
Genome-Wide Identification of Barley Long Noncoding RNAs and Analysis of Their Regulatory Interactions during Shoot and Grain Development


Abstract
:1. Introduction
2. Results
2.1. Identification and Basic Characterization of Barley LncRNAs
2.2. Evolutionary Conservation of Identified LncRNAs
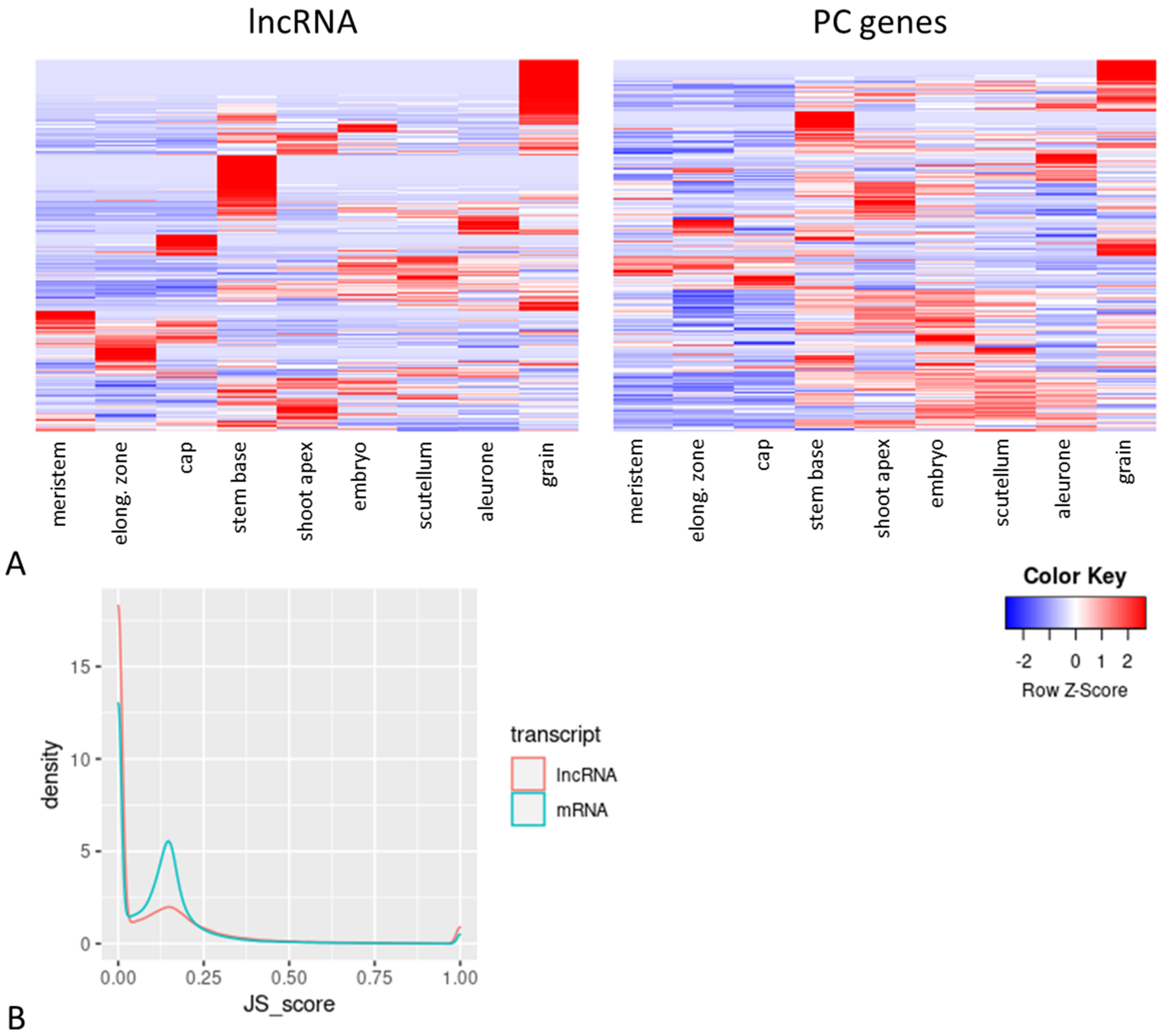
2.3. Tissue Specificity of Barley LncRNAs
2.4. Developmental Regulation of LncRNAs
2.5. Cis-Regulation of LncRNA Neighboring Genes
2.6. Trans-Regulation of PC Genes by LncRNAs
3. Discussion
3.1. Expression of Barley LncRNAs Is Tissue Specific
3.2. Stage of Development-Related lncRNAs
3.3. Cis-Regulation of PC Genes by LncRNAs
3.4. Trans-Regulation of PC Genes by LncRNAs
4. Materials and Methods
4.1. Data Retrieval
4.2. Transcriptome Assembly and Expression Analysis
4.3. Identification of lncRNA Transcripts
4.4. Evaluation of lncRNA Tissue Specificity
4.5. Analysis of Evolutionary Conservation of LncRNAs
4.6. Identification of Cis- and Trans-Acting LncRNAs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, J.; Wang, H.; Chua, N.H. Long noncoding RNA transcriptome of plants. Plant Biotechnol. J. 2015, 13, 319–328. [Google Scholar] [CrossRef]
- Ariel, F.; Romero-Barrios, N.; Jégu, T.; Benhamed, M.; Crespi, M. Battles and hijacks: Noncoding transcription in plants. Trends Plant Sci. 2015, 20, 362–371. [Google Scholar] [CrossRef]
- Matzke, M.A.; Birchler, J.A. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 2005, 6, 24–35. [Google Scholar] [CrossRef]
- Batista, P.J.; Chang, H.Y. Long Noncoding RNAs: Cellular Address Codes in Development and Disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-L.V.; Chekanova, J.A. Long Noncoding RNAs in Plants. In Long Non Coding RNA Biology; Rao, M.R.S., Ed.; Springer: Singapore, 2017; pp. 133–154. [Google Scholar] [CrossRef]
- Budak, H.; Kaya, S.B.; Cagirici, H.B. Long Non-coding RNA in Plants in the Era of Reference Sequences. Front. Plant Sci. 2020, 11, 276. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Liu, S.; Qi, H.; Cai, H.; Xu, M. Research Progress on Plant Long Non-Coding RNA. Plants 2020, 9, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Zheng, H.; Sui, N. Regulation mechanism of long non-coding RNA in plant response to stress. Biochem. Biophys. Res. Commun. 2018, 503, 402–407. [Google Scholar] [CrossRef]
- Gil-Humanes, J.; Wang, Y.; Liang, Z.; Shan, Q.; Ozuna, C.V.; Sanchez-Leon, S.; Baltes, N.J.; Starker, C.; Barro, F.; Gao, C.; et al. High-efficiency gene targeting in hexaploid wheat using DNA replicons and CRISPR/Cas9. Plant J. Cell Mol. Biol. 2017, 89, 1251–1262. [Google Scholar] [CrossRef] [Green Version]
- Gasparis, S.; Kala, M.; Przyborowski, M.; Lyznik, L.A.; Orczyk, W.; Nadolska-Orczyk, A. A simple and efficient CRISPR/Cas9 platform for induction of single and multiple, heritable mutations in barley (Hordeum vulgare L.). Plant Methods 2018, 14, 111. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Lu, P.; Xu, Y.; Li, Z.; Yu, S.; Liu, J.; Wang, H.; Chua, N.H.; Cao, P. PLncDB V2.0: A comprehensive encyclopedia of plant long noncoding RNAs. Nucleic Acids Res. 2021, 49, D1489–D1495. [Google Scholar] [CrossRef] [PubMed]
- Karlik, E.; Gozukirmizi, N. Expression analysis of lncRNA AK370814 involved in the barley vitamin B6 salvage pathway under salinity. Mol. Biol. Rep. 2018, 45, 1597–1609. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.-W.; Zhao, J.; Chen, Q.; Wu, F. Genome-wide characterization of drought stress responsive long non-coding RNAs in Tibetan wild barley. Environ. Exp. Bot. 2019, 164, 124–134. [Google Scholar] [CrossRef]
- Unver, T.; Tombuloglu, H. Barley long non-coding RNAs (lncRNA) responsive to excess boron. Genomics 2020, 112, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jiang, Q.; Jiang, P.; Zhang, W.; Huang, J.; Liu, C.; Halford, N.G.; Lu, R. Novel low-nitrogen stress-responsive long non-coding RNAs (lncRNA) in barley landrace B968 (Liuzhutouzidamai) at seedling stage. BMC Plant Biol. 2020, 20, 142. [Google Scholar] [CrossRef]
- Barakate, A.; Orr, J.; Schreiber, M.; Colas, I.; Lewandowska, D.; McCallum, N.; Macaulay, M.; Morris, J.; Arrieta, M.; Hedley, P.E.; et al. Barley Anther and Meiocyte Transcriptome Dynamics in Meiotic Prophase I. Front. Plant Sci. 2021, 11. [Google Scholar] [CrossRef]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [Green Version]
- Torkamaneh, D.; Boyle, B.; Belzile, F. Efficient genome-wide genotyping strategies and data integration in crop plants. Theor. Appl. Genet. 2018, 131, 499–511. [Google Scholar] [CrossRef]
- Lu, Z.; Xia, X.; Jiang, B.; Ma, K.; Zhu, L.; Wang, L.; Jin, B. Identification and characterization of novel lncRNAs in Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 2017, 488, 348–354. [Google Scholar] [CrossRef]
- Sreenivasulu, N.; Schnurbusch, T. A genetic playground for enhancing grain number in cereals. Trends Plant Sci. 2012, 17, 91–101. [Google Scholar] [CrossRef]
- Jenner, C.; Ugalde, T.; Aspinall, D. The Physiology of Starch and Protein Deposition in the Endosperm of Wheat. Funct. Plant Biol. 1991, 18, 211–226. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Waddington, S.R.; Cartwright, P.M.; Wall, P.C. A Quantitative Scale of Spike Initial and Pistil Development in Barley and Wheat. Ann. Bot. 1983, 51, 119–130. [Google Scholar] [CrossRef]
- Schwacke, R.; Ponce-Soto, G.Y.; Krause, K.; Bolger, A.M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M.E.; Usadel, B. MapMan4: A Refined Protein Classification and Annotation Framework Applicable to Multi-Omics Data Analysis. Mol. Plant 2019, 12, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Li, L.; Muehlbauer, G.J.; Fowler, J.E.; Evans, M.M.S. RNA Isolation and Analysis of LncRNAs from Gametophytes of Maize. In Plant Long Non-Coding RNAs: Methods and Protocols; Chekanova, J.A., Wang, H.-L.V., Eds.; Springer: New York, NY, USA, 2019; pp. 67–86. [Google Scholar] [CrossRef]
- Kang, C.; Liu, Z. An Easy-to-Follow Pipeline for Long Noncoding RNA Identification: A Case Study in Diploid Strawberry Fragaria vesca. In Plant Long Non-Coding RNAs: Methods and Protocols; Chekanova, J.A., Wang, H.-L.V., Eds.; Springer: New York, NY, USA, 2019; pp. 223–243. [Google Scholar] [CrossRef]
- Qi, S.; Akter, S.; Li, S. Identification of Novel lincRNA and Co-Expression Network Analysis Using RNA-Sequencing Data in Plants. In Plant Long Non-Coding RNAs: Methods and Protocols; Chekanova, J.A., Wang, H.-L.V., Eds.; Springer: New York, NY, USA, 2019; pp. 207–221. [Google Scholar] [CrossRef]
- Lemos, S.M.C.; Fonçatti, L.F.C.; Guyot, R.; Paschoal, A.R.; Domingues, D.S. Genome-Wide Screening and Characterization of Non-Coding RNAs in Coffea canephora. Non Coding RNA 2020, 6, 39. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsson, P.; Lipovich, L.; Grandér, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Gasparis, S.; Yanushevska, Y.; Nadolska-Orczyk, A. Bioinformatic identification and expression analysis of new microRNAs from wheat (Triticum aestivum L.). Acta Physiol. Plant. 2017, 39, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-C.; Liao, J.-Y.; Li, Z.-Y.; Yu, Y.; Zhang, J.-P.; Li, Q.-F.; Qu, L.-H.; Shu, W.-S.; Chen, Y.-Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; Liu, Z. Global identification and analysis of long non-coding RNAs in diploid strawberry Fragaria vesca during flower and fruit development. BMC Genom. 2015, 16, 815. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Quan, M.; Zhang, D. Genome-wide identification of novel long non-coding RNAs in Populus tomentosa tension wood, opposite wood and normal wood xylem by RNA-seq. Planta 2015, 241, 125–143. [Google Scholar] [CrossRef]
- Bian, J.; Deng, P.; Zhan, H.; Wu, X.; Nishantha, M.D.L.C.; Yan, Z.; Du, X.; Nie, X.; Song, W. Transcriptional Dynamics of Grain Development in Barley (Hordeum vulgare L.). Int. J. Mol. Sci. 2019, 20, 962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Digel, B.; Pankin, A.; von Korff, M. Global Transcriptome Profiling of Developing Leaf and Shoot Apices Reveals Distinct Genetic and Environmental Control of Floral Transition and Inflorescence Development in Barley. Plant Cell 2015, 27, 2318–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pefanis, E.; Wang, J.; Rothschild, G.; Lim, J.; Kazadi, D.; Sun, J.; Federation, A.; Chao, J.; Elliott, O.; Liu, Z.-P.; et al. RNA Exosome-Regulated Long Non-Coding RNA Transcription Controls Super-Enhancer Activity. Cell 2015, 161, 774–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 2013, 494, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Jabnoune, M.; Secco, D.; Lecampion, C.; Robaglia, C.; Shu, Q.; Poirier, Y. A Rice cis-Natural Antisense RNA Acts as a Translational Enhancer for Its Cognate mRNA and Contributes to Phosphate Homeostasis and Plant Fitness. Plant Cell 2013, 25, 4166–4182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierzbicki, A.T.; Haag, J.R.; Pikaard, C.S. Noncoding Transcription by RNA Polymerase Pol IVb/Pol V Mediates Transcriptional Silencing of Overlapping and Adjacent Genes. Cell 2008, 135, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Held, M.A.; Penning, B.; Brandt, A.S.; Kessans, S.A.; Yong, W.; Scofield, S.R.; Carpita, N.C. Small-interfering RNAs from natural antisense transcripts derived from a cellulose synthase gene modulate cell wall biosynthesis in barley. Proc. Natl. Acad. Sci. USA 2008, 105, 20534–20539. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, R.; Sugawara, H.; Shumway, M. International Nucleotide Sequence Database, C. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [Green Version]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Bolser, D.; Staines, D.M.; Pritchard, E.; Kersey, P. Ensembl Plants: Integrating Tools for Visualizing, Mining, and Analyzing Plant Genomics Data. In Plant Bioinformatics: Methods and Protocols; Edwards, D., Ed.; Springer: New York, NY, USA, 2016; pp. 115–140. [Google Scholar] [CrossRef]
- FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 January 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Griffiths-Jones, S. miRBase: MicroRNA sequences and annotation. Curr. Protoc. Bioinform. 2010, 29, 12.9.1–12.9.10. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef]
- Oróstica, K.Y.; Verdugo, R.A. chromPlot: Visualization of genomic data in chromosomal context. Bioinformatics 2016, 32, 2366–2368. [Google Scholar] [CrossRef] [Green Version]
- Camargo, A.; Vasconcelos, A.; Fiamenghi, M.; Pereira, G.; Carazzolle, M. tspex: A tissue-specificity calculator for gene expression data. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.11–11.12.34. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2012, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | No. Unique Hits in PLncDB | Total lncRNAs in PLncDB |
|---|---|---|
| Arabidopsis thaliana | 0 | 13,599 |
| Rice | 7 | 11,565 |
| Maize | 3 | 32,397 |
| Wheat | 861 | 43,659 |
| Barley | 7920 | 25,884 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasparis, S.; Przyborowski, M.; Nadolska-Orczyk, A. Genome-Wide Identification of Barley Long Noncoding RNAs and Analysis of Their Regulatory Interactions during Shoot and Grain Development. Int. J. Mol. Sci. 2021, 22, 5087. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105087
Gasparis S, Przyborowski M, Nadolska-Orczyk A. Genome-Wide Identification of Barley Long Noncoding RNAs and Analysis of Their Regulatory Interactions during Shoot and Grain Development. International Journal of Molecular Sciences. 2021; 22(10):5087. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105087
Chicago/Turabian StyleGasparis, Sebastian, Mateusz Przyborowski, and Anna Nadolska-Orczyk. 2021. "Genome-Wide Identification of Barley Long Noncoding RNAs and Analysis of Their Regulatory Interactions during Shoot and Grain Development" International Journal of Molecular Sciences 22, no. 10: 5087. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105087