid copolymer. J. Am. Chem. Soc. 2009, 131, 7484–7485. [Google Scholar] [CrossRef] [PubMed]
- Oluwole, A.O.; Danielczak, B.; Meister, A.; Babalola, J.O.; Vargas, C.; Keller, S. Solubilization of membrane proteins into functional lipid-bilayer nanodiscs using a diisobutylene/maleic acid copolymer. Angew. Chem. Int. Ed. 2017, 56, 1919–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sligar, S.G.; Denisov, I.G. Nanodiscs: A toolkit for membrane protein science. Protein Sci. 2020, 1–19. [Google Scholar] [CrossRef]
- Denisov, I.G.; Sligar, S.G. Nanodiscs in membrane biochemistry and biophysics. Chem. Rev. 2017, 117, 4669–4713. [Google Scholar] [CrossRef]
- Dörr, J.M.; Koorengevel, M.C.; Schäfer, M.; Prokofyev, A.V.; Scheidelaar, S.; van der Cruijsen, E.A.W.W.; Dafforn, T.R.; Baldus, M.; Killian, J.A. Detergent-free isolation, characterization, and functional reconstitution of a tetrameric K+ channel: The power of native nanodiscs. Proc. Natl. Acad. Sci. USA 2014, 111, 18607–18612. [Google Scholar] [CrossRef] [Green Version]
- Overduin, M.; Klumperman, B. Advancing membrane biology with poly(styrene-co-maleic acid)-based native nanodiscs. Eur. Polym. J. 2019, 110, 63–68. [Google Scholar] [CrossRef]
- Xue, M.; Cheng, L.; Faustino, I.; Guo, W.; Marrink, S.J. Molecular mechanism of lipid nanodisc formation by styrene-maleic acid copolymers. Biophys. J. 2018, 115, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Cuevas Arenas, R.; Danielczak, B.; Martel, A.; Porcar, L.; Breyton, C.; Ebel, C.; Keller, S. Fast collisional lipid transfer among polymer-bounded nanodiscs. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Danielczak, B.; Keller, S. Collisional lipid exchange among DIBMA-encapsulated nanodiscs (DIBMALPs). Eur. Polym. J. 2018, 109, 206–213. [Google Scholar] [CrossRef]
- Martinez, D.; Decossas, M.; Kowal, J.; Frey, L.; Stahlberg, H.; Dufourc, E.J.; Riek, R.; Habenstein, B.; Bibow, S.; Loquet, A. Lipid internal dynamics probed in nanodiscs. ChemPhysChem 2017, 18, 2651–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raschle, T.; Hiller, S.; Etzkorn, M.; Wagner, G. Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr. Opin. Struct. Biol. 2010, 20, 471–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagn, F.; Etzkorn, M.; Raschle, T.; Wagner, G. Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 2013, 135, 1919–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokogawa, M.; Fukuda, M.; Osawa, M. Nanodiscs for structural biology in a membranous environment. Chem. Pharm. Bull. 2019, 67, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efremov, R.G.; Gatsogiannis, C.; Raunser, S. Lipid Nanodiscs as a Tool for High-Resolution Structure Determination of Membrane Proteins by Single-Particle Cryo-EM, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 594. [Google Scholar]
- Kalienkova, V.; Alvadia, C.; Clerico Mosina, V.; Paulino, C. Single-particle cryo-EM of membrane proteins in lipid nanodiscs. Methods Mol. Biol. 2020, 2127, 245–273. [Google Scholar] [CrossRef] [PubMed]
- Zocher, M.; Roos, C.; Wegmann, S.; Bosshart, P.D.; Dötsch, V.; Bernhard, F.; Müller, D.J. Single-molecule force spectroscopy from nanodiscs: An assay to quantify folding, stability, and interactions of native membrane proteins. ACS Nano 2012, 6, 961–971. [Google Scholar] [CrossRef]
- Daury, L.; Orange, F.; Taveau, J.C.; Verchère, A.; Monlezun, L.; Gounou, C.; Marreddy, R.K.R.; Picard, M.; Broutin, I.; Pos, K.M.; et al. Tripartite assembly of RND multidrug efflux pumps. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Meusch, D.; Gatsogiannis, C.; Efremov, R.G.; Lang, A.E.; Hofnagel, O.; Vetter, I.R.; Aktories, K.; Raunser, S. Mechanism of Tc toxin action revealed in molecular detail. Nature 2014, 508, 61–65. [Google Scholar] [CrossRef]
- Gatsogiannis, C.; Lang, A.E.; Meusch, D.; Pfaumann, V.; Hofnagel, O.; Benz, R.; Aktories, K.; Raunser, S. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature 2013, 495, 520–523. [Google Scholar] [CrossRef]
- Gatsogiannis, C.; Merino, F.; Prumbaum, D.; Roderer, D.; Leidreiter, F.; Meusch, D.; Raunser, S. Membrane insertion of a Tc toxin in near-atomic detail. Nat. Struct. Mol. Biol. 2016, 23, 884–890. [Google Scholar] [CrossRef]
- Rantalainen, K.; Berndsen, Z.T.; Antanasijevic, A.; Schiffner, T.; Zhang, X.; Lee, W.H.; Torres, J.L.; Zhang, L.; Irimia, A.; Copps, J.; et al. HIV-1 envelope and MPER antibody structures in lipid assemblies. Cell Rep. 2020, 31, 107583. [Google Scholar] [CrossRef] [PubMed]
- Frey, L.; Lakomek, N.A.; Riek, R.; Bibow, S. Micelles, bicelles, and nanodiscs: Comparing the impact of membrane mimetics on membrane protein backbone dynamics. Angew. Chemie Int. Ed. 2017, 56, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Frey, L.; Hiller, S.; Riek, R.; Bibow, S. Lipid- and cholesterol-mediated time-scale-specific modulation of the outer membrane protein X dynamics in lipid bilayers. J. Am. Chem. Soc. 2018, 140, 15402–15411. [Google Scholar] [CrossRef] [Green Version]
- Morgado, L.; Zeth, K.; Burmann, B.M.; Maier, T.; Hiller, S. Characterization of the insertase BamA in three different membrane mimetics by solution NMR spectroscopy. J. Biomol. NMR 2015, 61, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Osawa, M.; Mita, K.; Toyonaga, S.; Machiyama, A.; Ueda, T.; Takeuchi, K.; Oiki, S.; Shimada, I. Functional equilibrium of the KcsA structure revealed by NMR. J. Biol. Chem. 2012, 287, 39634–39641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiura, C.; Kofuku, Y.; Ueda, T.; Mase, Y.; Yokogawa, M.; Osawa, M.; Terashima, Y.; Matsushima, K.; Shimada, I. NMR analyses of the interaction between CCR5 and its ligand using functional reconstitution of CCR5 in lipid bilayers. J. Am. Chem. Soc. 2010, 132, 6768–6777. [Google Scholar] [CrossRef] [PubMed]
- Raschle, T.; Hiller, S.; Yu, T.Y.; Rice, A.J.; Walz, T.; Wagner, G. Structural and functional characterization of the integral membrane protein VDAC-1 in lipid bilayer nanodiscs. J. Am. Chem. Soc. 2009, 131, 17777–17779. [Google Scholar] [CrossRef] [Green Version]
- Oesterhelt, D.; Stoeckenius, W. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat. New Biol. 1971, 233, 149–152. [Google Scholar] [CrossRef]
- Blaurock, A.E.; Stoeckenius, W. Structure of the purple membrane. Nat. New Biol. 1971, 233, 152–155. [Google Scholar] [CrossRef]
- Olive, J. The Structural Organization of Mammalian Retinal Disc Membrane. Int Rev Cytol. 1980, 64, 107–169. [Google Scholar] [CrossRef]
- Arnold, T.; Linke, D. The use of detergents to purify membrane proteins. Curr. Protoc. Protein Sci. 2008, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Linke, D. Chapter 34 Detergents. An Overview, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2009; Volume 463. [Google Scholar]
- Rémigy, H.W.; Caujolle-Bert, D.; Suda, K.; Schenk, A.; Chami, M.; Engel, A. Membrane protein reconstitution and crystallization by controlled dilution. FEBS Lett. 2003, 555, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Rigaud, J.L.; Pitard, B.; Levy, D. Reconstitution of membrane proteins into liposomes: Application to energy-transducing membrane proteins. Biochim. Biophys. Acta Bioenerg. 1995, 1231, 223–246. [Google Scholar] [CrossRef] [Green Version]
- Hasler, L.; Heymann, J.; Engel, A. 2D crystallization of membrane proteins: Rationales and examples. J. Struct. Biol. 1998, 121, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Jap, B.; Zulauf, M.; Scheybani, T.; Hefti, A. 2D crystallization: From art to science. Ultramicroscopy 1992, 46, 45–84. [Google Scholar] [CrossRef]
- Romsicki, Y.; Sharom, F.J. Phospholipid flippase activity of the reconstituted P-glycoprotein multidrug transporter. Biochemistry 2001, 40, 6937–6947. [Google Scholar] [CrossRef]
- Tunuguntla, R.; Bangar, M.; Kim, K.; Stroeve, P.; Ajo-Franklin, C.M.; Noy, A. Lipid bilayer composition can influence the orientation of proteorhodopsin in artificial membranes. Biophys. J. 2013, 105, 1388–1396. [Google Scholar] [CrossRef] [Green Version]
- Ritzmann, N.; Thoma, J.; Hirschi, S.; Kalbermatter, D.; Fotiadis, D.; Müller, D.J. Fusion domains guide the oriented insertion of light-driven proton pumps into liposomes. Biophys. J. 2017, 113, 1181–1186. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, M.; Xie, J.; Heacock, P.; Dowhan, W. To flip or not to flip: Lipid-protein charge interactions are a determinant of final membrane protein topology. J. Cell Biol. 2008, 182, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Dowhan, W.; Bogdanov, M. Lipid-dependent membrane protein topogenesis. Annu. Rev. Biochem. 2009, 78, 515–540. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, M.; Dowhan, W. Lipid-dependent generation of dual topology for a membrane protein. J. Biol. Chem. 2012, 287, 37939–37948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinschmidt, J.H.; Den Blaauwen, T.; Driessen, A.J.M.; Tamm, L.K. Outer membrane protein A of Escherichia coli inserts and folds into lipid bilayers by a concerted mechanism. Biochemistry 1999, 38, 5006–5016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, A.M.; Fleming, K.G. BamA alone accelerates outer membrane protein folding in vitro through a catalytic mechanism. Biochemistry 2015, 54, 6009–6011. [Google Scholar] [CrossRef] [PubMed]
- Gessmann, D.; Chung, Y.H.; Danoff, E.J.; Plummer, A.M.; Sandlin, C.W.; Zaccai, N.R.; Fleming, K.G. Outer membrane β-barrel protein folding is physically controlled by periplasmic lipid head groups and BamA. Proc. Natl. Acad. Sci. USA 2014, 111, 5878–5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffrin, B.; Calabrese, A.N.; Higgins, A.J.; Humes, J.R.; Ashcroft, A.E.; Kalli, A.C.; Brockwell, D.J.; Radford, S.E. Effects of periplasmic chaperones and membrane thickness on BamA-catalyzed outer-membrane Ppotein folding. J. Mol. Biol. 2017, 429, 3776–3792. [Google Scholar] [CrossRef] [PubMed]
- Fujiyoshi, Y. The structural study of membrane proteins by electron crystallography. Adv. Biophys. 1998, 35, 25–80. [Google Scholar] [CrossRef]
- Stahlberg, H.; Fotiadis, D.; Scheuring, S.; Rémigy, H.; Braun, T.; Mitsuoka, K.; Fujiyoshi, Y.; Engel, A. Two-dimensional crystals: A powerful approach to assess structure, function and dynamics of membrane proteins. FEBS Lett. 2001, 504, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Henderson, R.; Baldwin, J.M.; Ceska, T.A.; Zemlin, F.; Beckmann, E.; Downing, K.H. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 1990, 213, 899–929. [Google Scholar] [CrossRef]
- Mitsuoka, K.; Murata, K.; Walz, T.; Hirai, T.; Agre, P.; Heymann, J.B.; Engel, A.; Fujiyoshi, Y. The structure of aquaporin-1 at 4.5-Å resolution reveals short α- helices in the center of the monomer. J. Struct. Biol. 1999, 128, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Kühlbrandt, W.; Wang, D.N.; Fujiyoshi, Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature 1994, 367, 614–621. [Google Scholar] [CrossRef]
- Hutchings, J.; Stancheva, V.; Miller, E.A.; Zanetti, G. Subtomogram averaging of COPII assemblies reveals how coat organization dictates membrane shape. Nat. Commun. 2018, 9, 4154. [Google Scholar] [CrossRef] [PubMed]
- Kudryashev, M.; Castaño-Díez, D.; Deluz, C.; Hassaine, G.; Grasso, L.; Graf-Meyer, A.; Vogel, H.; Stahlberg, H. The Structure of the mouse serotonin 5-HT3 receptor in lipid vesicles. Structure 2016, 24, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaño-Díez, D.; Zanetti, G. In situ structure determination by subtomogram averaging. Curr. Opin. Struct. Biol. 2019, 58, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Opella, S.J. Solid-state NMR and membrane proteins. J. Magn. Reson. 2015, 253, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubeis, T.; Le Marchand, T.; Andreas, L.B.; Pintacuda, G. 1H magic-angle spinning NMR evolves as a powerful new tool for membrane proteins. J. Magn. Reson. 2018, 287, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Giller, K.; Hornig, S.; Martin-Eauclaire, M.F.; Pongs, O.; Becker, S.; Baldus, M. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature 2006, 440, 959–962. [Google Scholar] [CrossRef]
- Jekhmane, S.; Medeiros-Silva, J.; Li, J.; Kümmerer, F.; Müller-Hermes, C.; Baldus, M.; Roux, B.; Weingarth, M. Shifts in the selectivity filter dynamics cause modal gating in K+ channels. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Saurel, O.; Iordanov, I.; Nars, G.; Demange, P.; Le Marchand, T.; Andreas, L.B.; Pintacuda, G.; Milon, A. Local and global dynamics in Klebsiella pneumoniae outer membrane protein a in lipid bilayers probed at atomic resolution. J. Am. Chem. Soc. 2017, 139, 1590–1597. [Google Scholar] [CrossRef] [Green Version]
- Retel, J.S.; Nieuwkoop, A.J.; Hiller, M.; Higman, V.A.; Barbet-Massin, E.; Stanek, J.; Andreas, L.B.; Franks, W.T.; Van Rossum, B.J.; Vinothkumar, K.R.; et al. Structure of outer membrane protein G in lipid bilayers. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bippes, C.A.; Müller, D.J. High-resolution atomic force microscopy and spectroscopy of native membrane proteins. Rep. Prog. Phys. 2011, 74, 086601. [Google Scholar] [CrossRef]
- Engel, A.; Gaub, H.E. Structure and mechanics of membrane proteins. Annu. Rev. Biochem. 2008, 77, 127–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, A.; Schoenenberger, C.-A.; Müller, D.J. High resolution imaging of native biological sample surfaces using scanning probe microscopy. Curr. Opin. Struct. Biol. 1997, 7, 279–284. [Google Scholar] [CrossRef]
- Frederix, P.L.T.M.; Bosshart, P.D.; Engel, A. Atomic force microscopy of biological membranes. Biophys. J. 2009, 96, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufrêne, Y.F.; Ando, T.; Garcia, R.; Alsteens, D.; Martinez-Martin, D.; Engel, A.; Gerber, C.; Müller, D.J. Imaging modes of atomic force microscopy for application in molecular and cell biology. Nat. Nanotechnol. 2017, 12, 295–307. [Google Scholar] [CrossRef]
- Mari, S.A.; Pessoa, J.; Altieri, S.; Hensen, U.; Thomas, L.; Morais-Cabral, J.H.; Müller, D.J. Gating of the MlotiK1 potassium channel involves large rearrangements of the cyclic nucleotide-binding domains. Proc. Natl. Acad. Sci. USA 2011, 108, 20802–20807. [Google Scholar] [CrossRef] [Green Version]
- Philippsen, A.; Im, W.; Engel, A.; Schirmer, T.; Roux, B.; Müller, D.J. Imaging the electrostatic potential of transmembrane channels: Atomic probe microscopy of OmpF porin. Biophys. J. 2002, 82, 1667–1676. [Google Scholar] [CrossRef] [Green Version]
- Pfreundschuh, M.; Hensen, U.; Müller, D. Quantitative imaging of the electrostatic field and potential generated by a transmembrane protein pore at subnanometer resolution. Nano Lett. 2013, 13, 5585–5593. [Google Scholar] [CrossRef]
- Yamashita, H.; Voïtchovsky, K.; Uchihashi, T.; Contera, S.A.; Ryan, J.F.; Ando, T. Dynamics of bacteriorhodopsin 2D crystal observed by high-speed atomic force microscopy. J. Struct. Biol. 2009, 167, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Shibata, M.; Yamashita, H.; Uchihashi, T.; Kandori, H.; Ando, T. High-speed atomic force microscopy shows dynamic molecular processes in photoactivated bacteriorhodopsin. Nat. Nanotechnol. 2010, 5, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Oesterhelt, F.; Oesterhelt, D.; Pfeiffer, M.; Engel, A.; Gaub, H.E.; Müller, D.J. Unfolding pathways of individual bacteriorhodopsins. Science 2000, 288, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Kedrov, A.; Janovjak, H.; Sapra, K.T.; Müller, D.J. Deciphering molecular interactions of native membrane proteins by single-molecule force spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 233–260. [Google Scholar] [CrossRef] [PubMed]
- Thoma, J.; Sapra, K.T.; Müller, D.J. Single-molecule force spectroscopy of transmembrane β-barrel proteins. Annu. Rev. Anal. Chem. 2018, 11, 375–395. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Gottschalk, K.E.; Janovjak, H.; Müller, D.J.; Gaub, H.E. Bacteriorhodopsin folds into the membrane against an external force. J. Mol. Biol. 2006, 357, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Thoma, J.; Burmann, B.M.; Hiller, S.; Müller, D.J. Impact of holdase chaperones Skp and SurA on the folding of β-barrel outer-membrane proteins. Nat. Struct. Mol. Biol. 2015, 22, 795–802. [Google Scholar] [CrossRef]
- Serdiuk, T.; Balasubramaniam, D.; Sugihara, J.; Mari, S.A.; Kaback, H.R.; Müller, D.J. YidC assists the stepwise and stochastic folding of membrane proteins. Nat. Chem. Biol. 2016, 12, 911–917. [Google Scholar] [CrossRef]
- Alsteens, D.; Pfreundschuh, M.; Zhang, C.; Spoerri, P.M.; Coughlin, S.R.; Kobilka, B.K.; Müller, D.J. Imaging G protein-coupled receptors while quantifying their ligand-binding free-energy landscape. Nat. Methods 2015, 1, 12103–12108. [Google Scholar] [CrossRef] [Green Version]
- Harsman, A.; Niemann, M.; Pusnik, M.; Schmidt, O.; Burmann, B.M.; Hiller, S.; Meisinger, C.; Schneider, A.; Wagner, R. Bacterial origin of a mitochondrial outer membrane protein translocase: New perspectives from comparative single channel electrophysiology. J. Biol. Chem. 2012, 287, 31437–31445. [Google Scholar] [CrossRef] [Green Version]
- Hilty, C.; Winterhalter, M. Facilitated substrate transport through membrane proteins. Phys. Rev. Lett. 2001, 86, 5624–5627. [Google Scholar] [CrossRef]
- Wang, J.; Terrasse, R.; Bafna, J.A.; Benier, L.; Winterhalter, M. Electrophysiological characterization of transport across outer-membrane channels from gram-negative bacteria in presence of lipopolysaccharides. Angew. Chem. Int. Ed. 2020, 59, 8517–8521. [Google Scholar] [CrossRef] [Green Version]
- Kreir, M.; Farre, C.; Beckler, M.; George, M.; Fertig, N. Rapid screening of membrane protein activity: Electrophysiological analysis of OmpF reconstituted in proteoliposomes. Lab Chip 2008, 8, 587–595. [Google Scholar] [CrossRef]
- Zeidel, M.L.; Ambudkar, S.V.; Smith, B.L.; Agre, P. Reconstitution of functional water channels in liposomes containing purified red cell CHIP28 protein. Biochemistry 1992, 31, 7436–7440. [Google Scholar] [CrossRef] [PubMed]
- Posson, D.J.; Rusinova, R.; Andersen, O.S.; Nimigean, C.M. Stopped-flow fluorometric ion flux assay for ligand-gated ion channel studies. Methods Mol. Biol. 2018, 1684, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Akiyoshi, K. Biotransporting biocatalytic reactors toward therapeutic nanofactories. Adv. Sci. 2018, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, M.; Davidson, M.; Karlsson, R.; Karlsson, A.; Bergenholtz, J.; Konkoli, Z.; Jesorka, A.; Lobovkina, T.; Hurtig, J.; Voinova, M.; et al. Biomimetic nanoscale reactors and networks. Annu. Rev. Phys. Chem. 2004, 55, 613–649. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Sharma, S.K.; Dai, T.; Chung, H.; Yaroslavsky, A.; Garcia-Diaz, M.; Chang, J.; Chiang, L.Y.; Hamblin, M.R. Can nanotechnology potentiate photodynamic therapy? Nanotechnol. Rev. 2012, 1, 111–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Discher, D.E.; Ahmed, F. Polymersomes. Annu. Rev. Biomed. Eng. 2006, 8, 323–341. [Google Scholar] [CrossRef]
- Mecke, A.; Dittrich, C.; Meier, W. Biomimetic membranes designed from amphiphilic block copolymers. Soft Matter 2006, 2, 751–759. [Google Scholar] [CrossRef]
- Zhang, X.; Tanner, P.; Graff, A.; Palivan, C.G.; Meier, W. Mimicking the cell membrane with block copolymer membranes. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2293–2318. [Google Scholar] [CrossRef]
- Poschenrieder, S.T.; Schiebel, S.K.; Castiglione, K. Stability of polymersomes with focus on their use as nanoreactors. Eng. Life Sci. 2018, 18, 101–113. [Google Scholar] [CrossRef]
- Palivan, C.G.; Goers, R.; Najer, A.; Zhang, X.; Car, A.; Meier, W. Bioinspired polymer vesicles and membranes for biological and medical applications. Chem. Soc. Rev. 2016, 45, 377–411. [Google Scholar] [CrossRef] [Green Version]
- Kowal, J.; Wu, D.; Mikhalevich, V.; Palivan, C.G.; Meier, W. Hybrid polymer-lipid films as platforms for directed membrane protein insertion. Langmuir 2015, 31, 4868–4877. [Google Scholar] [CrossRef] [PubMed]
- Thoma, J.; Belegrinou, S.; Rossbach, P.; Grzelakowski, M.; Kita-Tokarczyk, K.; Meier, W. Membrane protein distribution in composite polymer–lipid thin films. Chem. Commun. 2012, 48, 8811. [Google Scholar] [CrossRef] [PubMed]
- Kaback, H.R. The role of the phosphoenolpyruvate-phosphotransferase system in the transport of sugars by isolated membrane preparations of Escherichia coli. J. Biol. Chem. 1968, 243, 3711–3724. [Google Scholar] [PubMed]
- Kaback, H.R. Transport studies in bacterial membrane vesicles. Science 1974, 186, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; Unwin, P.N.T. Three-dimensional model of purple membrane obtained by electron microscopy. Nature 1975, 257, 28–32. [Google Scholar] [CrossRef]
- Baldwin, J.; Henderson, R. Measurement and evaluation of electron diffraction patterns from two-dimensional crystals. Ultramicroscopy 1984, 14, 319–335. [Google Scholar] [CrossRef]
- Glaeser, R.M.; Jubb, J.S.; Henderson, R. Structural comparison of native and deoxycholate-treated purple membrane. Biophys. J. 1985, 48, 775–780. [Google Scholar] [CrossRef] [Green Version]
- Hoenger, A.; Ghosh, R.; Schoenenberger, C.A.; Aebi, U.; Engel, A. Direct in situ structural analysis of recombinant outer membrane porins expressed in an OmpA-deficient mutant Escherichia coli strain. J. Struct. Biol. 1993, 111, 212–221. [Google Scholar] [CrossRef]
- Briggs, J.A.G. Structural biology in situ-the potential of subtomogram averaging. Curr. Opin. Struct. Biol. 2013, 23, 261–267. [Google Scholar] [CrossRef]
- Galaz-Montoya, J.G.; Ludtke, S.J. The advent of structural biology in situ by single particle cryo-electron tomography. Biophys. Rep. 2017, 3, 17–35. [Google Scholar] [CrossRef] [Green Version]
- Gold, V.A.M.; Ieva, R.; Walter, A.; Pfanner, N.; Van Der Laan, M.; Kühlbrandt, W. Visualizing active membrane protein complexes by electron cryotomography. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nans, A.; Kudryashev, M.; Saibil, H.R.; Hayward, R.D. Structure of a bacterial type III secretion system in contact with a host membrane in situ. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Chen, M.; Yu, Z.; Bell, J.M.; Wang, H.; Forrester, I.; Villarreal, H.; Jakana, J.; Du, D.; Luisi, B.F.; et al. In situ structure and assembly of the multidrug efflux pump AcrAB-TolC. Nat. Commun. 2019, 10, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, D.; Chang, Y.; Jeong, K.C.; Vogel, J.P.; Jensen, G.J. In situ structure of the Legionella Dot/Icm type IV secretion system by electron cryotomography. EMBO Rep. 2017, 18, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Rapisarda, C.; Cherrak, Y.; Kooger, R.; Schmidt, V.; Pellarin, R.; Logger, L.; Cascales, E.; Pilhofer, M.; Durand, E.; Fronzes, R. In situ and high-resolution cryo- EM structure of a bacterial type VI secretion system membrane complex. EMBO J. 2019, 38, 1–18. [Google Scholar] [CrossRef]
- Zeev-Ben-Mordehai, T.; Vasishtan, D.; Siebert, C.A.; Whittle, C.; Grünewald, K. Extracellular vesicles: A platform for the structure determination of membrane proteins by cryo-EM. Structure 2014, 22, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.E.; Wang, S.; Munro, R.; Ritz, E.; Hung, I.; Gor’Kov, P.L.; Jiang, Y.; Liang, H.; Brown, L.S.; Ladizhansky, V. In situ structural studies of anabaena sensory rhodopsin in the E. coli membrane. Biophys. J. 2015, 108, 1683–1696. [Google Scholar] [CrossRef] [Green Version]
- Pinto, C.; Mance, D.; Julien, M.; Daniels, M.; Weingarth, M.; Baldus, M. Studying assembly of the BAM complex in native membranes by cellular solid-state NMR spectroscopy. J. Struct. Biol. 2019, 206, 1–11. [Google Scholar] [CrossRef]
- Baker, L.A.; Sinnige, T.; Schellenberger, P.; de Keyzer, J.; Siebert, C.A.; Driessen, A.J.M.; Baldus, M.; Grünewald, K. Combined 1H-detected solid-state NMR spectroscopy and electron cryotomography to study membrane proteins across resolutions in native environments. Structure 2018, 26, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Müller, D. AFM: A Nanotool in membrane biology. Biochemistry 2008, 47, 7986–7998. [Google Scholar] [CrossRef]
- Liu, L.N.; Scheuring, S. High-resolution AFM imaging of native biological membranes. Nanoscale Liq. Interfaces 2013, 657–681. [Google Scholar] [CrossRef]
- Worcester, D.L.; Miller, R.G.; Bryant, P.J. Atomic force microscopy of purple membranes. J. Microsc. 1988, 152, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.J.; Schabert, F.A.; Büldt, G.; Engel, A. Imaging purple membranes in aqueous solutions at sub-nanometer resolution by atomic force microscopy. Biophys. J. 1995, 68, 1681–1686. [Google Scholar] [CrossRef] [Green Version]
- Buzhynskyy, N.; Hite, R.K.; Walz, T.; Scheuring, S. The supramolecular architecture of junctional microdomains in native lens membranes. EMBO Rep. 2007, 8, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, R.P.; Buzhynskyy, N.; Prima, V.; Sturgis, J.N.; Scheuring, S. Supramolecular assembly of VDAC in native mitochondrial outer membranes. J. Mol. Biol. 2007, 369, 413–418. [Google Scholar] [CrossRef]
- Fotiadis, D.; Liang, Y.; Filipek, S.; Saperstein, D.A.; Engel, A.; Palczewski, K. Atomic-force microscopy: Rhodopsin dimers in native disc membranes. Nature 2003, 421, 127–128. [Google Scholar] [CrossRef]
- Phuthong, W.; Huang, Z.; Wittkopp, T.M.; Sznee, K.; Heinnickel, M.L.; Dekker, J.P.; Frese, R.N.; Prinz, F.B.; Grossman, A.R. The use of contact mode atomic force microscopy in aqueous medium for structural analysis of spinach photosynthetic complexes. Plant Physiol. 2015, 169, 1318–1332. [Google Scholar] [CrossRef] [Green Version]
- Thoma, J.; Manioglu, S.; Kalbermatter, D.; Bosshart, P.D.; Fotiadis, D.; Müller, D.J. Protein-enriched outer membrane vesicles as a native platform for outer membrane protein studies. Commun. Biol. 2018, 1, 23. [Google Scholar] [CrossRef]
- Thoma, J.; Sun, Y.; Ritzmann, N.; Müller, D.J. POTRA domains, extracellular lid, and membrane composition modulate the conformational stability of the β-barrel assembly factor BamA. Structure 2018, 26, 987–996. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Solubilized states of membrane proteins. (A) Membrane protein in a detergent micelle (cyan). (B) Membrane protein in a disc-shaped bicelle containing lipids (blue). (C) Membrane proteins in nanodiscs stabilized by membrane scaffold protein (MSP, green) and amphipathic polymers (purple).
Figure 1.
Solubilized states of membrane proteins. (A) Membrane protein in a detergent micelle (cyan). (B) Membrane protein in a disc-shaped bicelle containing lipids (blue). (C) Membrane proteins in nanodiscs stabilized by membrane scaffold protein (MSP, green) and amphipathic polymers (purple).

Figure 2.
Solubilization and purification of membrane proteins. (A) Native membrane containing a membrane protein of interest over a background of other membrane proteins in a diverse lipid composition. (B) Mixed membrane protein-detergent and lipid-detergent micelles resulting from solubilization of the membrane through the addition of detergent. (C) Purified membrane protein in detergent micelles after removal of undesired lipid components and protein contaminations.
Figure 2.
Solubilization and purification of membrane proteins. (A) Native membrane containing a membrane protein of interest over a background of other membrane proteins in a diverse lipid composition. (B) Mixed membrane protein-detergent and lipid-detergent micelles resulting from solubilization of the membrane through the addition of detergent. (C) Purified membrane protein in detergent micelles after removal of undesired lipid components and protein contaminations.

Figure 3.
Reconstitution of membrane proteins into liposomes. (A) Bottom-up assembly from a ternary mixture (left), driven by detergent removal, typically results in an alternating up-down orientation of membrane proteins (right). (B) Undirected insertion of membrane proteins into preformed liposomes. (C) Directed insertion of membrane proteins into preformed liposomes with soluble domains facing outwards.
Figure 3.
Reconstitution of membrane proteins into liposomes. (A) Bottom-up assembly from a ternary mixture (left), driven by detergent removal, typically results in an alternating up-down orientation of membrane proteins (right). (B) Undirected insertion of membrane proteins into preformed liposomes. (C) Directed insertion of membrane proteins into preformed liposomes with soluble domains facing outwards.


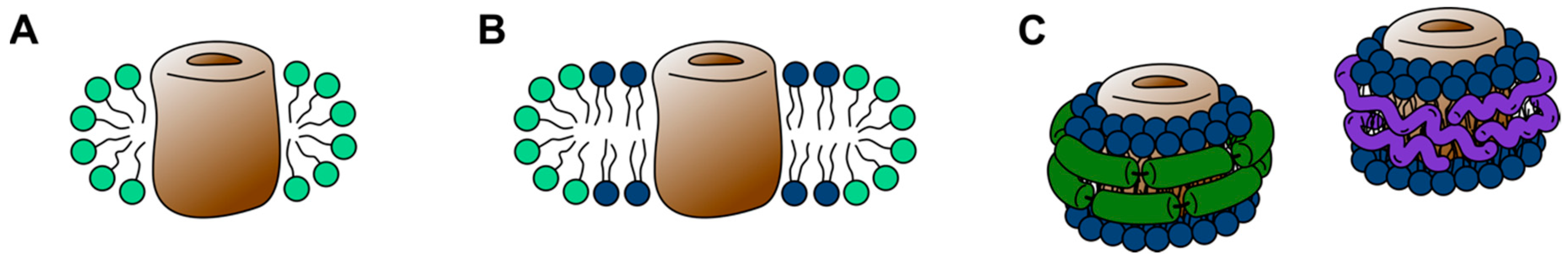{kind=link}
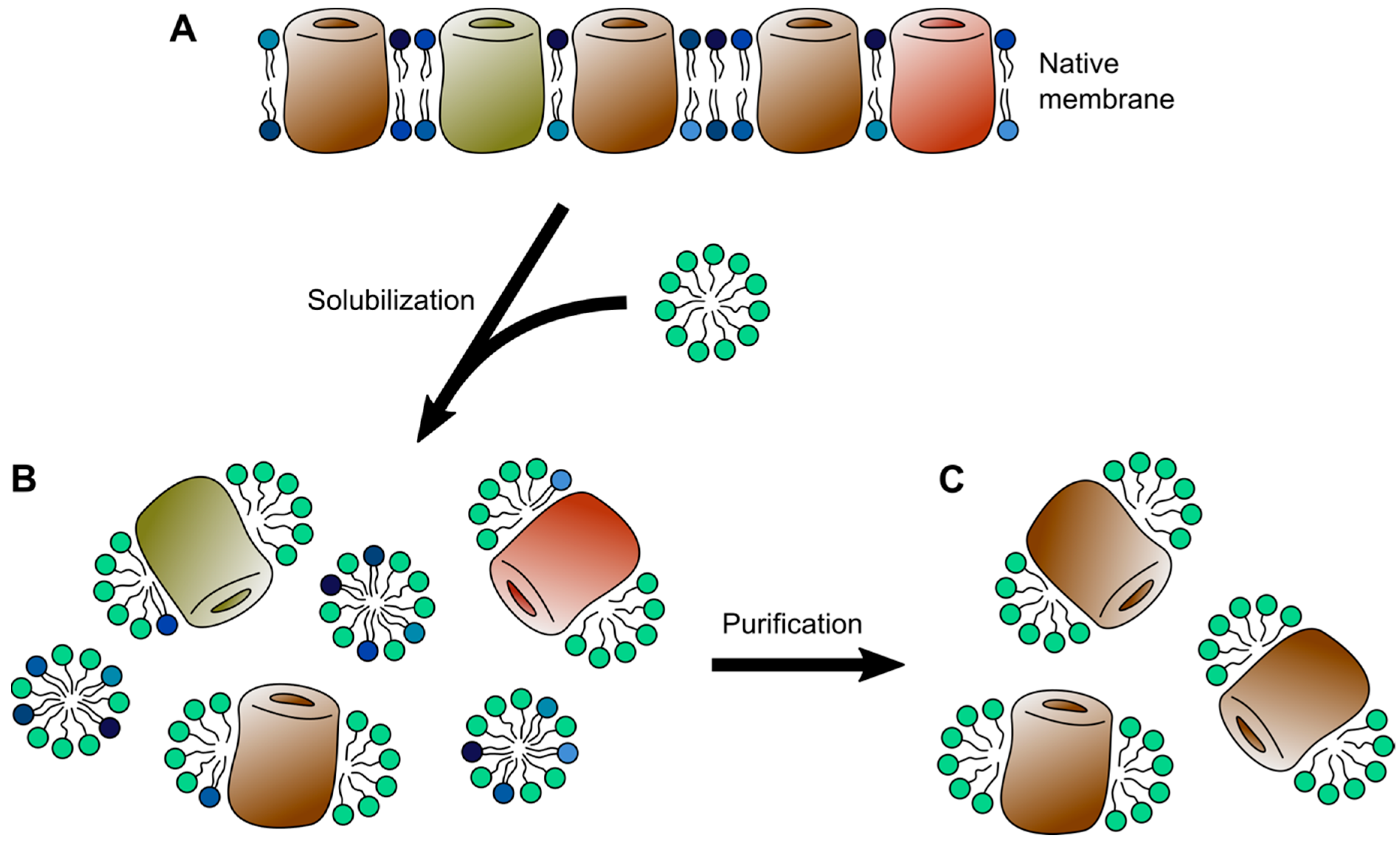{kind=link}
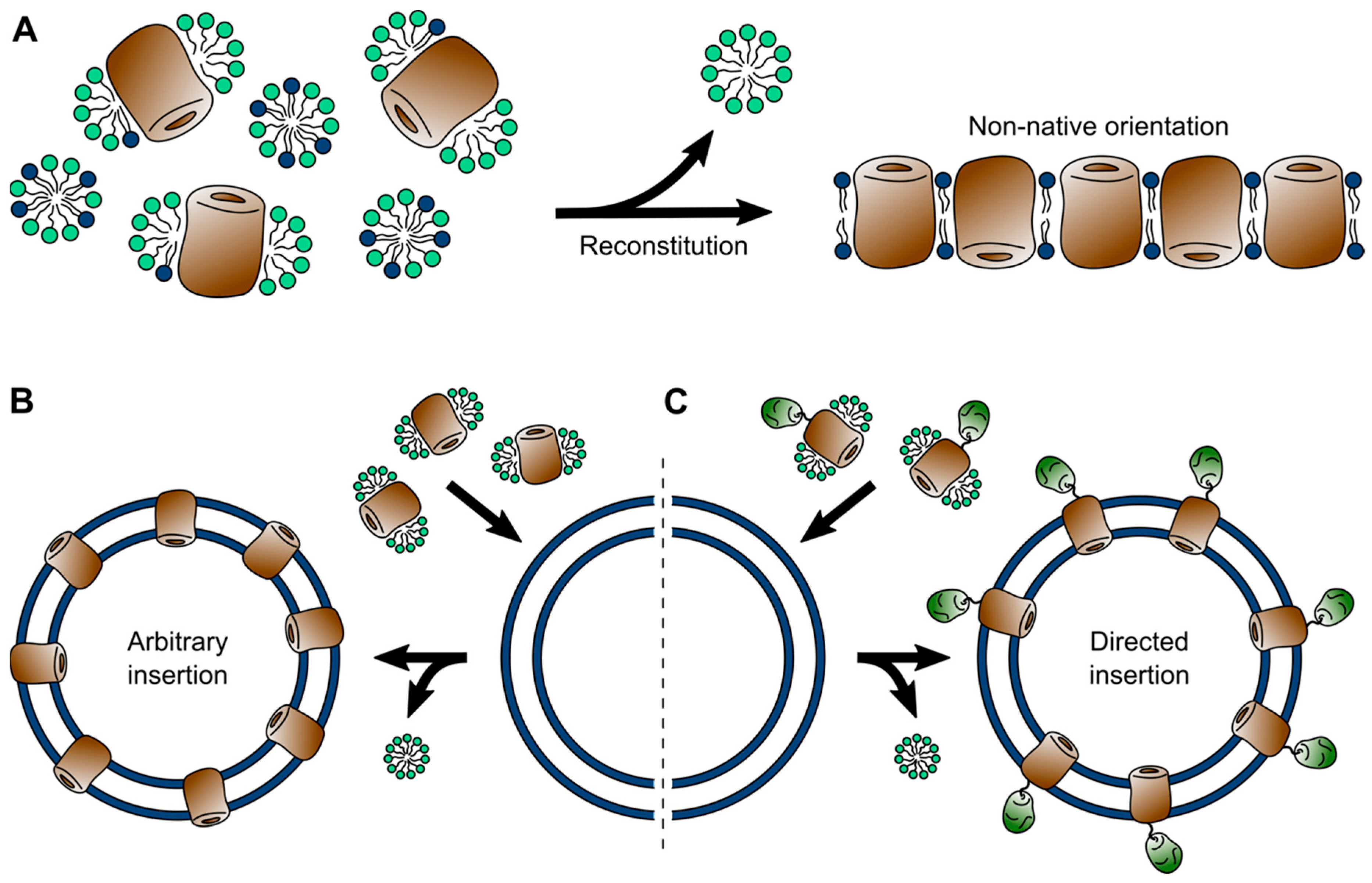{kind=link}
Table 1.
Pros and cons of different membrane systems.
| System | Pro | Contra | Suitable methods |
|---|---|---|---|
| Detergent micelles | Universally used; starting point for downstream applications | Can have denaturing effects; may disrupt complexes; de-lipidation of membrane proteins | Single-particle Cryo-EM; solution NMR; X-ray / neutron solution scattering; MS/MS |
| 3D crystals | Most prevalent system for structure determination; can include lipids (LCP) | Non-native crystal contacts; often requires protein engineering; proteins are “locked” in one state; crystallization artifacts | X-ray crystallography; Micro-ED |
| Bicelles | Lipid system; can be used for 3D crystallization; variety of shapes and sizes | Limited lipid diversity; altered lipid dynamics | Solution NMR; solid-state NMR; (X-ray crystallography) |
| Nanodiscs | Lipid system; broad range of lipid compositions; possible to extract native lipid composition | Limited size range; altered lipid dynamics; membrane asymmetry is lost | Single particle Cryo-EM; solution NMR; Cryo-electron tomography; AFM |
| Liposomes | Lipid system; Broad range of lipid compositions; high protein density possible; facilitate transmembrane transport studies | Often non-native protein orientation; not possible to create asymmetric bilayers | Electron crystallography; Cryo-ET; solid-state NMR; AFM; electrophysiology; fluorometry |
| Native membranes | Native environment | Often difficult to handle; low content of protein of interest over “contaminants” | Cryo-ET; solid-state NMR; AFM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Thoma, J.; Burmann, B.M. Fake It ‘Till You Make It—The Pursuit of Suitable Membrane Mimetics for Membrane Protein Biophysics. Int. J. Mol. Sci. 2021, 22, 50. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010050
AMA Style
Thoma J, Burmann BM. Fake It ‘Till You Make It—The Pursuit of Suitable Membrane Mimetics for Membrane Protein Biophysics. International Journal of Molecular Sciences. 2021; 22(1):50. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010050
Chicago/Turabian StyleThoma, Johannes, and Björn M. Burmann. 2021. "Fake It ‘Till You Make It—The Pursuit of Suitable Membrane Mimetics for Membrane Protein Biophysics" International Journal of Molecular Sciences 22, no. 1: 50. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010050
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.