Proinflammatory Cytokines Perturb Mouse and Human Pancreatic Islet Circadian Rhythmicity and Induce Uncoordinated β-Cell Clock Gene Expression via Nitric Oxide, Lysine Deacetylases, and Immunoproteasomal Activity

 , , ,
, , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
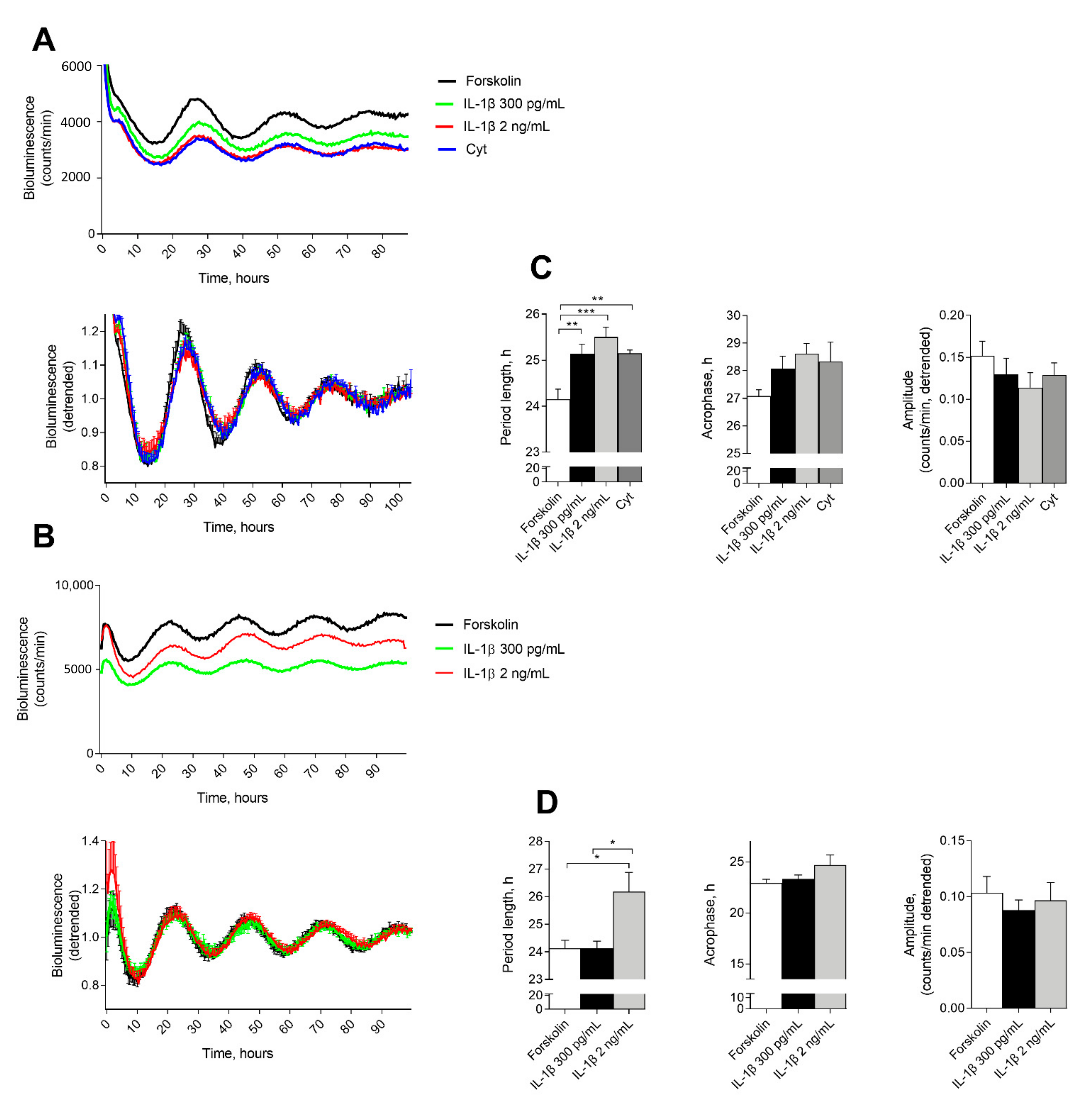
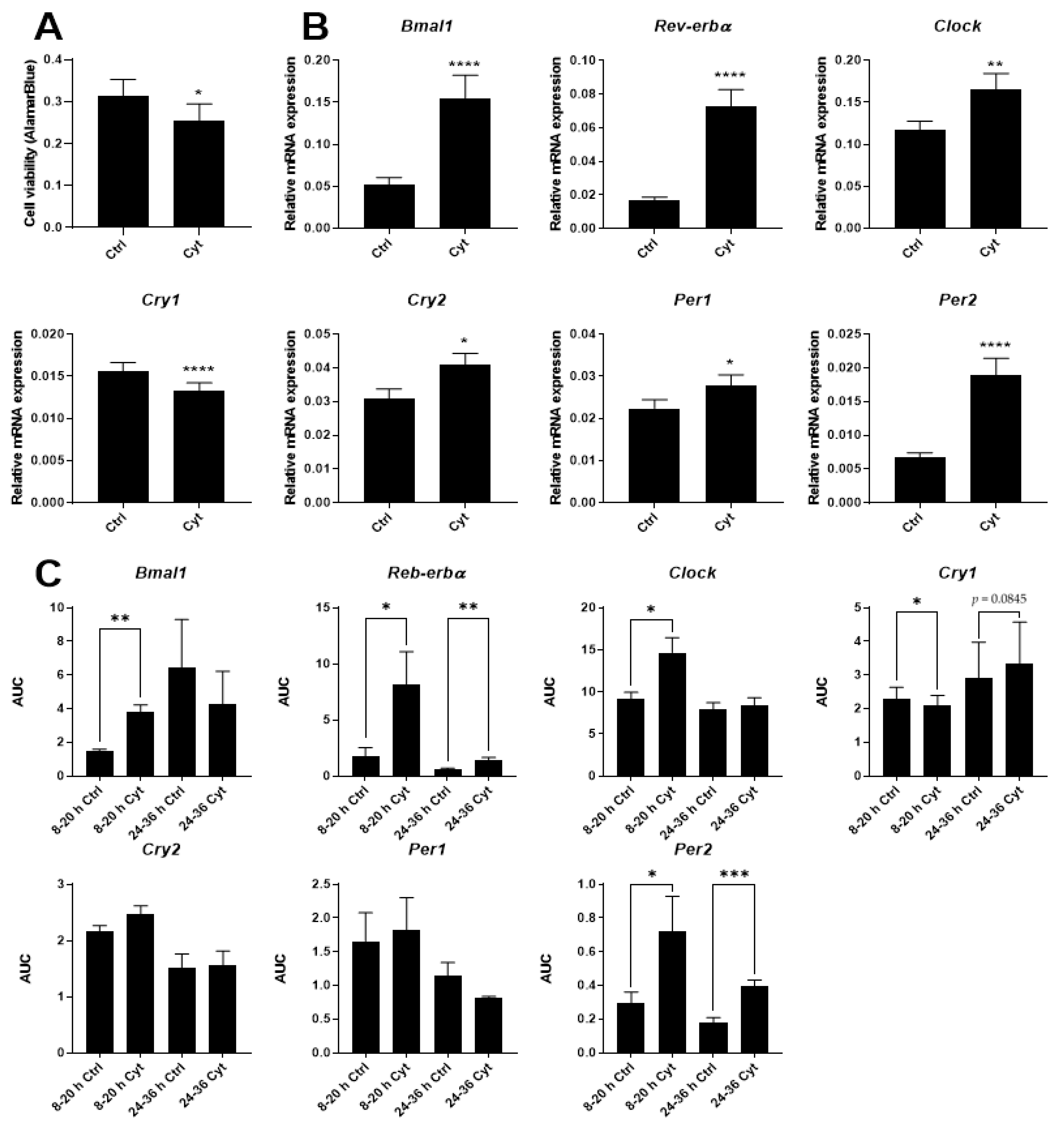
2.1. IL-1β Affects Circadian Clockwork in Murine and Human Pancreatic Islets
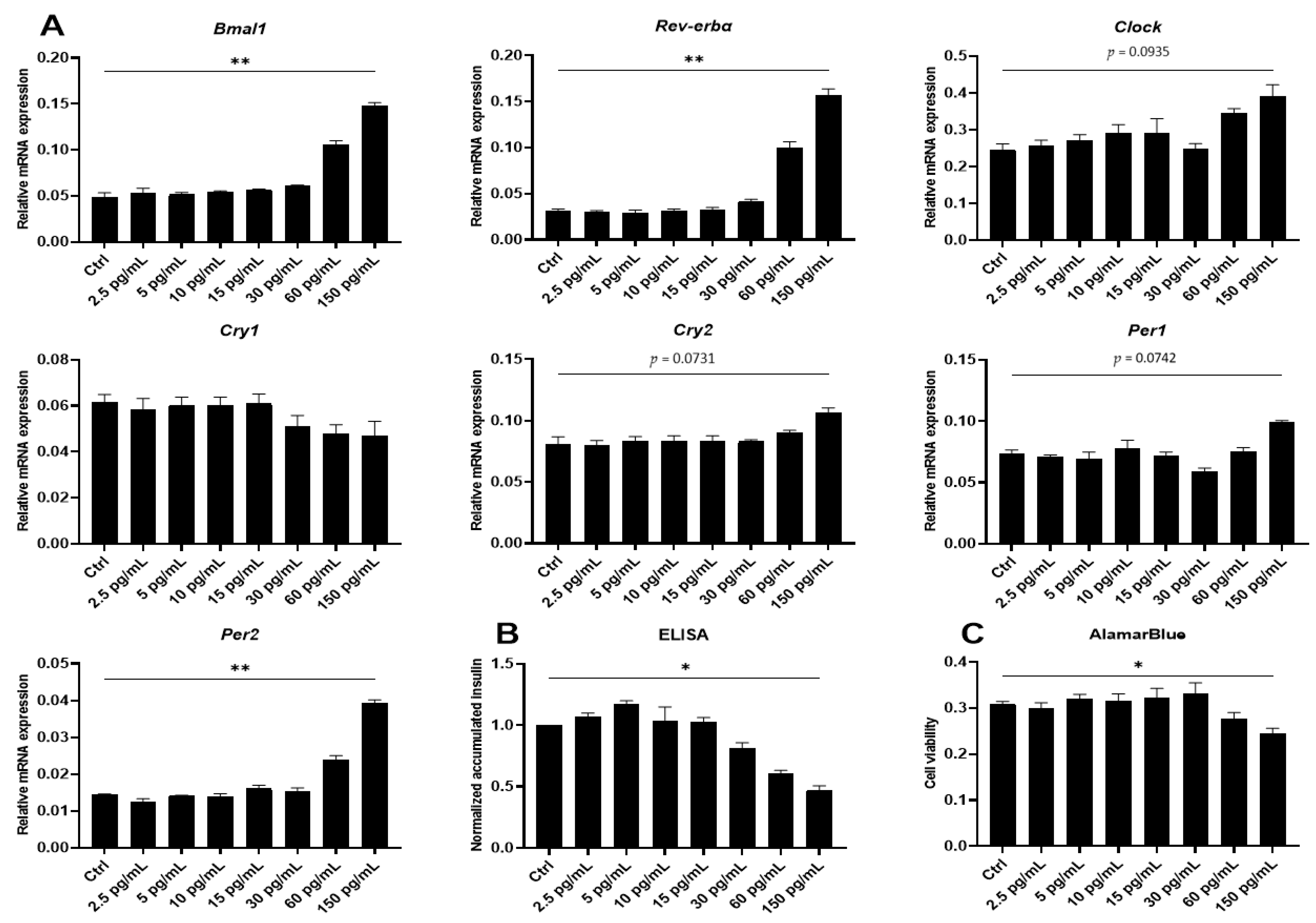
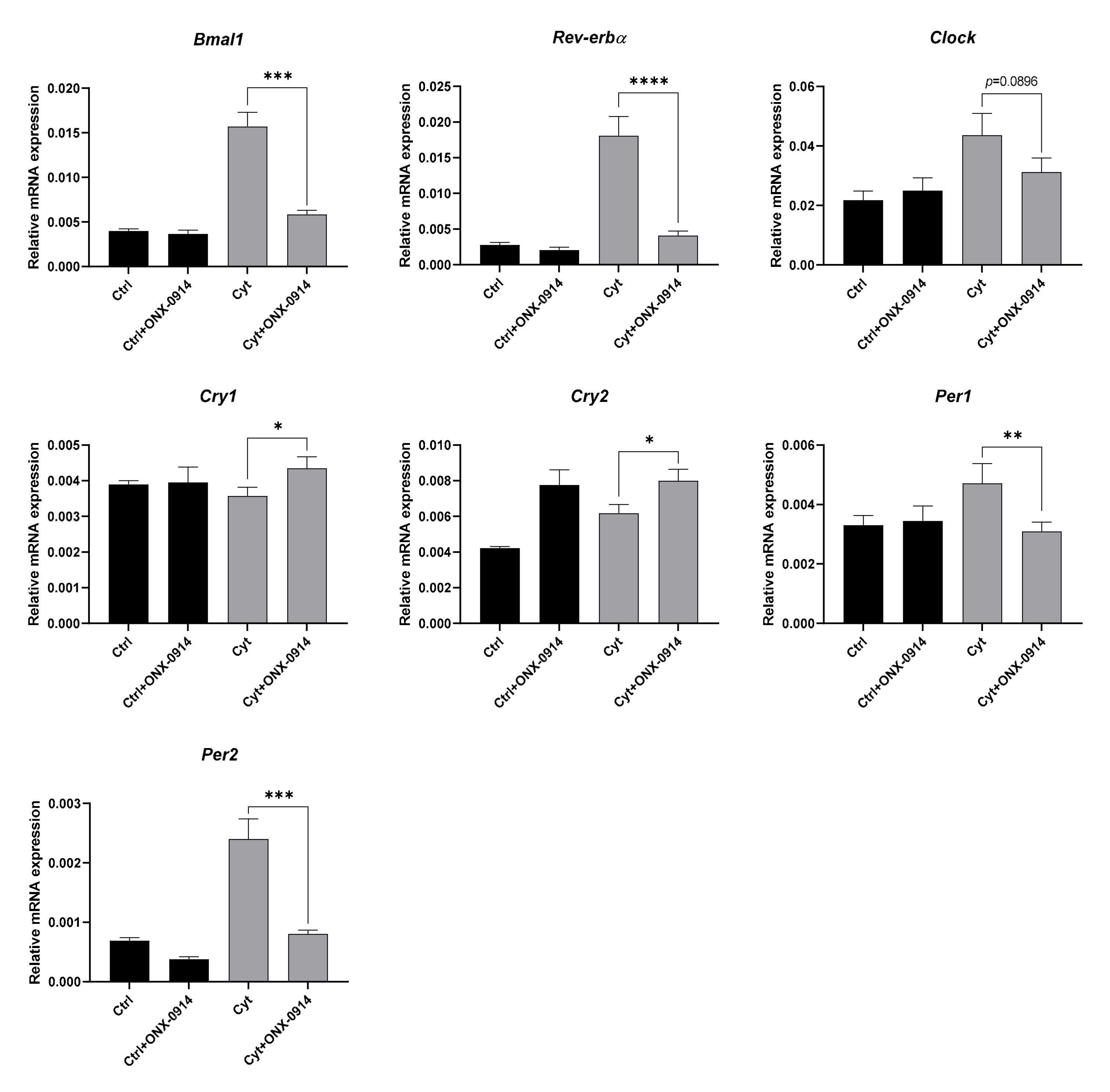
2.2. Proinflammatory Cytokines Induce Marked but Uncoordinated Expression of Core Clock Genes in Insulin Producing Cells
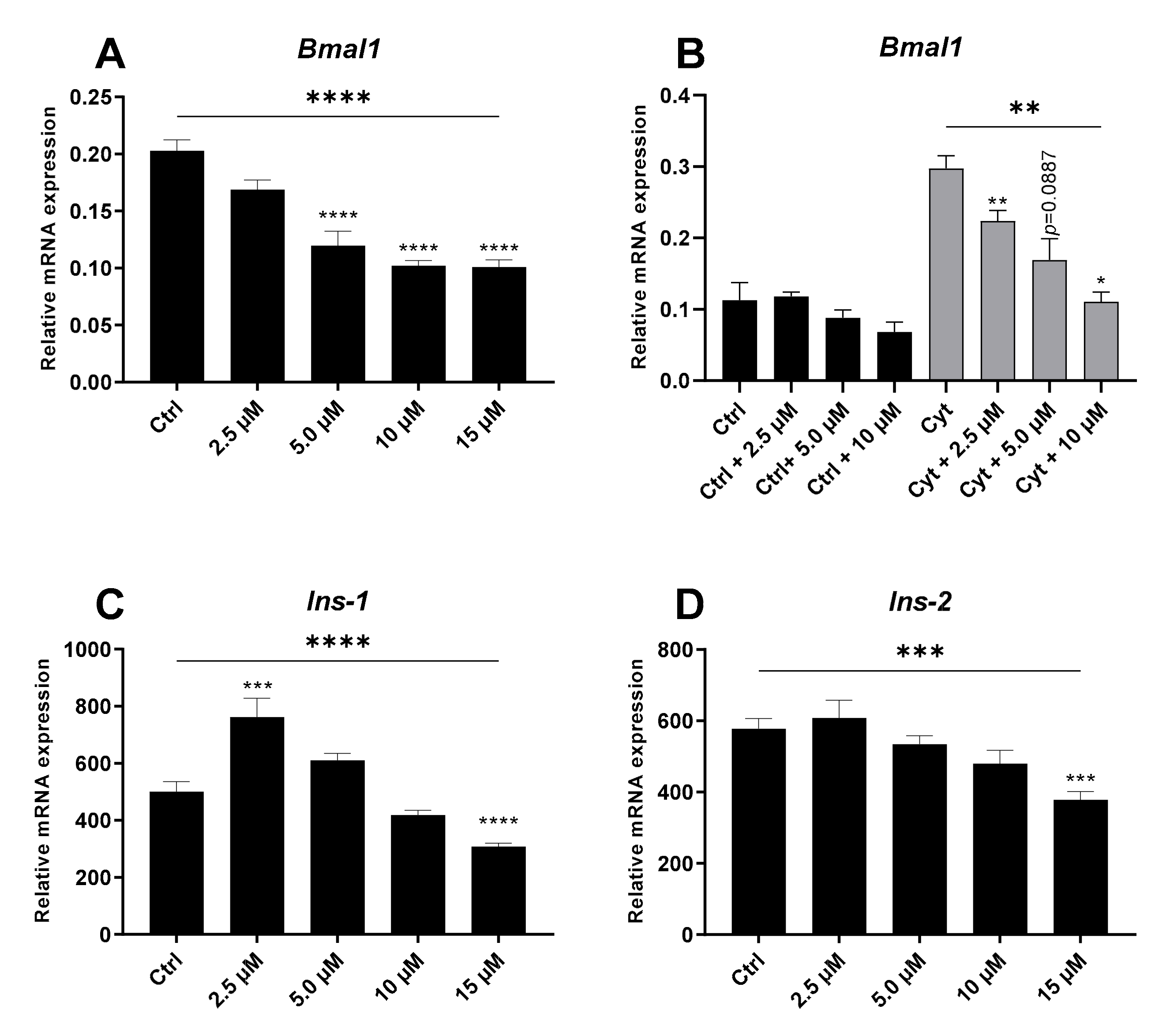
2.3. The REV-ERBα/β Agonist Inhibits Bmal1 in the Absence and Presence of Cytokines in Non-Synchronized Insulin-Producing Cells
2.4. The REV-ERBα/β Agonist Mimics Cytokine-Action in Non-Synchronized Insulin-Producing Cells
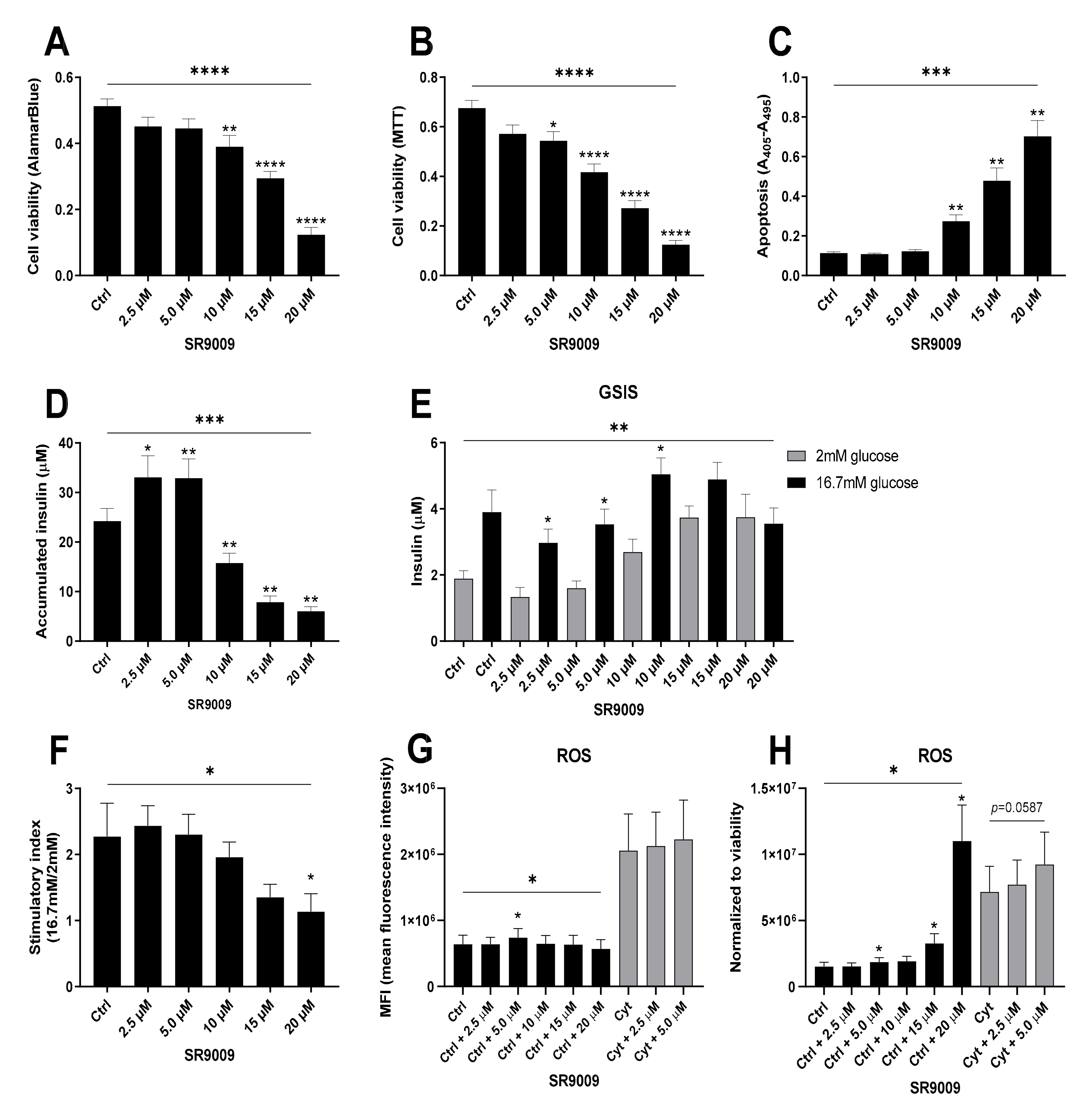
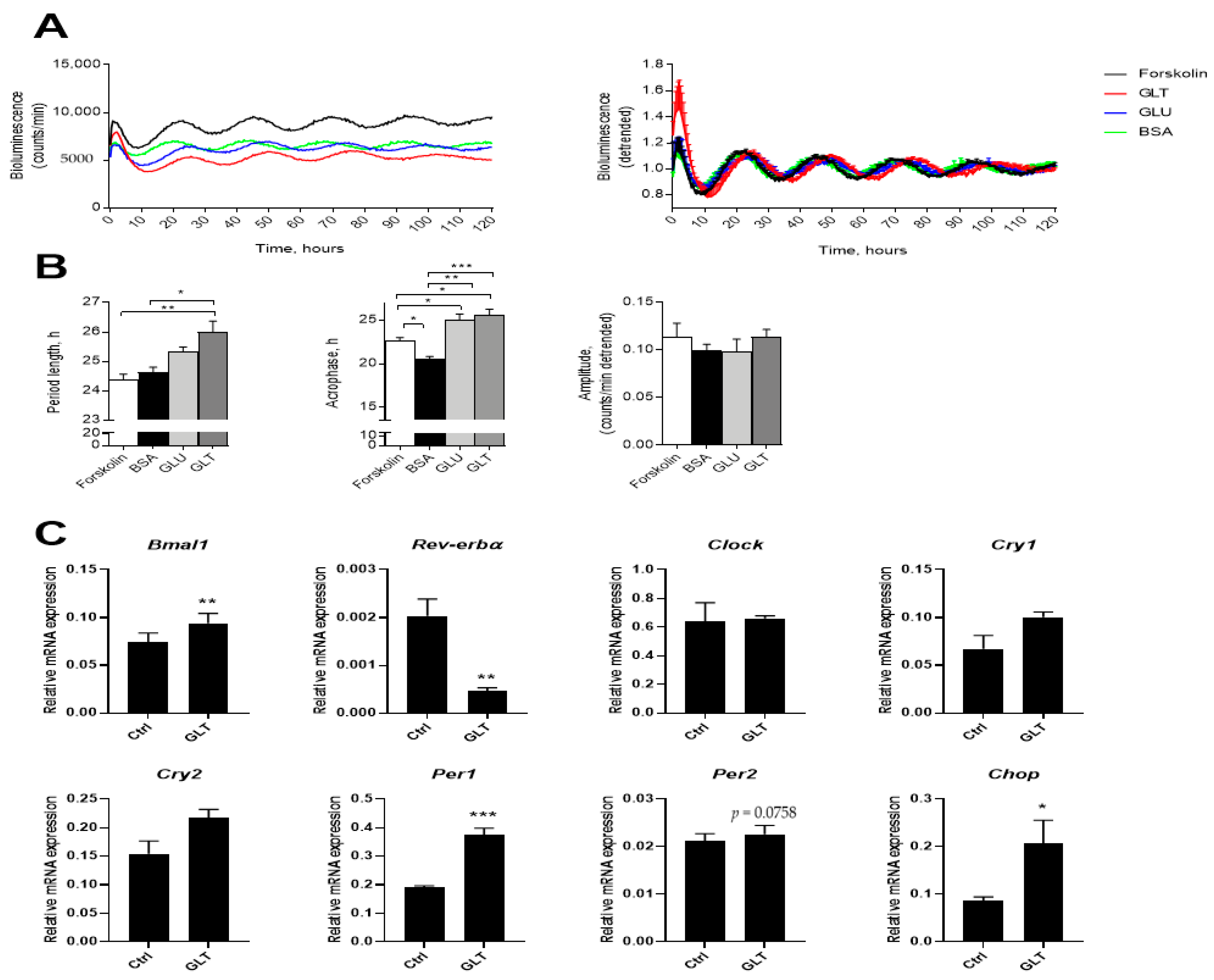
2.5. Metabolic Stress Perturbs the Intrinsic β-Cell Clock but Differently From Inflammatory Stress
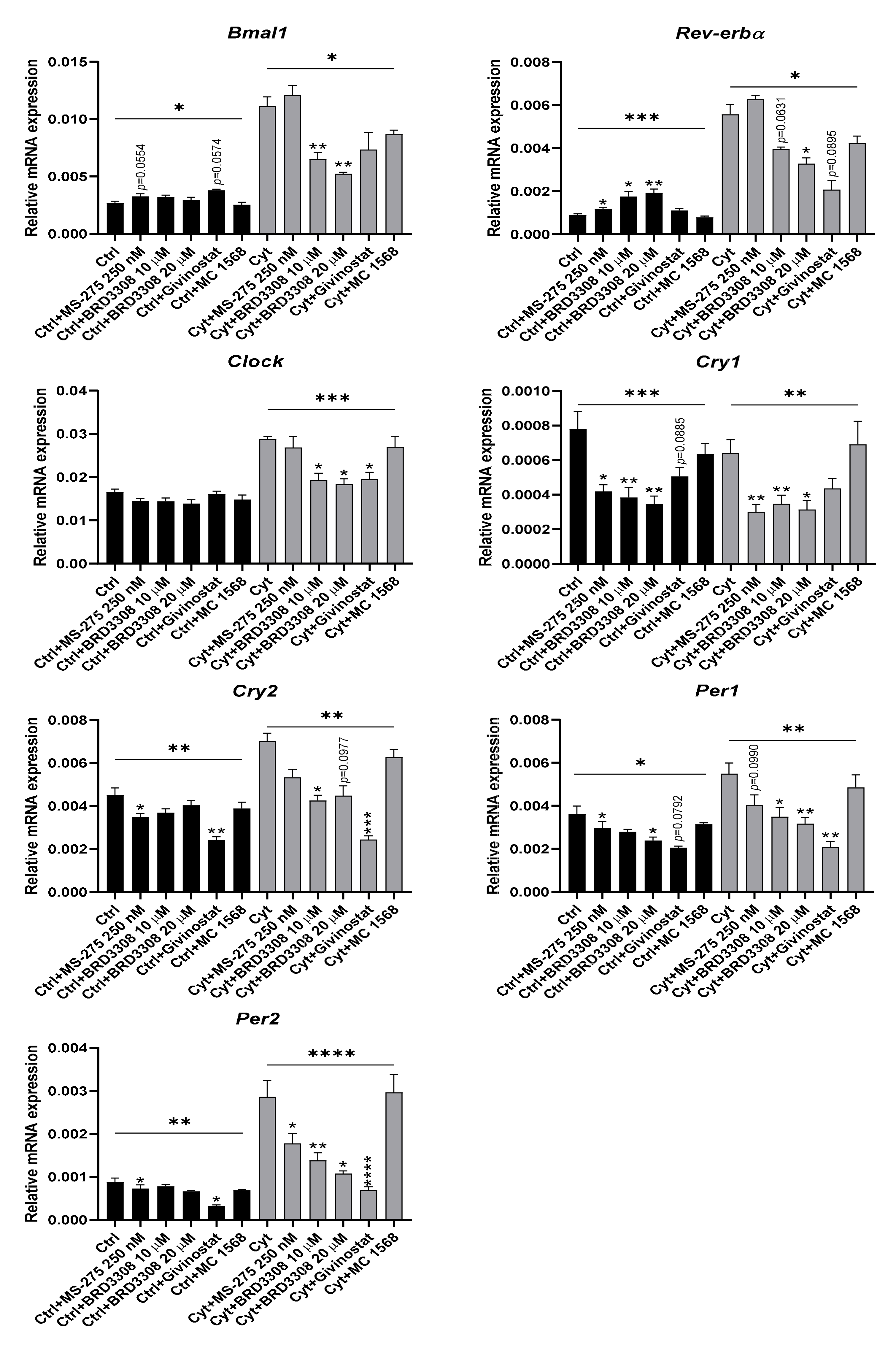
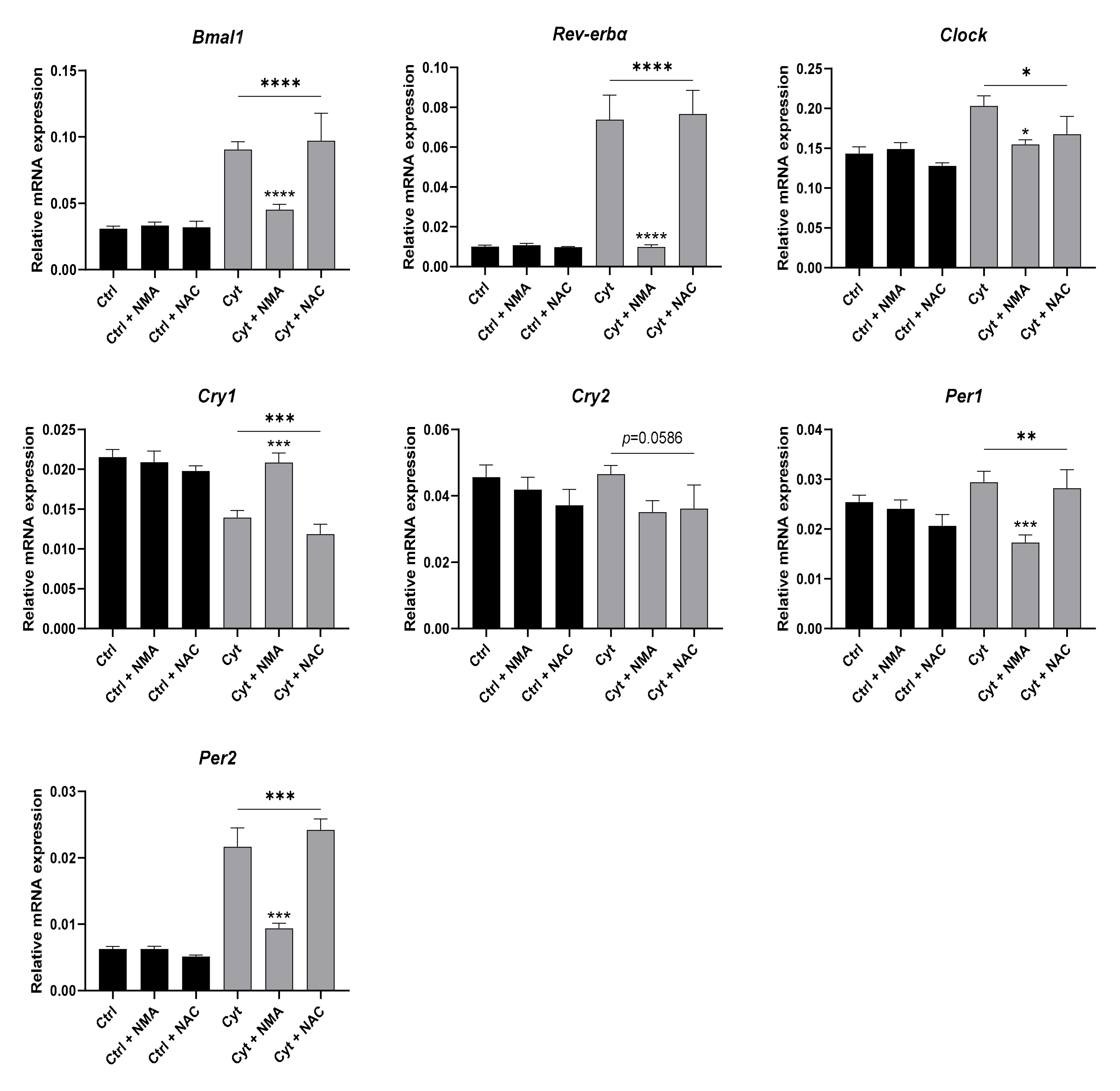
2.6. Proinflammatory Cytokine-Mediated Expression of Clock Genes Depends on Lysine Deacetylase Activity, Nitric Oxide, and the Immunoproteasome in Insulin Producing Cells
3. Discussion
4. Materials and Methods
4.1. Human Pancreatic Islets
4.2. Lentiviral Transduction of Human Islets
4.3. Animal Care, Mouse Strain, and Pancreatic islet Preparations
4.4. In Vitro Islet Synchronization and Circadian Bioluminescence Recording
4.5. Cell Culture
4.6. RT-qPCR
4.7. Nitric Oxide (NO) Assay
4.8. Insulin Secretion Assay
4.9. Apoptosis and Cell Viability Assays
4.10. Reactive Oxygen Species (ROS) Assay
4.11. Statistical Analysis
4.12. Inhibitor and Agonist Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Arg-1 | Arginase-1 |
| Bmal1 | Brain and muscle arnt-like 1 |
| Chop | C/EBP homologous protein |
| Clock | Circadian locomotor output cycles kaput |
| Cry | Cryptochrome |
| ECIT | European Consortium for islet transplantation |
| GLT | Glucolipotoxicity |
| GSIS | Glucose-stimulated insulin secretion |
| HDAC3 | Histone deacetylase 3 |
| Hprt1 | Hypoxanthine-guanine phosphoribosyltransferase |
| IFN-γ | Interferon-γ |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin 6 |
| Inos | Inducible nitric oxide synthase |
| Ins-1/2 | Insulin 1/2 |
| IκBα | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor α |
| KAT | Lysine acetyltransferase |
| KDAC | Lysine deacetylase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium-bromide |
| NAC | n-acetylcysteine |
| NF-κB | Nuclear factor of kappa light polypeptide gene enhancer in B-cells |
| NMA | NG-methyl-L-arginine |
| NO | Nitric oxide |
| Per | Period |
| Per2-luc | Per2-luciferase |
| Ppia | Peptidylprolyl isomerase A |
| PSMB8 | Inducible proteasome subunit β5i |
| Rev-erb | Reverse-erythroblastosis virus |
| RORα | Retinoic acid receptor-related orphan receptor α |
| ROS | Reactive oxygen species |
| SCN | Suprachiasmatic nucleus |
| SI | Stimulatory index |
| siRNA | Small interfering RNA |
| TNFα | Tumor necrosis factor α |
References
- Perelis, M.; Ramsey, K.M.; Marcheva, B.; Bass, J. Circadia Transcription from Beta Cell Function to Diabetes Pathophysiology. J. Biol. Rhythm. 2016, 31, 323–336. [Google Scholar] [CrossRef]
- Dibner, C.; Schibler, U. Circadian timing of metabolism in animal models and humans. J. Intern. Med. 2015, 277, 513–527. [Google Scholar] [CrossRef]
- Milev, N.B.; Reddy, A.B. Circadian redox oscillations and metabolism. Trends Endocrinol. Metab. 2015, 26, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Kondratova, A.A.; Kondratov, R.V. The circadian clock and pathology of the ageing brain. Nat. Rev. Neurosci. 2012, 13, 325–335. [Google Scholar] [CrossRef]
- Videnovic, A.; Lazar, A.S.; Barker, R.A.; Overeem, S. ‘The clocks that time us’—Circadian rhythms in neurodegenerative disorders. Nat. Rev. Neurol. 2014, 10, 683–693. [Google Scholar] [CrossRef]
- Dibner, C. The importance of being rhythmic: Living in harmony with your body clocks. Acta Physiol. 2020, 228, e13281. [Google Scholar] [CrossRef]
- Sinturel, F.; Petrenko, V.; Dibner, C. Circadian Clocks Make Metabolism Run. J. Mol. Biol. 2020, 432, 3680–3699. [Google Scholar] [CrossRef]
- Blancas-Velazquez, A.; Mendoza, J.; Garcia, A.N.; la Fleur, S.E. Diet-Induced Obesity and Circadian Disruption of Feeding Behavior. Front. Neurosci. 2017, 11, 23. [Google Scholar] [CrossRef]
- Takahashi, J.S. Molecular components of the circadian clock in mammals. Diabetes Obes. Metab. 2015, 17 (Suppl. 1), 6–11. [Google Scholar] [CrossRef]
- Cox, K.H.; Takahashi, J.S. Circadian clock genes and the transcriptional architecture of the clock mechanism. J. Mol. Endocrinol. 2019, 63, R93–R102. [Google Scholar] [CrossRef] [Green Version]
- Bass, J.; Takahashi, J.S. Circadian integration of metabolism and energetics. Science 2010, 330, 1349–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibner, C.; Schibler, U.; Albrecht, U. The mammalian circadian timing system: Organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 2010, 72, 517–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, C.; Brown, S.A.; Dibner, C. Human peripheral clocks: Applications for studying circadian phenotypes in physiology and pathophysiology. Front. Neurol. 2015, 6, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finger, A.M.; Dibner, C.; Kramer, A. Coupled network of the circadian clocks: A driving force of rhythmic physiology. FEBS Lett. 2020. [Google Scholar] [CrossRef]
- Perelis, M.; Marcheva, B.; Ramsey, K.M.; Schipma, M.J.; Hutchison, A.L.; Taguchi, A.; Peek, C.B.; Hong, H.; Huang, W.; Omura, C.; et al. Pancreatic beta cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science 2015, 350, aac4250. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, V.; Philippe, J.; Dibner, C. Time zones of pancreatic islet metabolism. Diabetes Obes. Metab. 2018, 20 (Suppl. 2), 116–126. [Google Scholar] [CrossRef] [Green Version]
- Basse, A.L.; Dalbram, E.; Larsson, L.; Gerhart-Hines, Z.; Zierath, J.R.; Treebak, J.T. Skeletal Muscle Insulin Sensitivity Show Circadian Rhythmicity Which Is Independent of Exercise Training Status. Front. Physiol. 2018, 9, 1198. [Google Scholar] [CrossRef]
- Saini, C.; Petrenko, V.; Pulimeno, P.; Giovannoni, L.; Berney, T.; Hebrok, M.; Howald, C.; Dermitzakis, E.T.; Dibner, C. A functional circadian clock is required for proper insulin secretion by human pancreatic islet cells. Diabetes Obes. Metab. 2016, 18, 355–365. [Google Scholar] [CrossRef]
- Petrenko, V.; Gandasi, N.R.; Sage, D.; Tengholm, A.; Barg, S.; Dibner, C. In pancreatic islets from type 2 diabetes patients, the dampened circadian oscillators lead to reduced insulin and glucagon exocytosis. Proc. Natl. Acad. Sci. USA 2020, 117, 2484–2495. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Moulik, M.; Fang, Z.; Saha, P.; Zou, F.; Xu, Y.; Nelson, D.L.; Ma, K.; Moore, D.D.; Yechoor, V.K. Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice. Mol. Cell Biol. 2013, 33, 2327–2338. [Google Scholar] [CrossRef] [Green Version]
- Stenvers, D.J.; Scheer, F.; Schrauwen, P.; la Fleur, S.E.; Kalsbeek, A. Circadian clocks and insulin resistance. Nat. Rev. Endocrinol. 2019, 15, 75–89. [Google Scholar] [CrossRef]
- Mason, I.C.; Qian, J.; Adler, G.K.; Scheer, F. Impact of circadian disruption on glucose metabolism: Implications for type 2 diabetes. Diabetologia 2020, 63, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Cermakian, N.; Westfall, S.; Kiessling, S. Circadian clocks and inflammation: Reciprocal regulation and shared mediators. Arch. Immunol. Ther. Exp. 2014, 62, 303–318. [Google Scholar] [CrossRef] [Green Version]
- Man, K.; Loudon, A.; Chawla, A. Immunity around the clock. Science 2016, 354, 999–1003. [Google Scholar] [CrossRef] [Green Version]
- Carter, S.J.; Durrington, H.J.; Gibbs, J.E.; Blaikley, J.; Loudon, A.S.; Ray, D.W.; Sabroe, I. A matter of time: Study of circadian clocks and their role in inflammation. J. Leukoc. Biol. 2016, 99, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Cable, E.J.; Onishi, K.G.; Prendergast, B.J. Circadian rhythms accelerate wound healing in female Siberian hamsters. Physiol. Behav. 2017, 171, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Vargas, N.N.; Guzman-Ruiz, M.; Fuentes, R.; Garcia, J.; Salgado-Delgado, R.; Basualdo Mdel, C.; Escobar, C.; Markus, R.P.; Buijs, R.M. Shift Work in Rats Results in Increased Inflammatory Response after Lipopolysaccharide Administration: A Role for Food Consumption. J. Biol. Rhythm. 2015, 30, 318–330. [Google Scholar] [CrossRef]
- Rijo-Ferreira, F.; Takahashi, J.S. Genomics of circadian rhythms in health and disease. Genome Med. 2019, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Hergenhan, S.; Holtkamp, S.; Scheiermann, C. Molecular Interactions Between Components of the Circadian Clock and the Immune System. J. Mol. Biol. 2020, 432, 3700–3713. [Google Scholar] [CrossRef]
- Scheiermann, C.; Gibbs, J.; Ince, L.; Loudon, A. Clocking in to immunity. Nat. Rev. Immunol. 2018, 18, 423–437. [Google Scholar] [CrossRef]
- Hong, H.K.; Maury, E.; Ramsey, K.M.; Perelis, M.; Marcheva, B.; Omura, C.; Kobayashi, Y.; Guttridge, D.C.; Barish, G.D.; Bass, J. Requirement for NF-kappaB in maintenance of molecular and behavioral circadian rhythms in mice. Genes Dev. 2018, 32, 1367–1379. [Google Scholar] [CrossRef]
- Simcox, J.A.; Mitchell, T.C.; Gao, Y.; Just, S.F.; Cooksey, R.; Cox, J.; Ajioka, R.; Jones, D.; Lee, S.H.; King, D.; et al. Dietary iron controls circadian hepatic glucose metabolism through heme synthesis. Diabetes 2015, 64, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.; Mandrup-Poulsen, T. An immune origin of type 2 diabetes? Diabetologia 2005, 48, 1038–1050. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dinarello, C.A.; Mandrup-Poulsen, T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat. Rev. Immunol. 2019, 19, 734–746. [Google Scholar] [CrossRef]
- Berchtold, L.A.; Prause, M.; Storling, J.; Mandrup-Poulsen, T. Cytokines and Pancreatic beta-Cell Apoptosis. Adv. Clin. Chem. 2016, 75, 99–158. [Google Scholar] [CrossRef]
- Borel, A.L.; Pepin, J.L.; Nasse, L.; Baguet, J.P.; Netter, S.; Benhamou, P.Y. Short sleep duration measured by wrist actimetry is associated with deteriorated glycemic control in type 1 diabetes. Diabetes Care 2013, 36, 2902–2908. [Google Scholar] [CrossRef] [Green Version]
- Larcher, S.; Gauchez, A.S.; Lablanche, S.; Pepin, J.L.; Benhamou, P.Y.; Borel, A.L. Impact of sleep behavior on glycemic control in type 1 diabetes: The role of social jetlag. Eur. J. Endocrinol. 2016, 175, 411–419. [Google Scholar] [CrossRef] [Green Version]
- The DCCT Research Group. Effects of Age, Duration and Treatment of Insulin-Dependent Diabetes Mellitus on Residual β-Cell Function: Observations During Eligibility Testing for the Diabetes Control and Complications Trial (DCCT). J. Clin. Endocrinol. Metab. 1987, 65, 30–36. [Google Scholar] [CrossRef]
- Javeed, N.; Brown, M.R.; Rakshit, K.; Her, T.; Sen, S.K.; Matveyenko, A.V. Pro-inflammatory Cytokine Interleukin 1β Disrupts β cell Circadian Clock Function and Regulation of Insulin Secretion. Endocrinology 2020, 162, bqaa084. [Google Scholar] [CrossRef]
- Mandrup-Poulsen, T.; Pickersgill, L.; Donath, M.Y. Blockade of interleukin 1 in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2010, 6, 158–166. [Google Scholar] [CrossRef]
- Balsalobre, A.; Damiola, F.; Schibler, U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 1998, 93, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Yagita, K.; Okamura, H. Forskolin induces circadian gene expression of rPer1, rPer2 and dbp in mammalian rat-1 fibroblasts. FEBS Lett. 2000, 465, 79–82. [Google Scholar] [CrossRef] [Green Version]
- Saadeh, M.; Ferrante, T.C.; Kane, A.; Shirihai, O.; Corkey, B.E.; Deeney, J.T. Reactive oxygen species stimulate insulin secretion in rat pancreatic islets: Studies using mono-oleoyl-glycerol. PLoS ONE 2012, 7, e30200. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.B.; Dos Santos, L.R.B.; Liu, Y.; Prentice, K.J.; Teudt, F.; Tonnesen, M.; Jonas, J.C.; Wheeler, M.B.; Mandrup-Poulsen, T. Glucolipotoxic conditions induce beta-cell iron import, cytosolic ROS formation and apoptosis. J. Mol. Endocrinol. 2018, 61, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [Green Version]
- Christensen, D.P.; Gysemans, C.; Lundh, M.; Dahllof, M.S.; Noesgaard, D.; Schmidt, S.F.; Mandrup, S.; Birkbak, N.; Workman, C.T.; Piemonti, L.; et al. Lysine deacetylase inhibition prevents diabetes by chromatin-independent immunoregulation and beta-cell protection. Proc. Natl. Acad. Sci. USA 2014, 111, 1055–1059. [Google Scholar] [CrossRef] [Green Version]
- Lundh, M.; Galbo, T.; Poulsen, S.S.; Mandrup-Poulsen, T. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes. Metab. 2015, 17, 703–707. [Google Scholar] [CrossRef]
- Broniowska, K.A.; Oleson, B.J.; Corbett, J.A. Beta-Cell responses to nitric oxide. Vitam. Horm. 2014, 95, 299–322. [Google Scholar] [CrossRef]
- Shi, G.; Xie, P.; Qu, Z.; Zhang, Z.; Dong, Z.; An, Y.; Xing, L.; Liu, Z.; Dong, Y.; Xu, G.; et al. Distinct Roles of HDAC3 in the Core Circadian Negative Feedback Loop Are Critical for Clock Function. Cell Rep. 2016, 14, 823–834. [Google Scholar] [CrossRef] [Green Version]
- Yakovlev, V.A.; Barani, I.J.; Rabender, C.S.; Black, S.M.; Leach, J.K.; Graves, P.R.; Kellogg, G.E.; Mikkelsen, R.B. Tyrosine nitration of IkappaBalpha: A novel mechanism for NF-kappaB activation. Biochemistry 2007, 46, 11671–11683. [Google Scholar] [CrossRef] [Green Version]
- Stratmann, M.; Suter, D.M.; Molina, N.; Naef, F.; Schibler, U. Circadian Dbp transcription relies on highly dynamic BMAL1-CLOCK interaction with E boxes and requires the proteasome. Mol. Cell 2012, 48, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, J.S.; Hong, H.K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775. [Google Scholar] [CrossRef]
- Khilji, M.S.; Verstappen, D.; Dahlby, T.; Burstein Prause, M.C.; Pihl, C.; Bresson, S.E.; Bryde, T.H.; Keller Andersen, P.A.; Klindt, K.; Zivkovic, D.; et al. The intermediate proteasome is constitutively expressed in pancreatic beta cells and upregulated by stimulatory, low concentrations of interleukin 1 beta. PLoS ONE 2020, 15, e0222432. [Google Scholar] [CrossRef]
- Spengler, M.L.; Kuropatwinski, K.K.; Comas, M.; Gasparian, A.V.; Fedtsova, N.; Gleiberman, A.S.; Gitlin, I.I.; Artemicheva, N.M.; Deluca, K.A.; Gudkov, A.V.; et al. Core circadian protein CLOCK is a positive regulator of NF-κB-mediated transcription. Proc. Natl. Acad. Sci. USA 2012, 109, E2457–E2465. [Google Scholar] [CrossRef] [Green Version]
- Bellet, M.M.; Zocchi, L.; Sassone-Corsi, P. The RelB subunit of NFkappaB acts as a negative regulator of circadian gene expression. Cell Cycle 2012, 11, 3304–3311. [Google Scholar] [CrossRef] [Green Version]
- Cavadini, G.; Petrzilka, S.; Kohler, P.; Jud, C.; Tobler, I.; Birchler, T.; Fontana, A. TNF-alpha suppresses the expression of clock genes by interfering with E-box-mediated transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 12843–12848. [Google Scholar] [CrossRef] [Green Version]
- Ortis, F.; Cardozo, A.K.; Crispim, D.; Störling, J.; Mandrup-Poulsen, T.; Eizirik, D.L. Cytokine-induced proapoptotic gene expression in insulin-producing cells is related to rapid, sustained, and nonoscillatory nuclear factor-kappaB activation. Mol. Endocrinol. 2006, 20, 1867–1879. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Lazar, M.A. The orphan nuclear receptor Rev-erbalpha recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol. Endocrinol. 2005, 19, 1452–1459. [Google Scholar] [CrossRef]
- Doi, M.; Hirayama, J.; Sassone-Corsi, P. Circadian regulator CLOCK is a histone acetyltransferase. Cell 2006, 125, 497–508. [Google Scholar] [CrossRef]
- Hirayama, J.; Sahar, S.; Grimaldi, B.; Tamaru, T.; Takamatsu, K.; Nakahata, Y.; Sassone-Corsi, P. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nature 2007, 450, 1086–1090. [Google Scholar] [CrossRef]
- Lundh, M.; Christensen, D.P.; Damgaard Nielsen, M.; Richardson, S.J.; Dahllof, M.S.; Skovgaard, T.; Berthelsen, J.; Dinarello, C.A.; Stevenazzi, A.; Mascagni, P.; et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia 2012, 55, 2421–2431. [Google Scholar] [CrossRef] [Green Version]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Sha, Y.; Marshall, H.E. S-nitrosylation in the regulation of gene transcription. Biochim. Biophys. Acta 2012, 1820, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Pardee, K.I.; Xu, X.; Reinking, J.; Schuetz, A.; Dong, A.; Liu, S.; Zhang, R.; Tiefenbach, J.; Lajoie, G.; Plotnikov, A.N.; et al. The structural basis of gas-responsive transcription by the human nuclear hormone receptor REV-ERBbeta. PLoS Biol. 2009, 7, e43. [Google Scholar] [CrossRef]
- Lundh, M.; Bugliani, M.; Dahlby, T.; Chou, D.H.; Wagner, B.; Ghiasi, S.M.; De Tata, V.; Chen, Z.; Lund, M.N.; Davies, M.J.; et al. The immunoproteasome is induced by cytokines and regulates apoptosis in human islets. J. Endocrinol. 2017, 233, 369–379. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Pati, P.; Xu, Y.; Chen, F.; Stepp, D.W.; Huo, Y.; Rudic, R.D.; Fulton, D.J.R. Endotoxin Disrupts Circadian Rhythms in Macrophages via Reactive Oxygen Species. PLoS ONE 2016, 11, e0155075. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Mandrup-Poulsen, T. A choice of death—The signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 2001, 44, 2115–2133. [Google Scholar] [CrossRef]
- Liu, D.; Pavlovic, D.; Chen, M.C.; Flodström, M.; Sandler, S.; Eizirik, D.L. Cytokines induce apoptosis in beta-cells isolated from mice lacking the inducible isoform of nitric oxide synthase (iNOS−/−). Diabetes 2000, 49, 1116. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Sandler, S.; Welsh, N.; Cetkovic-Cvrlje, M.; Nieman, A.; Geller, D.A.; Pipeleers, D.G.; Bendtzen, K.; Hellerström, C. Cytokines suppress human islet function irrespective of their effects on nitric oxide generation. J. Clin. Investig. 1994, 93, 1968–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, Y.; Yokota, A.; Shiraishi, T.; Yamada, S.; Haraguchi, A.; Shinozaki, A.; Shibata, S. In vitro and in vivo Phase Changes of the Mouse Circadian Clock by Oxidative Stress. J. Circadian Rhythm. 2016, 14, 4. [Google Scholar] [CrossRef] [Green Version]
- Fanjul-Moles, M.L.; Lopez-Riquelme, G.O. Relationship between Oxidative Stress, Circadian Rhythms, and AMD. Oxid. Med. Cell Longev. 2016, 2016, 7420637. [Google Scholar] [CrossRef] [Green Version]
- Wilking, M.; Ndiaye, M.; Mukhtar, H.; Ahmad, N. Circadian rhythm connections to oxidative stress: Implications for human health. Antioxid. Redox Signal. 2013, 19, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Ghiasi, S.M.; Hansen, J.B.; Christensen, D.P.; Tyrberg, B.; Mandrup-Poulsen, T. The Connexin 43 Regulator Rotigaptide Reduces Cytokine-Induced Cell Death in Human Islets. Int. J. Mol. Sci 2020, 21, 4311. [Google Scholar] [CrossRef]
- Amador, A.; Wang, Y.; Banerjee, S.; Kameneka, T.M.; Solt, L.A.; Burris, T.P. Pharmacological and Genetic Modulation of REV-ERB Activity and Expression Affects Orexigenic Gene Expression. PLoS ONE 2016, 11, e0151014. [Google Scholar] [CrossRef]
- Das, V.; Kc, R.; Li, X.; Varma, D.; Qiu, S.; Kroin, J.S.; Forsyth, C.B.; Keshavarzian, A.; van Wijnen, A.J.; Park, T.J.; et al. Pharmacological targeting of the mammalian clock reveals a novel analgesic for osteoarthritis-induced pain. Gene 2018, 655, 1–12. [Google Scholar] [CrossRef]
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 553, 351–355. [Google Scholar] [CrossRef]
- Rakshit, K.; Matveyenko, A.V. Induction of Core Circadian Clock Transcription Factor Bmal1 Enhances β Cell Function and Protects Against Obesity-Induced Glucose Intolerance. Diabetes 2020. [Google Scholar] [CrossRef]
- Sakai, K.; Matsumoto, K.; Nishikawa, T.; Suefuji, M.; Nakamaru, K.; Hirashima, Y.; Kawashima, J.; Shirotani, T.; Ichinose, K.; Brownlee, M.; et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2003, 300, 216–222. [Google Scholar] [CrossRef]
- Marcheva, B.; Ramsey, K.M.; Buhr, E.D.; Kobayashi, Y.; Su, H.; Ko, C.H.; Ivanova, G.; Omura, C.; Mo, S.; Vitaterna, M.H.; et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010, 466, 627–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, J.; Block, G.D.; Colwell, C.S.; Matveyenko, A.V. Consequences of exposure to light at night on the pancreatic islet circadian clock and function in rats. Diabetes 2013, 62, 3469–3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012, 485, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Wang, Y.; Solt, L.A.; Griffett, K.; Kazantzis, M.; Amador, A.; El-Gendy, B.M.; Huitron-Resendiz, S.; Roberts, A.J.; Shin, Y.; et al. Pharmacological targeting of the mammalian clock regulates sleep architecture and emotional behaviour. Nat. Commun. 2014, 5, 5759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierickx, P.; Emmett, M.J.; Jiang, C.; Uehara, K.; Liu, M.; Adlanmerini, M.; Lazar, M.A. SR9009 has REV-ERB–independent effects on cell proliferation and metabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 12147–12152. [Google Scholar] [CrossRef] [Green Version]
- Pulimeno, P.; Mannic, T.; Sage, D.; Giovannoni, L.; Salmon, P.; Lemeille, S.; Giry-Laterriere, M.; Unser, M.; Bosco, D.; Bauer, C.; et al. Autonomous and self-sustained circadian oscillators displayed in human islet cells. Diabetologia 2013, 56, 497–507. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, V.; Saini, C.; Giovannoni, L.; Gobet, C.; Sage, D.; Unser, M.; Heddad Masson, M.; Gu, G.; Bosco, D.; Gachon, F.; et al. Pancreatic alpha- and beta-cellular clocks have distinct molecular properties and impact on islet hormone secretion and gene expression. Genes Dev. 2017, 31, 383–398. [Google Scholar] [CrossRef]
- Ghiasi, S.M.; Dahlby, T.; Hede Andersen, C.; Haataja, L.; Petersen, S.; Omar-Hmeadi, M.; Yang, M.; Pihl, C.; Bresson, S.E.; Khilji, M.S.; et al. Endoplasmic Reticulum Chaperone Glucose-Regulated Protein 94 Is Essential for Proinsulin Handling. Diabetes 2019, 68, 747–760. [Google Scholar] [CrossRef]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [Green Version]
- Lindelov Vestergaard, A.; Heiner Bang-Berthelsen, C.; Floyel, T.; Lucien Stahl, J.; Christen, L.; Taheri Sotudeh, F.; de Hemmer Horskjaer, P.; Stensgaard Frederiksen, K.; Greek Kofod, F.; Bruun, C.; et al. MicroRNAs and histone deacetylase inhibition-mediated protection against inflammatory beta-cell damage. PLoS ONE 2018, 13, e0203713. [Google Scholar] [CrossRef]
- Haase, T.N.; Rasmussen, M.; Jaksch, C.A.; Gaarn, L.W.; Petersen, C.K.; Billestrup, N.; Nielsen, J.H. Growth arrest specific protein (GAS) 6: A role in the regulation of proliferation and functional capacity of the perinatal rat beta cell. Diabetologia 2013, 56, 763–773. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersen, P.A.K.; Petrenko, V.; Rose, P.H.; Koomen, M.; Fischer, N.; Ghiasi, S.M.; Dahlby, T.; Dibner, C.; Mandrup-Poulsen, T. Proinflammatory Cytokines Perturb Mouse and Human Pancreatic Islet Circadian Rhythmicity and Induce Uncoordinated β-Cell Clock Gene Expression via Nitric Oxide, Lysine Deacetylases, and Immunoproteasomal Activity. Int. J. Mol. Sci. 2021, 22, 83. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010083
Andersen PAK, Petrenko V, Rose PH, Koomen M, Fischer N, Ghiasi SM, Dahlby T, Dibner C, Mandrup-Poulsen T. Proinflammatory Cytokines Perturb Mouse and Human Pancreatic Islet Circadian Rhythmicity and Induce Uncoordinated β-Cell Clock Gene Expression via Nitric Oxide, Lysine Deacetylases, and Immunoproteasomal Activity. International Journal of Molecular Sciences. 2021; 22(1):83. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010083
Chicago/Turabian StyleAndersen, Phillip Alexander Keller, Volodymyr Petrenko, Peter Horskjær Rose, Melissa Koomen, Nico Fischer, Seyed Mojtaba Ghiasi, Tina Dahlby, Charna Dibner, and Thomas Mandrup-Poulsen. 2021. "Proinflammatory Cytokines Perturb Mouse and Human Pancreatic Islet Circadian Rhythmicity and Induce Uncoordinated β-Cell Clock Gene Expression via Nitric Oxide, Lysine Deacetylases, and Immunoproteasomal Activity" International Journal of Molecular Sciences 22, no. 1: 83. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010083