
CACNA1A Mutations Causing Early Onset Ataxia: Profiling Clinical, Dysmorphic and Structural-Functional Findings

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Clinical Description Of Patients
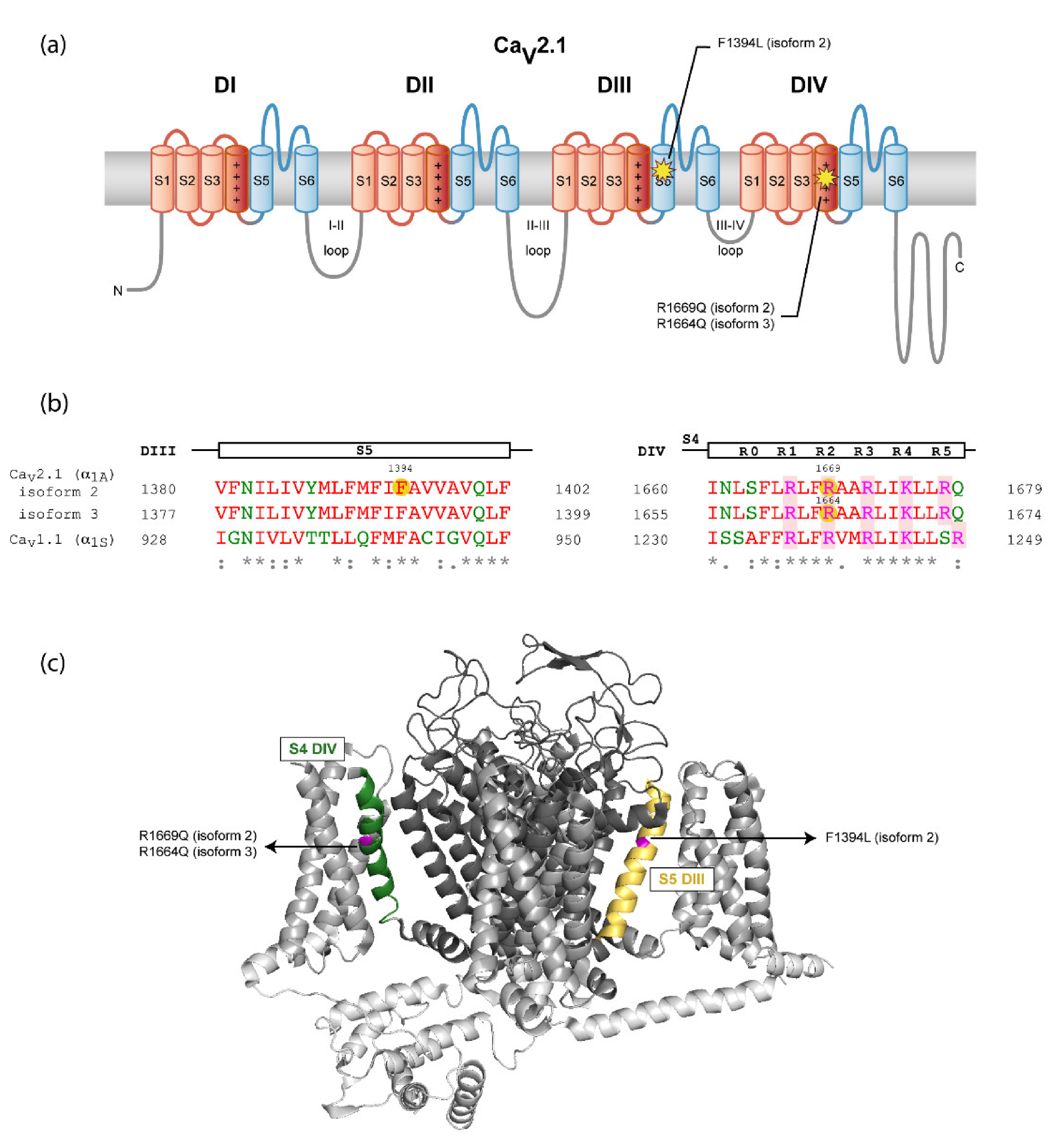
2.2. Molecular Characterization of Patients
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pietrobon, D. CaV2.1 channelopathies. Pflügers Arch. Eur. J. Physiol. 2010, 460, 375–393. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Ion channel voltage sensors: Structure, function, and pathophysiology. Neuron 2010, 67, 915–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolphin, A.C. Voltage-gated calcium channels and their auxiliary subunits: Physiology and pathophysiology and pharmacology. J. Physiol. 2016, 594, 5369–5390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumkin, L.; Michelson, M.; Leshinsky-Silver, E.; Kivity, S.; Lev, D.; Lerman-Sagie, T. congenital ataxia, mental retardation, and dyskinesia associated with a novel CACNA1A mutation. J. Child Neurol. 2010, 25, 892–897. [Google Scholar] [CrossRef]
- Calandriello, L.; Veneziano, L.; Francia, A.; Sabbadini, G.; Colonnese, C.; Mantuano, E.; Jodice, C.; Trettel, F.; Viviani, P.; Manfredi, M.; et al. Acetazolamide-responsive episodic ataxia in an Italian family refines gene mapping on chromosome 19p13. Brain 1997, 120, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- Myers, C.T.; McMahon, J.M.; Schneider, A.L.; Petrovski, S.; Allen, A.S.; Carvill, G.L.; Zemel, M.; Saykally, J.E.; LaCroix, A.J.; Heinzen, E.L.; et al. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am. J. Hum. Genet. 2016, 99, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Riant, F.; Lescoat, C.; Vahedi, K.; Kaphan, E.; Toutain, A.; Soisson, T.; Wiener-Vacher, S.R.; Tournier-Lasserve, E. Identification of CACNA1A large deletions in four patients with episodic ataxia. Neurogenetics 2009, 11, 101–106. [Google Scholar] [CrossRef]
- Bertini, E.; Zanni, G.; Boltshauser, E. Nonprogressive congenital ataxias. Handb. Clin. Neurol. 2018, 155, 91–103. [Google Scholar] [CrossRef]
- Humbertclaude, V.; Riant, F.; Krams, B.; Zimmermann, V.; Nagot, N.; Annequin, D.; Echenne, B.; Tournier-Lasserve, E.; Roubertie, A.; Bonnemains, C.; et al. Cognitive impairment in children with CACNA 1A mutations. Dev. Med. Child Neurol. 2020, 62, 330–337. [Google Scholar] [CrossRef]
- Indelicato, E.; Nachbauer, W.; Karner, E.; Eigentler, A.; Wagner, M.; Unterberger, I.; Poewe, W.; Delazer, M.; Boesch, S. The neuropsychiatric phenotype in CACNA1A mutations: A retrospective single center study and review of the literature. Eur. J. Neurol. 2018, 26, 66.e7. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yan, Z.; Li, Z.; Qian, X.; Lu, S.; Dong, M.; Zhou, Q.; Yan, N. Structure of the voltage-gated calcium channel Cav1.1 at 3.6 Å resolution. Nat. Cell Biol. 2016, 537, 191–196. [Google Scholar] [CrossRef]
- Segarra, N.G.; Gautschi, I.; Mittaz-Crettol, L.; Zetchi, C.K.; Al-Qusairi, L.; Van Bemmelen, M.X.; Maeder, P.; Bonafe, L.; Schild, L.; Roulet-Perez, E. Congenital ataxia and hemiplegic migraine with cerebral edema associated with a novel gain of function mutation in the calcium channel CACNA1A. J. Neurol. Sci. 2014, 342, 69–78. [Google Scholar] [CrossRef]
- Gripp, K.W.; Baker, L.; Telegrafi, A.; Monaghan, K.G. The role of objective facial analysis using FDNA in making diagnoses following whole exome analysis. Report of two patients with mutations in the BAF complex genes. Am. J. Med. Genet. Part A 2016, 170, 1754–1762. [Google Scholar] [CrossRef]
- Tavano, A.; Grasso, R.; Gagliardi, C.; Triulzi, F.; Bresolin, N.; Fabbro, F.; Borgatti, R. Disorders of cognitive and affective development in cerebellar malformations. Brain 2007, 130, 2646–2660. [Google Scholar] [CrossRef] [Green Version]
- Whitney, E.R.; Kemper, T.L.; Bauman, M.L.; Rosene, D.L.; Blatt, G.J. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: A stereological experiment using calbindin-D28k. Cerebellum 2008, 7, 406–416. [Google Scholar] [CrossRef]
- Vahedi, K.; Denier, C.; Ducros, A.; Bousson, V.; Levy, C.; Chabriat, H.; Haguenau, M.; Tournier-Lasserve, E.; Bousser, M.G. CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology 2000, 55, 1040–1042. [Google Scholar] [CrossRef]
- Jiang, X.; Raju, P.K.; D’Avanzo, N.; Lachance, M.; Pepin, J.; Dubeau, F.; Mitchell, W.G.; Bello-Espinosa, L.E.; Pierson, T.M.; Minassian, B.A.; et al. Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia 2019, 60, 1881–1894. [Google Scholar] [CrossRef]
- Travaglini, L.; Nardella, M.; Bellacchio, E.; D’Amico, A.; Capuano, A.; Frusciante, R.; Di Capua, M.; Cusmai, R.; Barresi, S.; Morlino, S.; et al. Missense mutations of CACNA1A are a frequent cause of autosomal dominant nonprogressive congenital ataxia. Eur. J. Paediatr. Neurol. 2017, 21, 450–456. [Google Scholar] [CrossRef]
- Bahamonde, M.I.; Serra, S.A.; Drechsel, O.; Rahman, R.; Marcé-Grau, A.; Prieto, M.; Ossowski, S.; Macaya, A.; Fernández-Fernández, J.M. A single amino acid deletion (ΔF1502) in the S6 segment of CaV2.1 domain III associated with congenital ataxia increases channel activity and promotes Ca2+ influx. PLoS ONE 2015, 10, e0146035. [Google Scholar] [CrossRef]
- Gandini, M.A.; Souza, I.A.; Ferron, L.; Innes, A.M.; Zamponi, G.W. The de novo CACNA1A pathogenic variant Y1384C associated with hemiplegic migraine, early onset cerebellar atrophy and developmental delay leads to a loss of Cav2.1 channel function. Mol. Brain 2021, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van den Maagdenberg, A.M.; Kors, E.E.; Brunt, E.R.; van Paesschen, W.; Pascual, J.; Ravine, D.; Keeling, S.; Vanmolkot, K.R.; Vermeulen, F.L.; Terwindt, G.M.; et al. Episodic ataxia type 2. Three novel truncating mutations and one novel missense mutation in the CACNA1A gene. J. Neurol. 2002, 249, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Jaudon, F.; Baldassari, S.; Musante, I.; Thalhammer, A.; Zara, F.; Cingolani, L.A. Targeting alternative splicing as a potential therapy for episodic ataxia type 2. Biomedicines 2020, 8, 332. [Google Scholar] [CrossRef] [PubMed]
- Zafeiriou, D.; Lehmann-Horn, F.; Vargiami, E.; Teflioudi, E.; Ververi, A.; Jurkat-Rott, K. Episodic ataxia type 2 showing ictal hyperhidrosis with hypothermia and interictal chronic diarrhea due to a novel CACNA1A mutation. Eur. J. Paediatr. Neurol. 2009, 13, 191–193. [Google Scholar] [CrossRef]
- Choi, K.-D.; Kim, J.-S.; Kim, H.-J.; Jung, I.; Jeong, S.-H.; Lee, S.-H.; Kim, D.U.; Kim, S.-H.; Choi, S.Y.; Shin, J.-H.; et al. Genetic variants associated with episodic ataxia in Korea. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rajakulendran, S.; Graves, T.D.; Labrum, R.W.; Kotzadimitriou, D.; Eunson, L.; Davis, M.B.; Davies, R.; Wood, N.W.; Kullmann, D.M.; Hanna, M.G.; et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J. Physiol. 2010, 588, 1905–1913. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular findings among patients referred for clinical whole-exome sequencing. J. Am. Med. Assoc. 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [Green Version]
- Ducros, A.; Denier, C.; Joutel, A.; Cecillon, M.; Lescoat, C.; Vahedi, K.; Darcel, F.; Vicaut, E.; Bousser, M.-G.; Tournier-Lasserve, E. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N. Engl. J. Med. 2001, 345, 17–24. [Google Scholar] [CrossRef]
- Kors, E.E.; Melberg, A.; Vanmolkot, K.R.J.; Kumlien, E.; Haan, J.; Raininko, R.; Flink, R.; Ginjaar, H.B.; Frants, R.R.; Ferrari, M.D.; et al. Childhood epilepsy, familial hemiplegic migraine, cerebellar ataxia, and a new CACNA1A mutation. Neurology 2004, 63, 1136–1137. [Google Scholar] [CrossRef]
- Müllner, C.; Broos, L.A.M.; van den Maagdenberg, A.M.J.M.; Striessnig, J. Familial hemiplegic migraine type 1 mutations K1336E, W1684R, and V1696I alter CaV2.1 Ca2+ channel gating: Evidence for β-subunit isoform-specific effects. J. Biol. Chem. 2004, 279, 51844–51850. [Google Scholar] [CrossRef] [Green Version]
- Garza-López, E.; Sandoval, A.; González-Ramírez, R.; Gandini, M.A.; Van Den Maagdenberg, A.; De Waard, M.; Felix, R. Familial hemiplegic migraine type 1 mutations W1684R and V1696I alter G protein-mediated regulation of CaV2.1 voltage-gated calcium channels. Biochim. Biophys. Acta 2012, 1822, 1238–1246. [Google Scholar] [CrossRef]
- Denier, C.; Ducros, A.; Durr, A.; Eymard, B.; Chassande, B.; Tournier-Lasserve, E. Missense CACNA1A mutation causing episodic ataxia type 2. Arch. Neurol. 2001, 58, 292–295. [Google Scholar] [CrossRef]
- Luo, X.; Rosenfeld, J.A.; Yamamoto, S.; Harel, T.; Zuo, Z.; Hall, M.; Wierenga, K.J.; Pastore, M.T.; Bartholomew, D.; Delgado, M.R.; et al. Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet. 2017, 13, e1006905. [Google Scholar] [CrossRef]
- Tonelli, A.; D’Angelo, M.G.; Salati, R.; Villa, L.; Germinasi, C.; Frattini, T.; Meola, G.; Turconi, A.C.; Bresolin, N.; Bassi, M.T. Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J. Neurol. Sci. 2006, 241, 13–17. [Google Scholar] [CrossRef]
- Miki, T.; Zwingman, T.A.; Wakamori, M.; Lutz, C.M.; Cook, S.A.; Hosford, D.A.; Herrup, K.; Fletcher, C.F.; Mori, Y.; Frankel, W.N.; et al. Two novel alleles of tottering with distinct Ca(v)2.1 calcium channel neuropathologies. Neuroscience 2008, 155, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Blumkin, L.; Leshinsky-Silver, E.; Michelson, M.; Zerem, A.; Kivity, S.; Lev, D.; Lerman-Sagie, T. Paroxysmal tonic upward gaze as a presentation of de-novo mutations in CACNA1A. Eur. J. Paediatr. Neurol. 2015, 19, 292–297. [Google Scholar] [CrossRef]
- Tantsis, E.M.; Gill, D.; Griffiths, L.; Gupta, S.; Lawson, J.; Maksemous, N.; Ouvrier, R.; Riant, F.; Smith, R.; Troedson, C.; et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev. Med. Child. Neurol. 2016, 58, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Camia, F.; Pisciotta, L.; Morana, G.; Schiaffino, M.C.; Renna, S.; Carrera, P.; Ferrari, M.; Baglietto, M.G.; Veneselli, E.; Siri, L.; et al. Combined early treatment in hemiplegic attacks related to CACNA1A encephalopathy with brain oedema: Blocking the cascade? Cephalalgia 2016, 37, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; De Diego, V.; Muchart, J.; Cuadras, D.; Felipe, A.; Macaya, A.; Velázquez, R.; Poo, M.P.; Fons, C.; O’Callaghan, M.M.; et al. Phosphomannomutase deficiency (PMM2-CDG): Ataxia and cerebellar assessment. Orphanet J. Rare Dis. 2015, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gripp, K.W.; Slavotinek, A.M.; Hall, J.G.; Allanson, J.E. Handbook of Physical Measurements; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- De Diego, V.; Martínez-Monseny, A.F.; Muchart, J.; Cuadras, D.; Montero, R.; Artuch, R.; Pérez-Cerdá, C.; Pérez, B.; Pérez-Dueñas, B.; Poretti, A.; et al. Longitudinal volumetric and 2D assessment of cerebellar atrophy in a large cohort of children with phosphomannomutase deficiency (PMM2-CDG). J. Inherit. Metab. Dis. 2017, 40, 709–713. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Galgonek, J.; Vymětal, J.; Jakubec, D.; Vondrášek, J. Amino Acid Interaction (INTAA) web server. Nucleic Acids Res. 2017, 45, W388–W392. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
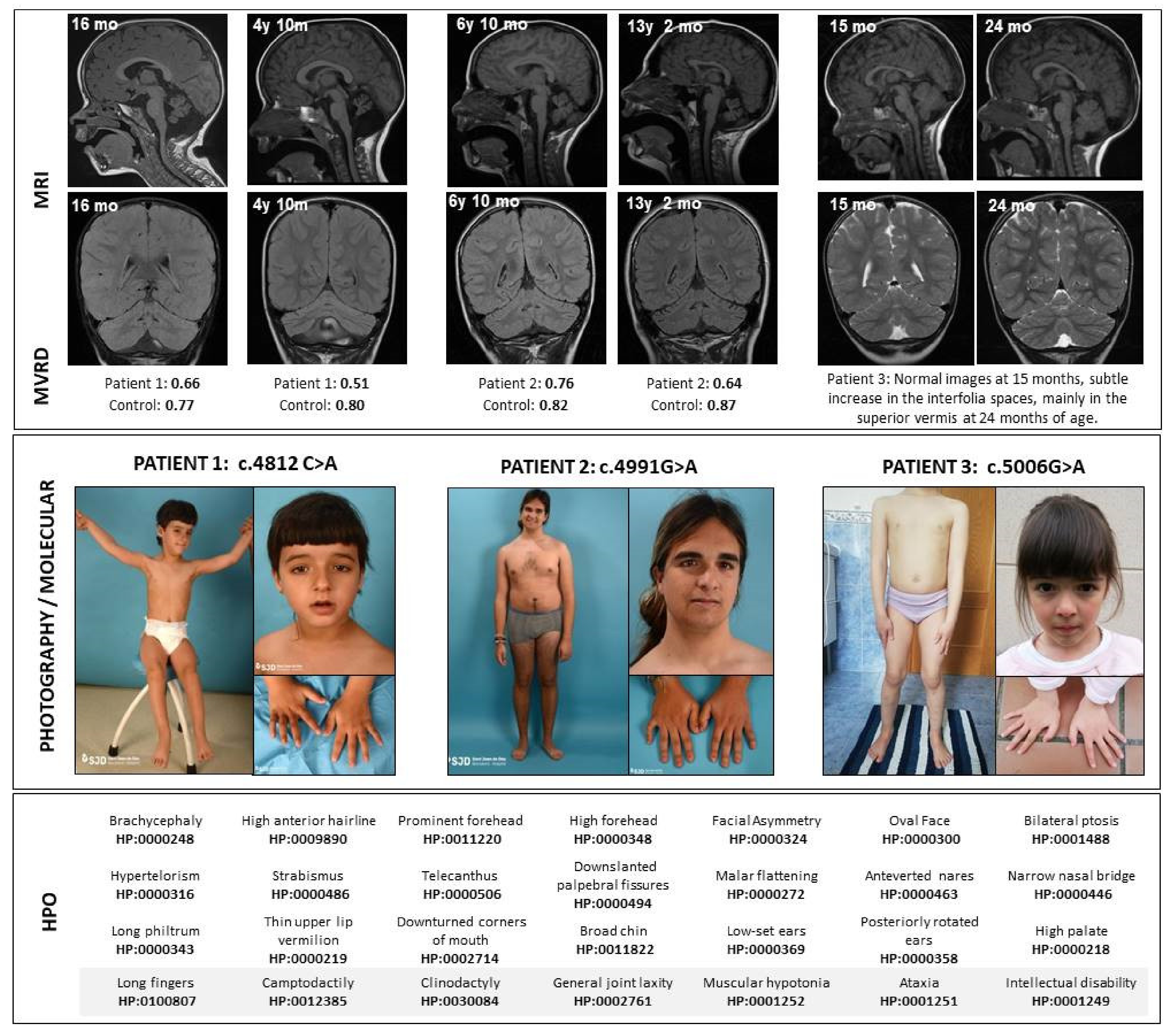
| Patient/Gender | 1 Female | 2 Male | 3 Female |
|---|---|---|---|
| Age | 8 years | 20 years | 7 years |
| Molecular findings/ inheritance | c.4182C>A p.Phe1394Leu/De novo | c.4991G>A p.Arg1664Gln/De novo | c.5006G>A p.Arg1669Gln/De novo |
| Pregnancy & delivery/GA | Twin pregnancy/HELLP syndrome/C-section/ 35 weeks | Normal/eutocic/41 weeks | Normal pregnancy/forceps/40 + 6 weeks |
| Somatometry at birth (SD) | Weight 2150 g (+1.0SD) Height 43.5cm (+0.7 SD) OFC 32 cm (+0.5 SD) | Weight 3520 g (+0,1 SD) Height 51 cm (+0.1 SD) OFC 36 cm (+0.4 SD) | Weight 3400 g (+0.3 SD) Height 51 cm (+0.6 SD) OFC 35 cm (+0.1 SD) |
| Somatometry at last evaluation (SD) | 19.5 kg (−1.5 SD) 1.16 m (−0.5 SD) OFC 52.5 cm (+0.5 SD) | 77.2 kg (+0.2 SD) 176 m (−0.2 SD) OFC 59 cm (+1.5 SD) | 22.6 kg (−0.89 9 SD) 1.23 m (−0.69 7 SD) OFC 53.5 cm (+1.2 SD) |
| Initial neurological symptoms | Hypotonia Severe developmental delay | Hypotonia Developmental delay | Hypotonia Development delay |
| Cerebellar syndrome | Truncal ataxia and stereotypes Strabismus, terminal nystagmus | Mild ataxia/Dysarthria Oculomotor apraxia Nystagmus | Mild ataxia |
| Neurodevelopment | Sitting position: 27 months Walk only with stroller: 5 years No speech (guttural sounds) Special schooling | Sitting position: 8 months Independent walking: 30 months Language delay Occupational school | Sitting position: 9 months Independent walking: 30 months Language Delay Ordinary school with support |
| Other neurological symptoms | Intellectual disability Autistic traits Attention deficit (treated with guanfacine) | Mild intellectual disability Uncontrolled lateral head movements without consciousness abnormalities. | Mild intellectual disability Brunet−Lézine (30 months) 67 * WIPPSI IV (5.5 years): verbal IQ 70, performance IQ 68. ADHD (MPD) |
| Cranial magnetic resonance | 6 months: ventricular enlargement and increased extra−axial spaces. Normal posterior fossa structures. 16 months: cerebellar atrophy: increased interfolia spaces mainly in vermis. MVRD = 0.66 (mean for controls 0.77). 4 years 10 months: progression of generalized cerebellar atrophy. MVRD = 0.51 (mean for controls 0.80) | 6 years 10 months: cerebellar atrophy, with a MVRD = 0.76 (mean for controls 0. 82). 13 years 2 months: progressive generalized cerebellar atrophy. MVRD = 0.64 (mean for controls 0.87) | 15 months: no signs of cerebellar atrophy. 24 months: cerebellar atrophy: increase of interfolia spaces in the superior vermis |
| ICARS assessment | No collaboration, no comprehension | 17 years: 14/100 19 years: 12/100 | 4 years: 28/100 6 years: 19/100 |
| Acetazolamide therapy(at least during 8 months) | 12 mg/kg/day in two doses Improvement in muscle tone and communication intention. No objective positive response in the long term. | 250 mg /12 h Improvement in motor symptoms. Abolished stereotyped episodes. Withdrawn due to lithiasis | 12 mg/kg/day in two doses. No objective positive response |
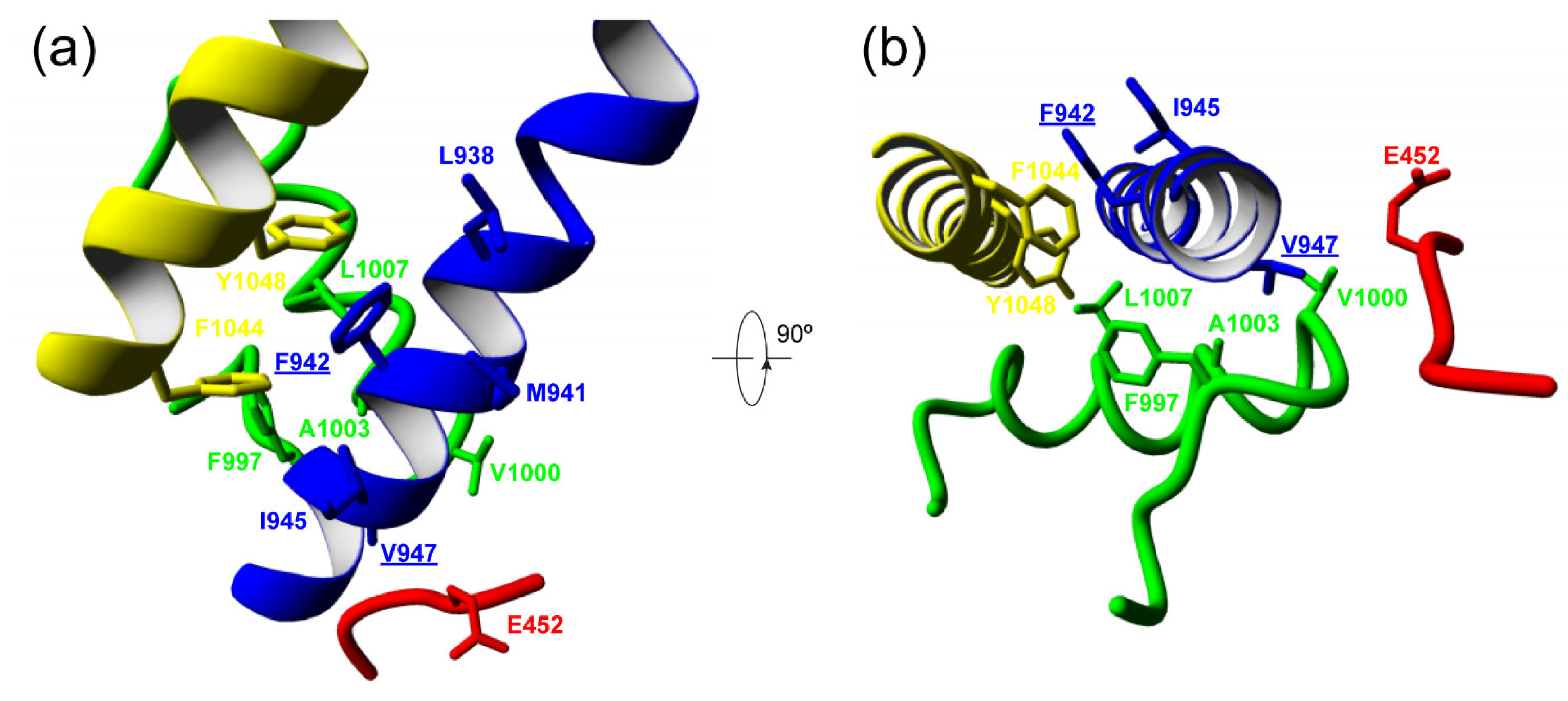
| E452 | V452 | F997 | V1000 | A1003 | ||
| V947 | −3.98 | −3.96 | −2.02 | −3.06 | −2.08 | |
| M947 | 47.27 | 30.3 | −1.09 | −1.93 | −0.67 | |
| Δ | +51.25 | +34.26 | +0.93 | +1.13 | +1.41 | |
| L938 | M941 | I945 | L1007 | F1044 | Y1048 | |
| F942 | −5.54 | −8.22 | −4.72 | −2.96 | −2.34 | −9.41 |
| L942 | −3.74 | −3.30 | −3.71 | −2.94 | 7.26 | 37.47 |
| Δ | +1.8 | +4.92 | +1.01 | +0.02 | +9.6 | +46.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Monseny, A.F.; Edo, A.; Casas-Alba, D.; Izquierdo-Serra, M.; Bolasell, M.; Conejo, D.; Martorell, L.; Muchart, J.; Carrera, L.; Ortez, C.I.; et al. CACNA1A Mutations Causing Early Onset Ataxia: Profiling Clinical, Dysmorphic and Structural-Functional Findings. Int. J. Mol. Sci. 2021, 22, 5180. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105180
Martínez-Monseny AF, Edo A, Casas-Alba D, Izquierdo-Serra M, Bolasell M, Conejo D, Martorell L, Muchart J, Carrera L, Ortez CI, et al. CACNA1A Mutations Causing Early Onset Ataxia: Profiling Clinical, Dysmorphic and Structural-Functional Findings. International Journal of Molecular Sciences. 2021; 22(10):5180. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105180
Chicago/Turabian StyleMartínez-Monseny, Antonio F., Albert Edo, Dídac Casas-Alba, Mercè Izquierdo-Serra, Mercè Bolasell, David Conejo, Loreto Martorell, Jordi Muchart, Laura Carrera, Carlos I. Ortez, and et al. 2021. "CACNA1A Mutations Causing Early Onset Ataxia: Profiling Clinical, Dysmorphic and Structural-Functional Findings" International Journal of Molecular Sciences 22, no. 10: 5180. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105180