
Ectopic Overexpression of Histone H3K4 Methyltransferase CsSDG36 from Tea Plant Decreases Hyperosmotic Stress Tolerance in Arabidopsis thaliana

 ,
,
Abstract
:1. Introduction
2. Results
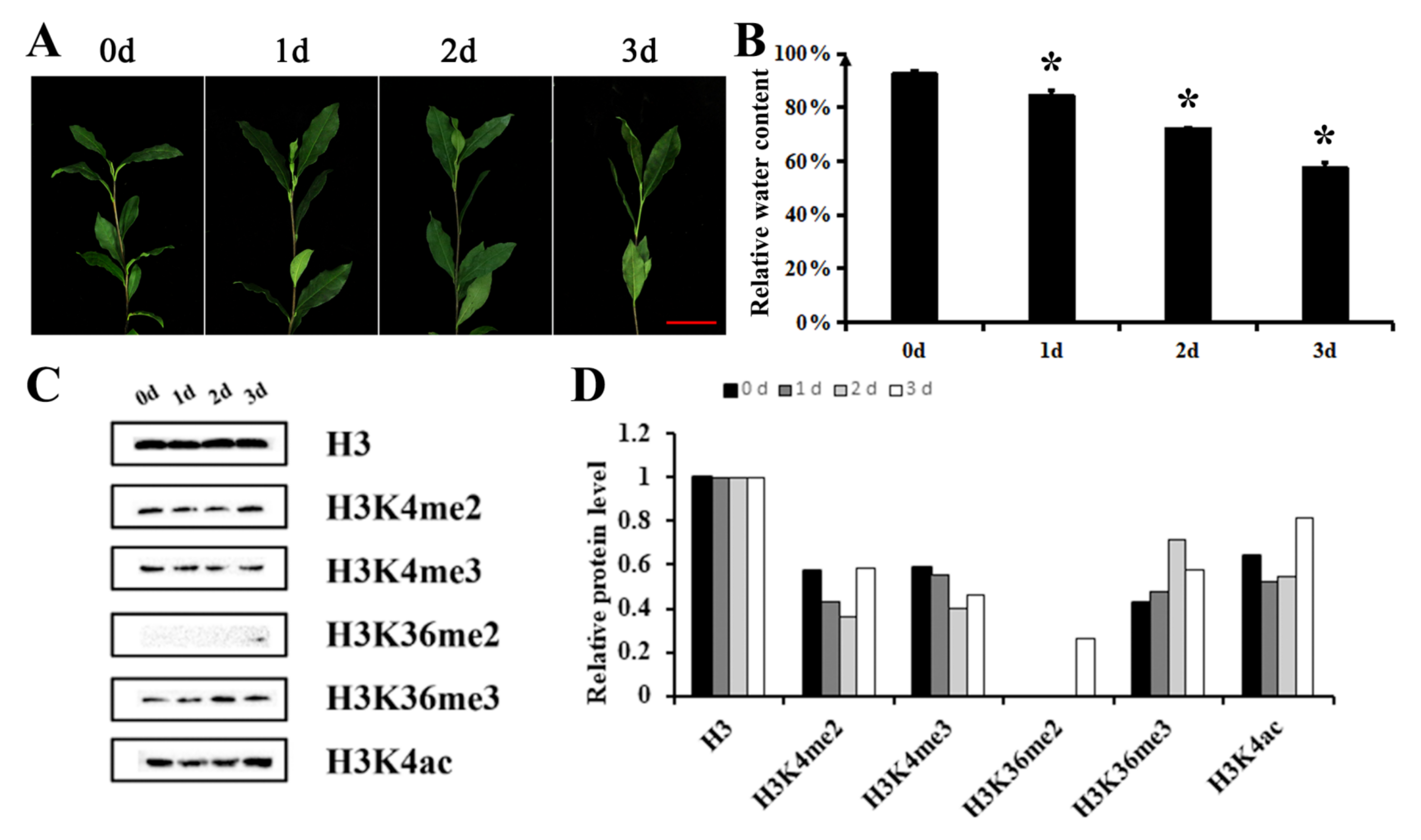
2.1. Histone H3K4 Methylation in Tea Leaves Is Significantly Down-Regulated under Drought Stress
2.2. Amino Acid Sequence Analysis of CsSDG36 Gene
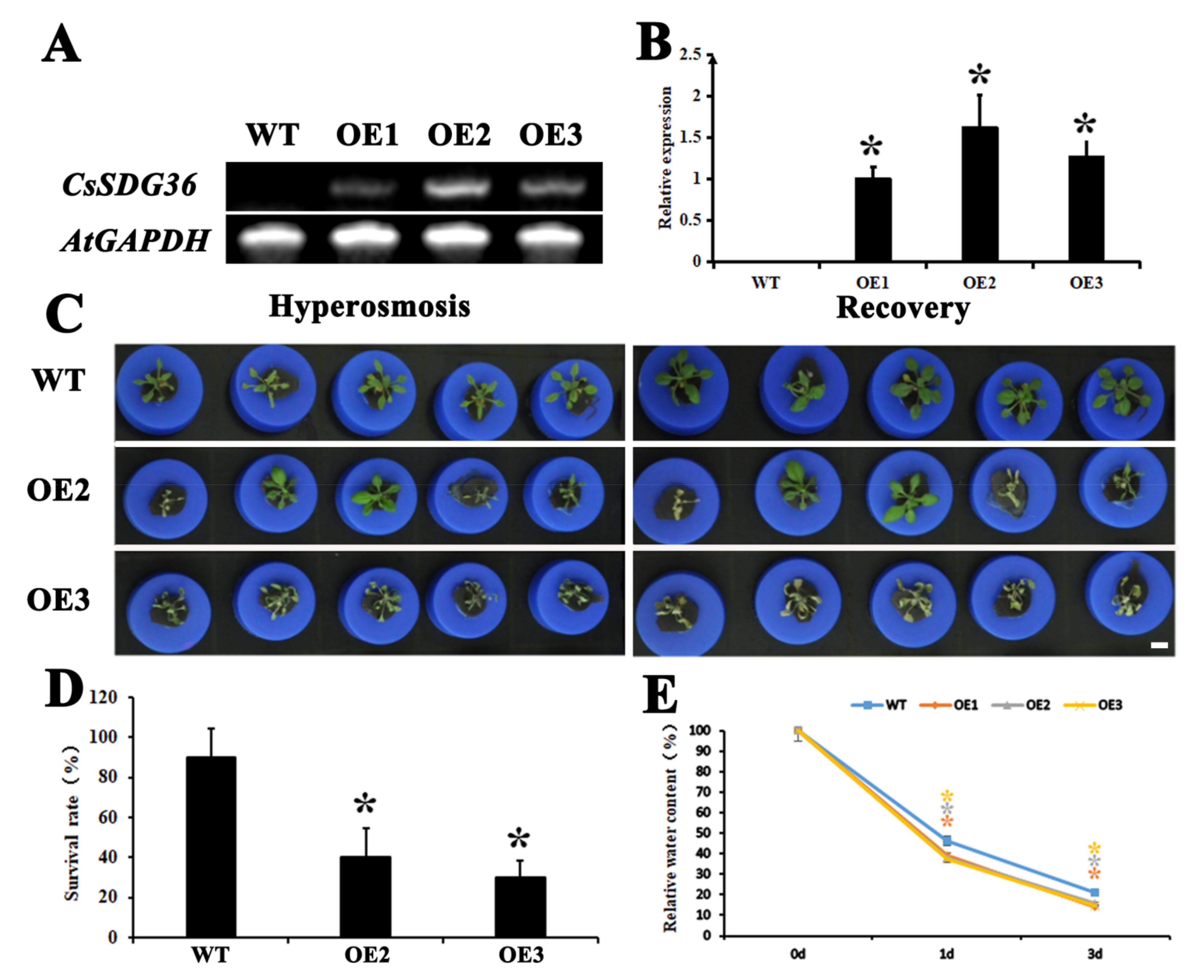
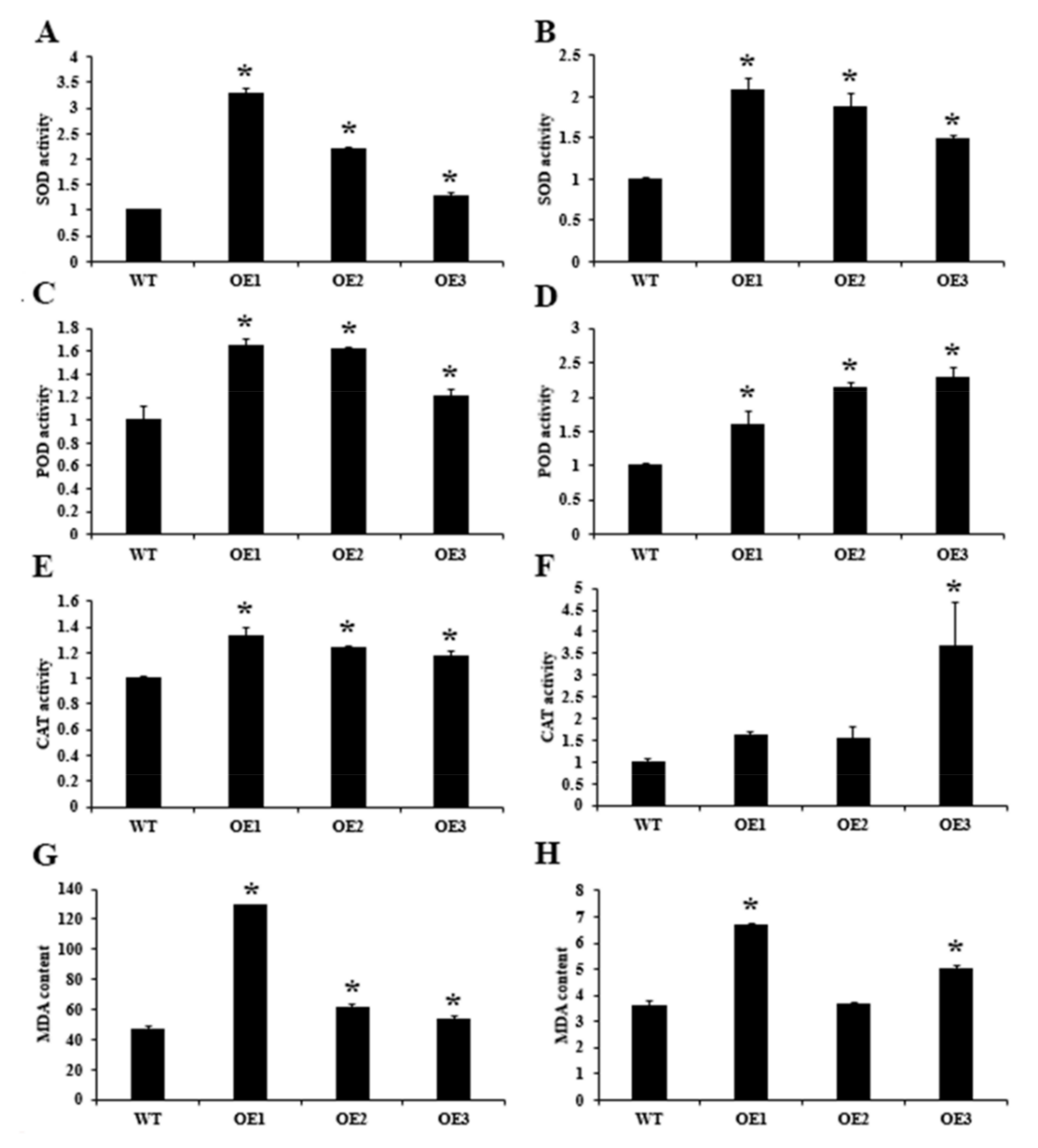
2.3. Phenotypic Profiling of CsSDG36 Protein in Arabidopsis thaliana
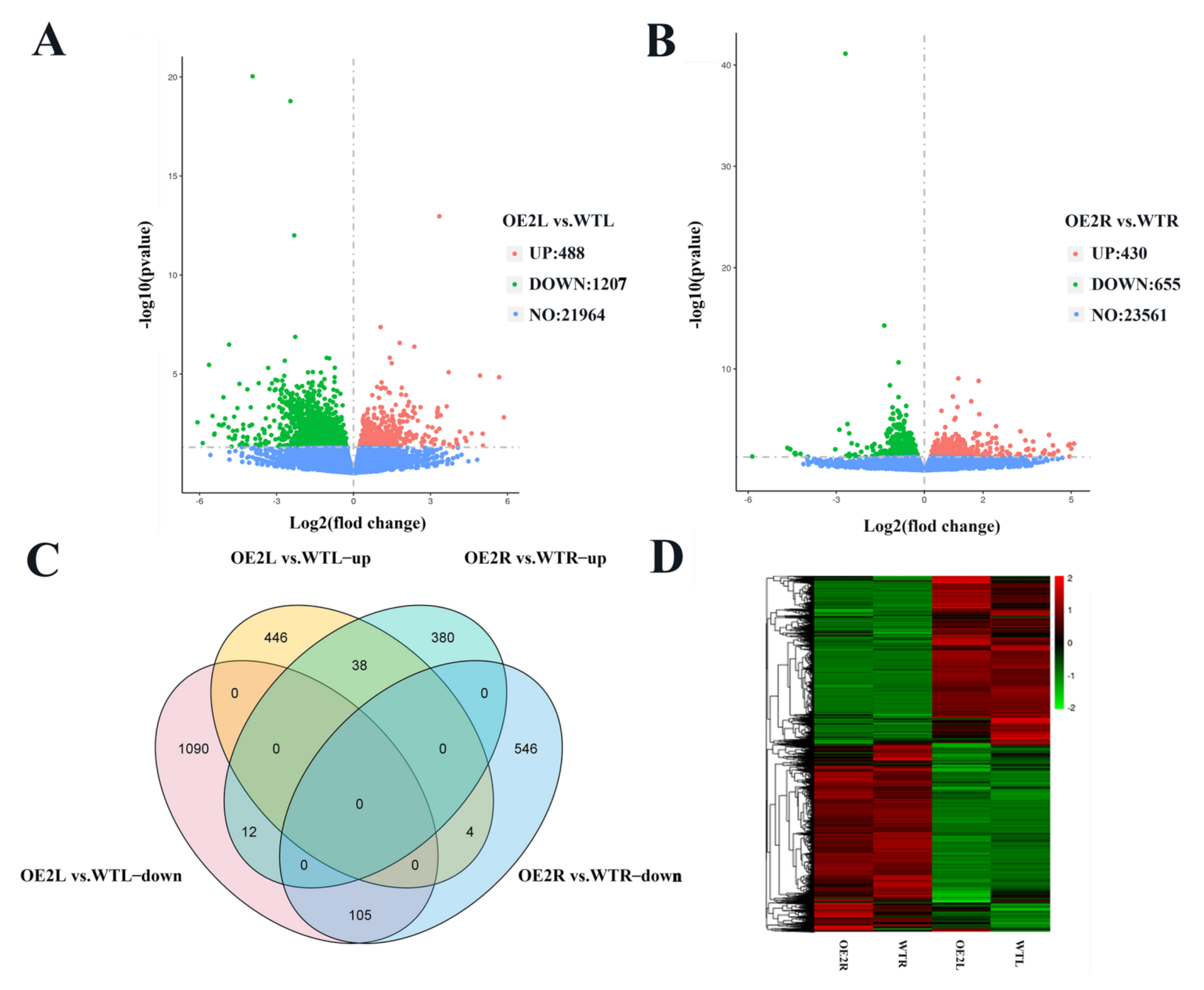
2.4. CsSDG36 Protein Is Associated with the Chromatin Assembly, Microtubule Assembly, and Stomatal Development Pathways
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Treatment
4.2. Western Blotting
4.3. Bioinformatics Analysis
4.4. Overexpression of CsSDG36 in Wild-Type Arabidopsis thaliana
4.5. Phenotypic Identification of CsSDG36 Over-Expression Lines
4.6. Physiological Analysis
4.7. Statistical Analysis of Stomatal Density
4.8. RNA Sequencing Analysis of CsSDG36 Overexpression Line 2 and Wild Type
4.9. qRT-PCR Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, K.; Sridhar, V.V.; Zhu, J.; Kapoor, A.; Zhu, J.-K. Distinctive core histone post-translational modification patterns in arabidopsis thaliana. PLoS ONE 2007, 2, e1210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Bernatavichute, Y.V.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 inArabidopsis thaliana. Genome Biol. 2009, 10, R62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, W.; Zhong, X.; You, J.; Xiong, L. Genome-wide profiling of histone h3k4-tri-methylation and gene expression in rice under drought stress. Plant Mol. Biol. 2013, 81, 175–188. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Yan, S.; Wang, P.; Zhu, W.; Wang, X.; Shen, Y.; Shao, K.; Xin, H.; Li, S.; Li, L. Comparative analysis of genome-wide chromosomal histone modification patterns in maize cultivars and their wild relatives. PLoS ONE 2014, 9, e97364. [Google Scholar] [CrossRef] [Green Version]
- Ng, W.K.; Wang, T.; Chandrasekharan, M.B.; Aramayo, R.; Kertbundit, S.; Hall, T.C. Plant set domain-containing proteins: Structure, function and regulation. Biochim. Biophys. Acta 2007, 1769, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumbusch, L.O.; Thorstensen, T.; Krauss, V.; Fischer, A.; Naumann, K.; Assalkhou, R.; Schulz, I.; Reuter, G.; Aalen, R.B. The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res. 2001, 29, 4319–4333. [Google Scholar] [CrossRef]
- Alvarez-Venegas, R.; Avramova, Z. SET-domain proteins of the Su (var) 3- 9, E (z) and trithorax families. Gene 2002, 285, 25–37. [Google Scholar] [CrossRef]
- Veerappan, C.S.; Avramova, Z.; Moriyama, E.N. Evolution of SET-domain protein families in the unicellular and multicellular Ascomycota fungi. BMC Evol. Biol. 2008, 8, 190. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Mo, H.; Fan, D.; Cao, Y.; Cui, S.; Ma, L. Overexpression of a histone H3K4 demethylase, JMJ15, accelerates flowering time in Arabidopsis. Plant Cell Rep. 2012, 31, 1297–1308. [Google Scholar] [CrossRef]
- Cartagena, J.A.; Matsunaga, S.; Seki, M.; Kurihara, D.; Yokoyama, M.; Shinozaki, K.; Fujimoto, S.; Azumi, Y.; Uchiyama, S.; Fukui, K. The Arabidopsis SDG4 contributes to the regulation of pollen tube growth by methylation of histone H3 lysines 4 and 36 in mature pollen. Dev. Biol. 2008, 315, 355–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, G.; Ma, D.P.; Li, J. The histone methyltransferase sdg8 regulates shoot branching in arabidopsis. Biochem. Biophys. Res. Commun. 2008, 373, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Feng, H.; Yu, Y.; Dong, A.; Shen, W.; He, Y. Sdg2-mediated h3k4 methylation is required for proper arabidopsis root growth and development. PLoS ONE 2013, 8, e56537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malapeira, J.; Khaitova, L.C.; Mas, P. Ordered changes in histone modifications at the core of the arabidopsis circadian clock. Proc. Natl. Acad. Sci. USA 2012, 109, 21540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, K.; Daniel, B.; Arp, S.; Kermode, A.R.; Berger, F. Evolutionarily conserved histone methylation dynamics during seed life-cycle transitions. PLoS ONE 2012, 7, e51532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Venegas, R.; Pien, S.; Sadder, M.; Witmer, X.; Grossniklaus, U.; Avramova, Z. ATX-1, an Arabidopsis homolog of trithorax, activates flower homeotic genes. Curr. Biol. 2003, 13, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Avramova, Z.; Fromm, M. The arabidopsis trithorax-like factor atx1 functions in dehydration stress responses via aba-dependent and aba-independent pathways. Plant J. Cell Mol. Biol. 2011, 66, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, A.; Yin, H.; Meng, Q.; Yu, X.; Huang, S.; Wang, J.; Ahmad, R.; Liu, B.; Xu, Z. Trithorax-group proteins arabidopsis trithorax4 (atx4) and atx5 function in abscisic acid and dehydration stress responses. New Phytol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Venegas, R.; Sadder, M.; Hlavacka, A.; Baluska, F.; Xia, Y.; Lu, G.; Firsov, A.; Sarath, G.; Moriyama, H.; Dubrovsky, J.G. The arabidopsis homolog of trithorax, atx1, binds phosphatidylinositol 5-phosphate, and the two regulate a common set of target genes. Proc. Natl. Acad. Sci. USA 2006, 103, 6049–6054. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; To, T.K.; Ishida, J.; Morosawa, T.; Kawashima, M.; Matsui, A.; Toyoda, T.; Kimura, H.; Shinozaki, K.; Seki, M. Alterations of lysine modifications on the histone h3 n-tail under drought stress conditions in arabidopsis thaliana. Plant Cell Physiol. 2008, 49, 1580–1588. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; To, T.K.; Ishida, J.; Matsui, A.; Kimura, H.; Seki, M. Transition of chromatin status during the process of recovery from drought stress in Arabidopsis thaliana. Plant Cell Physiol. 2012, 53, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Van, D.K.; Ding, Y.; Malkaram, S.; Riethoven, J.M.; Liu, R.; Yang, J.; Laczko, P.; Chen, H.; Xia, Y.; Ladunga, I.; et al. Dynamic changes in genome-wide histone H3 lysine 4 methylation patterns in response to dehydration stress in Arabidopsis thaliana. BMC Plant Biol. 2010, 10, 238. [Google Scholar]
- Zhao, Z.; Yu, Y.; Meyer, D.; Wu, C.; Shen, W.H. Prevention of early flowering by expression of flowering locus c requires methylation of histone h3k36. Nat. Cell Biol. 2005, 7, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mukherjee, I.; Thum, K.E.; Tanurdzic, M.; Katari, M.S.; Obertello, M.; Edwards, M.B.; McCombie, W.R.; Martienssen, R.A.; Coruzzi, G.M. The histone methyltransferase SDG8 mediates the epigenetic modification of light and carbon responsive genes in plants. Genome Biol. 2015, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, P.; Jin, J.; Ye, S.; Mu, C.; Gao, J.; Feng, H.; Shen, W.H.; Yu, Y.; Dong, A. H3k36 methylation is critical for brassinosteroid-regulated plant growth and development in rice. Plant J. 2012, 70, 340–347. [Google Scholar] [CrossRef]
- Wei, G.; Liu, K.; Shen, T.; Shi, J.; Liu, B.; Han, M.; Peng, M.; Fu, H.; Song, Y.; Zhu, J.; et al. Position-specific intron retention is mediated by the histone methyltransferase SDG725. BMC Biol. 2018, 16, 44. [Google Scholar] [CrossRef]
- Sun, C.; Fang, J.; Zhao, T.; Xu, B.; Zhang, F.; Liu, L.; Tang, J.; Zhang, G.; Deng, X.; Chen, F.; et al. The histone methyltransferase SDG724 mediates H3K36me2/3 deposition at MADS50 and RFT1 and promotes flowering in rice. Plant Cell 2012, 24, 3235–3247. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Wang, X.; Su, L.; Zhai, J.X.; Cao, S.Y.; Zhang, D.F.; Liu, C.Y.; Bi, Y.P.; Qian, Q.; Cheng, Z.K.; et al. Sdg714, a histone h3k9 methyltransferase, is involved in tos17 DNA methylation and transposition in rice. Plant Cell 2007, 19, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Ding, B.; Zhu, Y.; Bu, Z.Y.; Shen, W.H.; Yu, Y.; Dong, A.W. Sdg714 regulates specific gene expression and consequently affects plant growth via h3k9 dimethylation. J. Integr. Plant Biol. 2010, 52, 420–430. [Google Scholar] [CrossRef]
- Papaefthimiou, D.; Tsaftaris, A.S. Characterization of a drought inducible trithorax-like h3k4 methyltransferase from barley. Biol. Plant. 2012, 56, 683–692. [Google Scholar] [CrossRef]
- Wang, W.; Xin, H.; Wang, M.; Ma, Q.; Wang, L.; Kaleri, N.A.; Wang, Y.; Li, X. Transcriptomic analysis reveals the molecular mechanisms of drought-stress-induced decreases in camellia sinensis leaf quality. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Chen, Q.; Guo, F.; Wang, M.; Zhao, H.; Wang, Y.; Ni, D.; Wang, P. (z)-3-hexen-1-ol accumulation enhances hyperosmotic stress tolerance in camellia sinensis. Plant Mol. Biol. 2020, 103, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Feng, Z.G.; Yang, H.M.; Zhu, X.P.; Liu, J.; Yuan, H.Y. A novel cold-regulated gene from camellia sinensis, cscor1, enhances salt- and dehydration-tolerance in tobacco. Biochem. Biophys. Res. Commun. 2010, 394, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Singh, S.; Sharma, S.; Kumar, S. A stress-responsive late embryogenesis abundant protein 7 (cslea7) of tea [camellia sinensis (l.) o. kuntze] encodes for a chaperone that imparts tolerance to escherichia coli against stresses. Mol. Biol. Rep. 2014, 41, 7191–7200. [Google Scholar] [CrossRef] [PubMed]
- Gupta, O.P.; Meena, N.L.; Sharma, I.; Sharma, P. Differential regulation of micrornas in response to osmotic, salt and cold stresses in wheat. Mol. Biol. Rep. 2014, 41, 4623–4629. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Molina, L.; Mongrand, S.; McLachlin, D.T.; Chait, B.T.; Chua, N.H. Abi5 acts downstream of abi3 to execute an aba-dependent growth arrest during germination. Plant J. 2010, 32, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Hu, S.; Guo, F.; Zhao, H.; Wang, M.; Ni, D.; Wang, Y.; Wang, P. Characterization of the SET DOMAIN GROUP gene family members in Camellia sinensis and functional analysis of the SDG43 gene in abiotic stresses. Environ. Exp. Bot. 2021, 182, 104306. [Google Scholar] [CrossRef]
- Springer, N.M.; Napoli, C.A.; Selinger, D.A.; Pandey, R.; Cone, K.C.; Chandler, V.L. Comparative analysis of set domain proteins in maize and arabidopsis reveals multiple duplications preceding the divergence of monocots and dicots. Plant Physiol. 2003, 132, 907–925. [Google Scholar] [CrossRef] [Green Version]
- Berr, A.; Shafiq, S.; Shen, W.H. Histone modifications in transcriptional activation during plant development. BBA Gene Regul. Mech. 2011, 1809, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Luo, J.; Cui, Z.; Xue, M.; Wang, L.; Zhang, X.; Pawlowski, W.P.; He, Y. ATX3, ATX4, and ATX5 encode putative h3k4 methyltransferases and are critical for plant development. Plant Physiol. 2017, 174, 1795–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, A.; Alvarez-Venegas, R.; Yilmaz, M.; Le, O.; Hou, G.; Sadder, M.; Al-Abdallat, A.; Xia, Y.; Lu, G.; Ladunga, I.; et al. The highly similar arabidopsis homologs of trithorax ATX1 and ATX2 encode proteins with divergent biochemical functions. Plant Cell 2008, 20, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.Y.; Hasegawa, P.M.; Michaelbart, M.V. Regulation of stomatal density by the GTL1 transcription factor for improving water use efficiency. Plant Signal. Behav. 2011, 6, 1069–1071. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Liu, H.; Ji, H.; Wang, Y.; Dong, B.; Qiao, Y.; Liu, M.; Li, X. The wheat GT factor TaGT2L1D negatively regulates drought tolerance and plant development. Sci. Rep. 2016, 6, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Sack, F.D. The too many mouths and four lips mutations affect stomatal production in arabidopsis. Plant Cell Online 1995, 7, 2227–2239. [Google Scholar]
- Berger, D.; Altmann, T. A subtilisin-like serine protease involved in the regulation of stomatal density and distribution in arabidopsis thaliana. Genes Dev. 2000, 14, 1119–1131. [Google Scholar] [PubMed]
- Yoo, C.Y.; Pence, H.E.; Jin, J.B.; Miura, K.; Gosney, M.J.; Hasegawa, P.M.; Mickelbart, M.V. The Arabidopsis GTL1 transcription factor regulates water use effificiency and drought tolerance by modulating stomatal density via transrepression of SDD1. Plant Cell 2010, 22, 4128–4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, K.; Yokoo, T.; Kajita, R.; Onishi, T.; Yahata, S.; Peterson, K.M.; Torii, K.U.; Kakimoto, T. Epidermal cell density is autoregulated via a secretory peptide, epidermal patterning factor 2 in arabidopsis leaves. Plant Cell Physiol. 2009, 50, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Han, L.; Qin, L.; Zhao, D. Saccharomyces cerevisiae gene tps1 improves drought tolerance in Zea mays l. by increasing the expression of sdd1 and reducing stomatal density. Plant Cell Tissue Organ Cult. PCTOC 2015, 120, 779–789. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, L.; Han, L.; Xiang, Y.; Zhao, D. Overexpression of maize sdd1 (zmsdd1) improves drought resistance in Zea mays l. by reducing stomatal density. Plant Cell Tissue Organ Cult. PCTOC 2015, 122, 147–159. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; Pilon-Smits, E.; Huang, W.; Wang, P.; Wang, M.; Guo, F.; Wang, Y.; Li, R.; Zhao, H.; et al. Transcriptome-Wide Analysis of Nitrogen-Regulated Genes in Tea Plant (Camellia sinensis L. O. Kuntze) and Characterization of Amino Acid Transporter CsCAT9.1. Plants 2020, 9, 1218. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Analysis | |
|---|---|
| Molecular mass | 118,249.89 Da |
| Theoretical isoelectric point (PI) | 8.87 |
| Positively changed residues (Asp + Glu) | 121 |
| Negatively charged residues (Arg + Lys) | 153 |
| Unstable coefficient | 45.41 (unstable) |
| Aliphatic amino acid | 85.5% |
| Aromatic amino acid | 8.0% |
| Heterocyclic amino acid | 6.5% |
| Acidic amino acid | 11.6% |
| Basic amino acid | 16.9% |
| GRAVY | −0.440 (hydrophilic) |
| Transmembrane structure | Do not cross the membrane |
| Subcellular localization | nucleus |
| Gene Name | Primer Sequence (5′-3′) |
|---|---|
| CsSDG36-F | AAGAGGTGGTTGTGATTGGAGAGG |
| CsSDG36-R | AGTAGAGGGTGGTTGGGTTAGTGC |
| TYXL+CsSDG36-F | TATGACCATGATTACGAATTCAAGAGGTGGTTGTGATTGGAGAGG |
| TYXL+CsSDG36-R | ACGACGGCCAGTGCCAAGCTTAGTAGAGGGTGGTTGGGTTAGTGC |
| Gene Name | Gene ID | Primer Sequence |
|---|---|---|
| BASL | AT5G60880 | CGATGTGGTTAAAGAGGGTA |
| CCGCTAGATTTATCAGAGGC | ||
| SBT1.2 | AT1G04110 | ACAGGAGGAGATAAAGGAAGT |
| ACCGTGGCATTAACATAAGC | ||
| EPF2 | AT1G34245 | GCGTGTTCTTTGGTCGTTAA |
| CGTGATAGTATCTCCCTCTGC | ||
| TCX3 | AT3G22760 | CGGGAAAGATTCAGGACAAA |
| GCGAGTAGCCAGGACAACAT | ||
| CHAL | AT2G30370 | CTACTCTTCTTCGTCCTCTGTG |
| TACTGTCCTTGTCCTCGTGT | ||
| TMM | AT1G80080 | AAGATCGCTTGATTTGAGTGG |
| AAGACGGGAATGGACCTGAT | ||
| SPCH | AT5G53210 | TCATAGGAGGAGTTGTGGAG |
| CTGTGGGATGAGTGGTAGTT | ||
| ERL1 | AT5G62230 | CTTGCCAACAACCGTTTAGT |
| AGAAGTTATTGCCAGACAGA | ||
| EPFL9 | AT4G12970 | ATGAAGCATGAAATGATGAACA |
| GGGTCATTTCCTTCGACTG | ||
| AtGADPH | AT3G04120 | TTGGTGACAACAGGTCAAGCA |
| AAACTTGTCGCTCAATGCAATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Guo, L.; Yuan, Y.; Hu, S.; Guo, F.; Zhao, H.; Yun, Z.; Wang, Y.; Wang, M.; Ni, D.; et al. Ectopic Overexpression of Histone H3K4 Methyltransferase CsSDG36 from Tea Plant Decreases Hyperosmotic Stress Tolerance in Arabidopsis thaliana. Int. J. Mol. Sci. 2021, 22, 5064. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105064
Chen Q, Guo L, Yuan Y, Hu S, Guo F, Zhao H, Yun Z, Wang Y, Wang M, Ni D, et al. Ectopic Overexpression of Histone H3K4 Methyltransferase CsSDG36 from Tea Plant Decreases Hyperosmotic Stress Tolerance in Arabidopsis thaliana. International Journal of Molecular Sciences. 2021; 22(10):5064. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105064
Chicago/Turabian StyleChen, Qinghua, Linghui Guo, Yanwen Yuan, Shuangling Hu, Fei Guo, Hua Zhao, Zhenyu Yun, Yu Wang, Mingle Wang, Dejiang Ni, and et al. 2021. "Ectopic Overexpression of Histone H3K4 Methyltransferase CsSDG36 from Tea Plant Decreases Hyperosmotic Stress Tolerance in Arabidopsis thaliana" International Journal of Molecular Sciences 22, no. 10: 5064. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105064