
Method for Rapid Analysis of Mutant RNA Polymerase Activity on Templates Containing Unnatural Nucleotides

 and
and
Abstract
:1. Introduction
2. Results
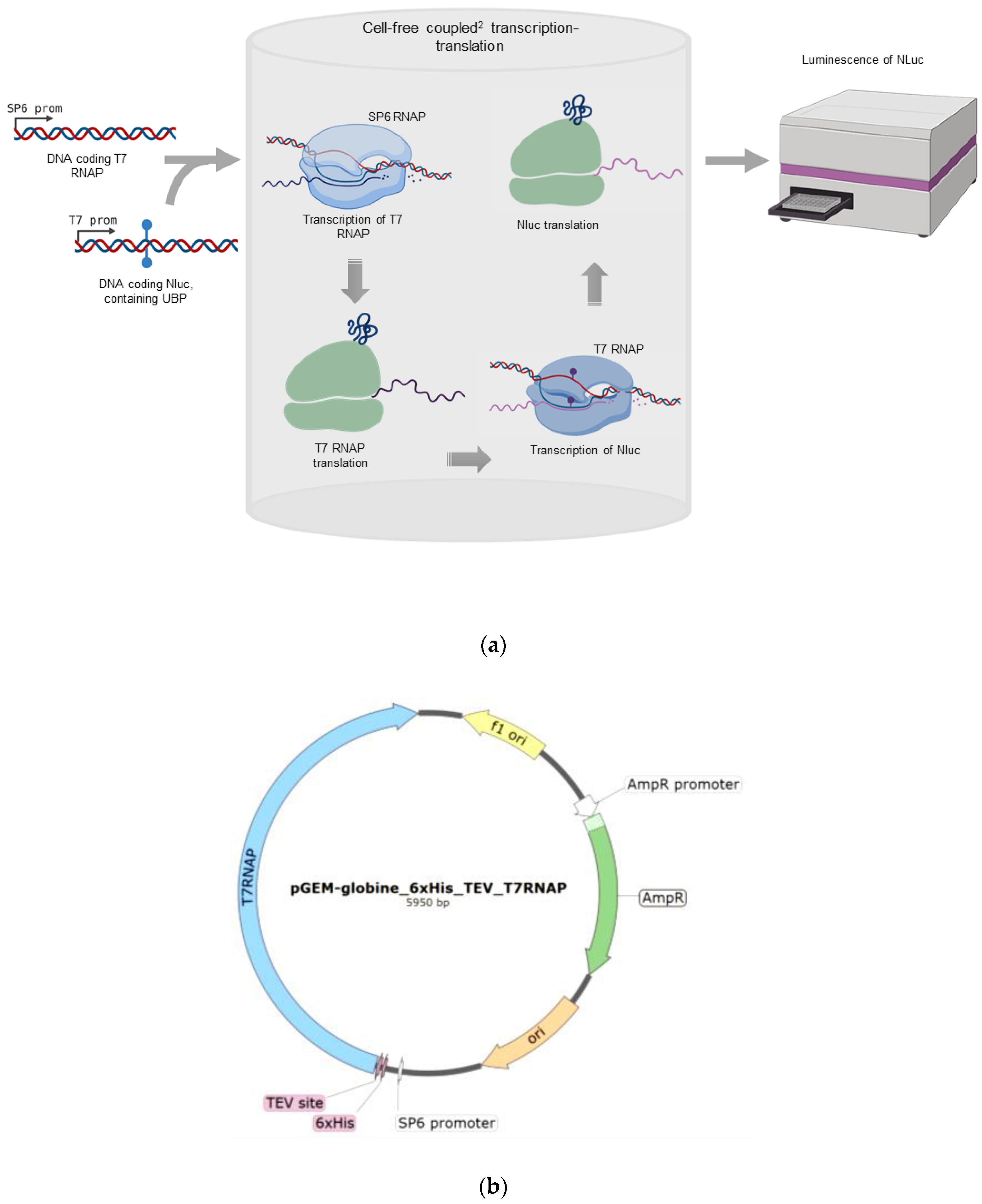
2.1. Overview of the Method
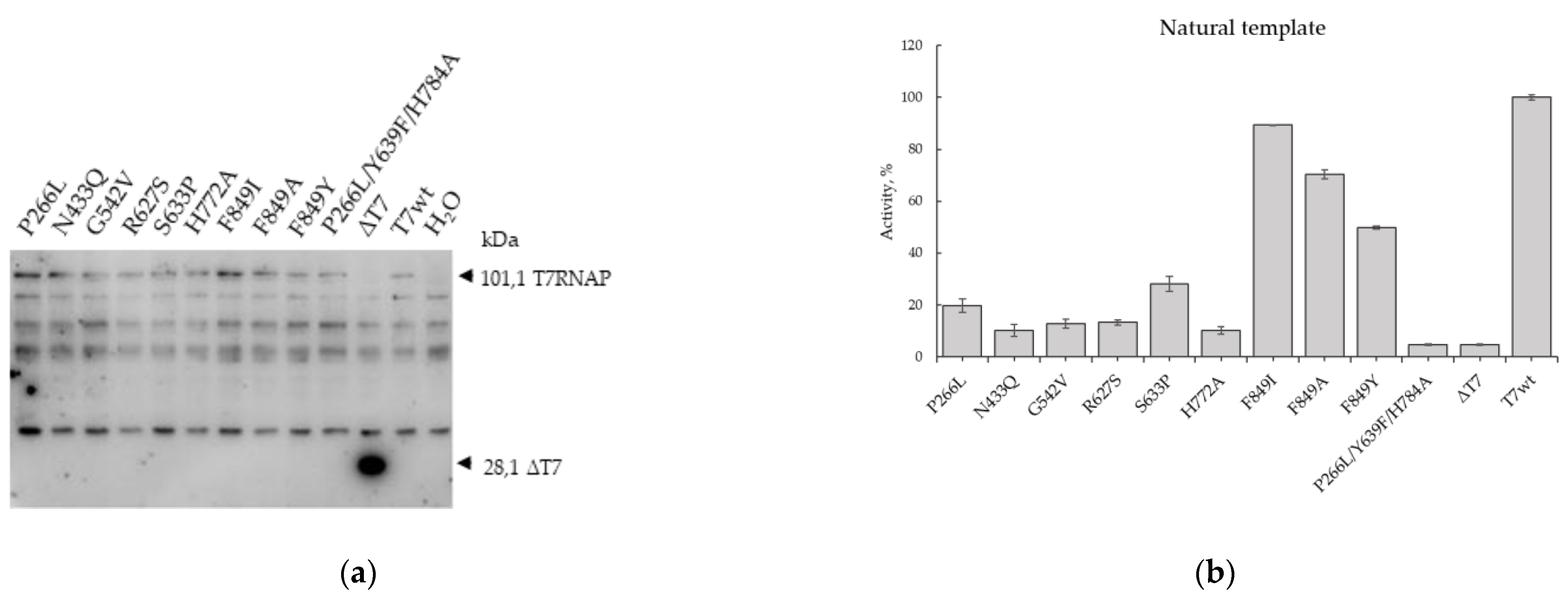
2.2. Testing of T7 RNAP Mutants on a Natural Template
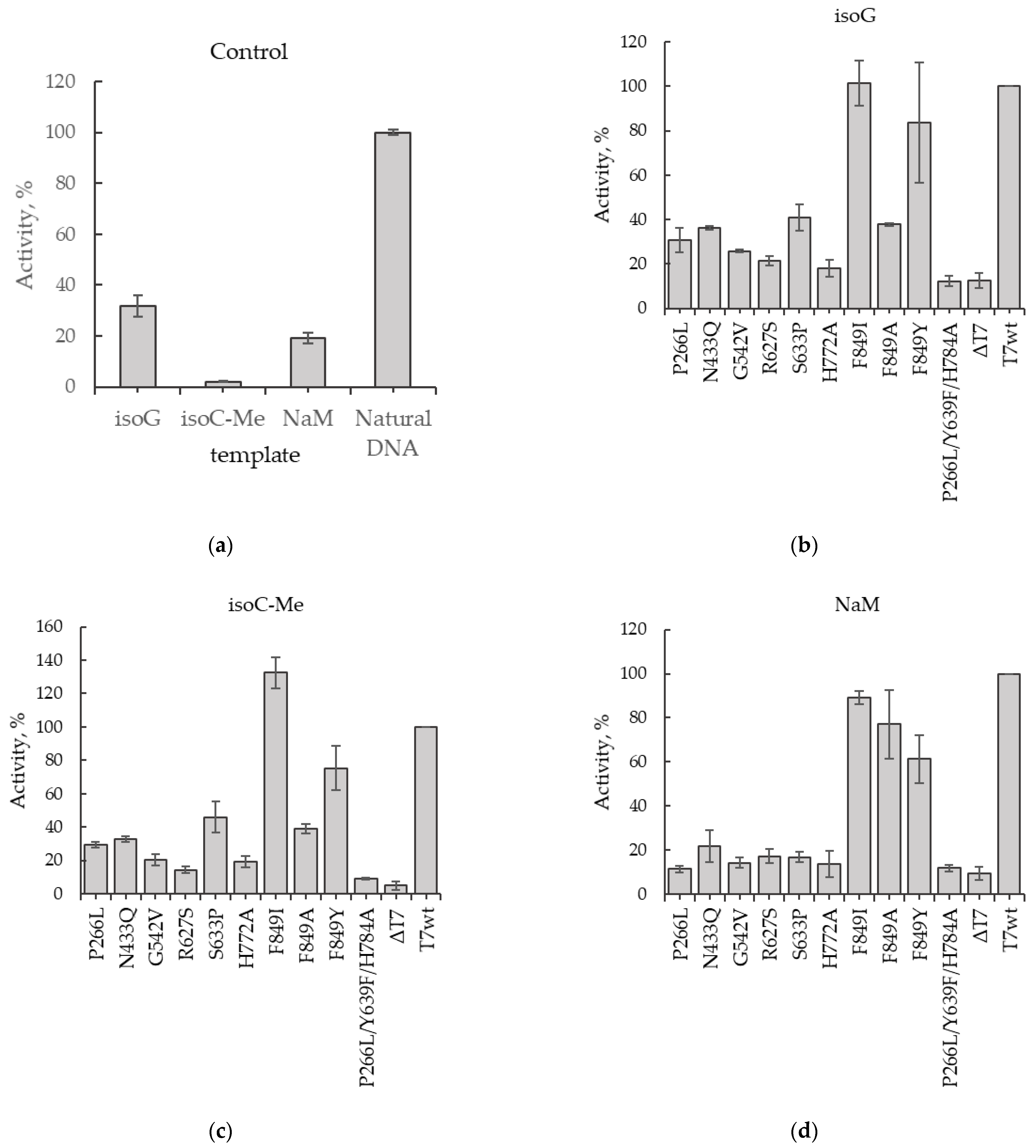
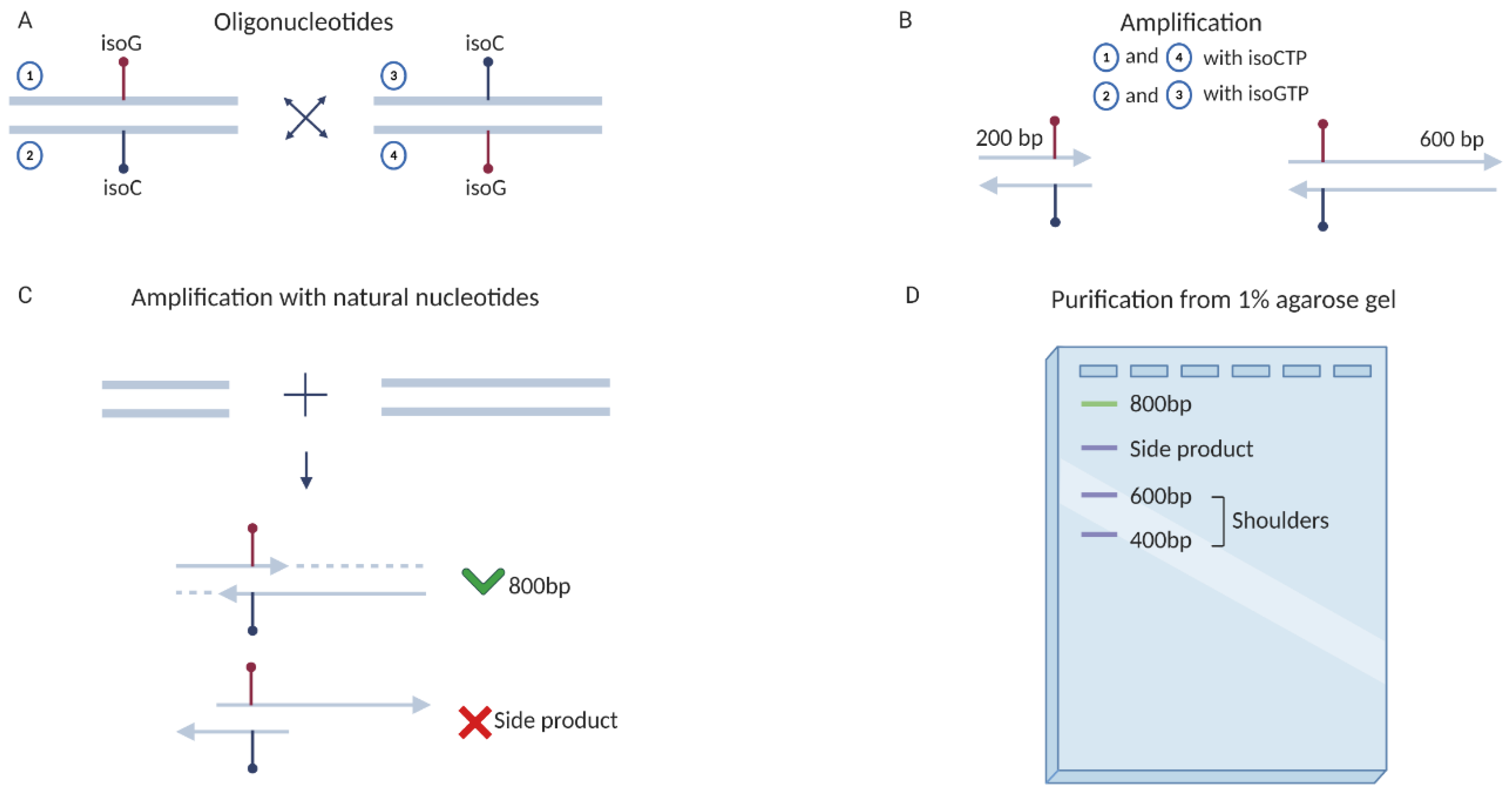
2.3. Obtaining DNA Templates Containing UBPs and Testing of T7 RNAP Mutants Activities
3. Discussion
4. Materials and Methods
4.1. Obtaining Constructs Encoding Mutant Forms of T7 RNAP
4.2. T7 RNAP and Nluc PCR Products Obtaining
4.3. The Coupled2 Transcription-Translation System
4.4. Western-Blot Analysis
4.5. Obtaining Unnatural Templates
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CDS | CoDing Sequence |
| dNTPs | deoxyribonucleotide triphosphate |
| isoC | isocytosine (2-amino-4-ketopyrimidine) |
| isoG | isoguanine (6-amino-2-ketopurine) |
| Nluc | nanoluciferase |
| NTPs | ribonucleotide triphosphate |
| PCR | Polymerase Chain Reaction |
| RNAP | RNA polymerase |
| UBP | unnatural base pair |
| 5′UTR | 5′-untranslated regions |
| WGE | wheat germ extract |
| Wt | wild type |
References
- Bain, J.D.; Switzer, C.; Chamberlin, A.R.; Benner, S.A. Ribosome-mediated incorporation of a non-standard amino acid into a peptide through expansion of the genetic code. Nature 1992, 356, 537–539. [Google Scholar] [CrossRef]
- Hirao, I.; Ohtsuki, T.; Fujiwara, T.; Mitsui, T.; Yokogawa, T.; Okuni, T.; Nakayama, H.; Takio, K.; Yabuki, T.; Kigawa, T.; et al. An unnatural base pair for incorporating amino acid analogs into proteins. Nat. Biotechnol. 2002, 20, 177–182. [Google Scholar] [CrossRef]
- Hirao, I.; Harada, Y.; Kimoto, M.; Mitsui, T.; Fujiwara, T.; Yokoyama, S. A two-unnatural-base-pair system toward the expansion of the genetic code. J. Am. Chem. Soc. 2004, 126, 13298–13305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ptacin, J.L.; Fischer, E.C.; Aerni, H.R.; Caffaro, C.E.; San Jose, K.; Feldman, A.W.; Turner, C.R.; Romesberg, F.E. A semi-synthetic organism that stores and retrieves increased genetic information. Nature 2017, 551, 644–647. [Google Scholar] [CrossRef] [Green Version]
- Fischer, E.C.; Hashimoto, K.; Zhang, Y.; Feldman, A.W.; Dien, V.T.; Karadeema, R.J.; Adhikary, R.; Ledbetter, M.P.; Krishnamurthy, R.; Romesberg, F.E. New codons for efficient production of unnatural proteins in a semisynthetic organism. Nat. Chem. Biol. 2020, 16, 570–576. [Google Scholar] [CrossRef]
- Neumann, H.; Wang, K.; Davis, L.; Garcia-Alai, M.; Chin, J.W. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 2010, 464, 441–444. [Google Scholar] [CrossRef] [Green Version]
- Berry, M.J.; Howard, M.T. Reprogramming the Ribosome for Selenoprotein Expression: RNA Elements and Protein Factors. In Recoding: Expansion of Decoding Rules Enriches Gene Expression; Atkins, J.F., Gesteland, R.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; p. 31. [Google Scholar]
- Hao, B.; Gong, W.; Ferguson, T.K.; James, C.M.; Krzycki, J.A.; Chan, M.K. A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science 2002, 296, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, G.; James, C.M.; Krzycki, J.A. Pyrrolysine encoded by UAG in Archaea: Charging of a UAG-decoding specialized tRNA. Science 2002, 296, 1459–1462. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Kramer, F.R. Molecular beacons: Probes that fluoresce upon hybridization. Nat. Biotechnol. 1996, 14, 303–308. [Google Scholar] [CrossRef]
- Collins, M.L.; Irvine, B.; Tyner, D.; Fine, E.; Zayati, C.; Chang, C.A.; Horn, T.; Ahle, D.; Detmer, J.; Shen, L.P.; et al. A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/mL. Nucleic Acids Res. 1997, 25, 2979–2984. [Google Scholar] [CrossRef] [Green Version]
- Sherrill, C.B.; Marshall, D.J.; Moser, M.J.; Larsen, C.A.; Daudé-Snow, L.; Prudent, J.R. Nucleic acid analysis using an expanded genetic alphabet to quench fluorescence. J. Am. Chem. Soc. 2004, 126, 4550–4556. [Google Scholar] [CrossRef]
- Johnson, S.C.; Marshall, D.J.; Harms, G.; Miller, C.M.; Sherrill, C.B.; Beaty, E.L.; Lederer, S.A.; Roesch, E.B.; Madsen, G.; Hoffman, G.L.; et al. Multiplexed genetic analysis using an expanded genetic alphabet. Clin. Chem. 2004, 50, 2019–2027. [Google Scholar] [CrossRef]
- Moser, M.J.; Christensen, D.R.; Norwood, D.; Prudent, J.R. Multiplexed detection of anthrax-related toxin genes. J. Mol. Diagn. 2006, 8, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, M.; Mitsui, T.; Yokoyama, S.; Hirao, I. A unique fluorescent base analogue for the expansion of the genetic alphabet. J. Am. Chem. Soc. 2010, 132, 4988–4989. [Google Scholar] [CrossRef]
- Kimoto, M.; Mitsui, T.; Yamashige, R.; Sato, A.; Yokoyama, S.; Hirao, I. A new unnatural base pair system between fluorophore and quencher base analogues for nucleic acid-based imaging technology. J. Am. Chem. Soc. 2010, 132, 15418–15426. [Google Scholar] [CrossRef]
- Yamashige, R.; Kimoto, M.; Takezawa, Y.; Sato, A.; Mitsui, T.; Yokoyama, S.; Hirao, I. Highly specific unnatural base pair systems as a third base pair for PCR amplification. Nucleic Acids Res. 2012, 40, 2793–2806. [Google Scholar] [CrossRef] [Green Version]
- Someya, T.; Ando, A.; Kimoto, M.; Hirao, I. Site-specific labeling of RNA by combining genetic alphabet expansion transcription and copper-free click chemistry. Nucleic Acids Res. 2015, 43, 6665–6676. [Google Scholar] [CrossRef] [PubMed]
- Riedl, J.; Ding, Y.; Fleming, A.M.; Burrows, C.J. Identification of DNA lesions using a third base pair for amplification and nanopore sequencing. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Malyshev, D.A.; Romesberg, F.E. The expanded genetic alphabet. Angew. Chemie Int. Ed. 2015, 54, 11930–11944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashige, R.; Kimoto, M.; Okumura, R.; Hirao, I. Visual detection of amplified DNA by polymerase chain reaction using a genetic alphabet expansion system. J. Am. Chem. Soc. 2018, 140, 14038–14041. [Google Scholar] [CrossRef]
- Feldman, A.W.; Dien, V.T.; Karadeema, R.J.; Fischer, E.C.; You, Y.; Anderson, B.A.; Krishnamurthy, R.; Chen, J.S.; Li, L.; Romesberg, F.E. Optimization of replication, transcription, and translation in a semi-synthetic organism. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef]
- Switzer, C.; Moroney, S.E.; Benner, S.A. Enzymatic incorporation of a new base pair into DNA and RNA. J. Am. Chem. Soc. 1989, 111, 8322–8323. [Google Scholar] [CrossRef]
- Piccirilli, J.A.; Krauch, T.; Moroney, S.E.; Benner, S.A. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature 1990, 343, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Sismour, A.M.; Benner, S.A. The use of thymidine analogs to improve the replication of an extra DNA base pair: A synthetic biological system. Nucleic Acids Res. 2005, 33, 5640–5646. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.Y.; Moroney, S.E.; Benner, S.A. Enzymatic recognition of the base pair between isocytidine and isoguanosine. Biochemistry 1993, 32, 10489–10496. [Google Scholar] [CrossRef]
- Horlacher, J.; Hottiger, M.; Podust, V.N.; Hubscher, U.; Benner, S.A. Recognition by viral and cellular DNA polymerases of nucleosides bearing bases with nonstandard hydrogen bonding patterns. Proc. Natl. Acad. Sci. USA 2006, 92, 6329–6333. [Google Scholar] [CrossRef] [Green Version]
- Benner, S.A. Understanding nucleic acids using synthetic chemistry. Acc. Chem. Res. 2004, 37, 784–797. [Google Scholar] [CrossRef]
- Martinot, T.A.; Benner, S.A. Artificial genetic systems: Exploiting the ‘aromaticity’ formalism to improve the tautomeric ratio for isoguanosine derivatives. J. Org. Chem. 2004, 69, 3972–3975. [Google Scholar] [CrossRef]
- Yang, Z.; Hutter, D.; Sheng, P.; Sismour, A.M.; Benner, S.A. Artificially expanded genetic information system: A new base pair with an alternative hydrogen bonding pattern. Nucleic Acids Res. 2006, 34, 6095–6101. [Google Scholar] [CrossRef] [Green Version]
- Hoshika, S.; Leal, N.A.; Kim, M.J.; Kim, M.S.; Karalkar, N.B.; Kim, H.J.; Bates, A.M.; Watkins, N.E., Jr.; SantaLucia, H.A.; Meyer, A.J.; et al. Hachimoji DNA and RNA: System with eight building blocks. Science 2019, 363, 884–887. [Google Scholar] [CrossRef]
- Johar, Z.; Zahn, A.; Leumann, C.J.; Jaun, B. Solution structure of a DNA duplex containing a biphenyl pair. Chemistry 2008, 14, 1080–1086. [Google Scholar] [CrossRef]
- Leconte, A.M.; Hwang, G.T.; Matsuda, S.; Capek, P.; Hari, Y.; Romesberg, F.E. Discovery, characterization, and optimization of an unnatural base pair for expansion of the genetic alphabet. J. Am. Chem. Soc. 2008, 130, 2336–2343. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.J.; Hwang, G.T.; Ordoukhanian, P.; Romesberg, F.E. Optimization of an unnatural base pair toward natural-like replication. J. Am. Chem. Soc. 2009, 131, 3246–3252. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, S.; Fillo, J.D.; Henry, A.A.; Rai, P.; Wilkens, S.J.; Dwyer, T.J.; Geierstanger, B.H.; Wemmer, D.E.; Schultz, P.G.; Spraggon, G.; et al. Efforts toward expansion of the genetic alphabet: Structure and replication of unnatural base pairs. J. Am. Chem. Soc. 2007, 129, 10466–10473. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.J.; Romesberg, F.E. Major groove derivatization of an unnatural base pair. Chembiochem 2009, 10, 2394–2400. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Sismour, A.M.; Sheng, P.; Puskar, N.L.; Benner, S.A. Enzymatic incorporation of a third nucleobase pair. Nucleic Acids Res. 2007, 35, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, F.; Alvarado, J.B.; Benner, S.A. Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J. Am. Chem. Soc. 2011, 133, 15105–15112. [Google Scholar] [CrossRef] [Green Version]
- Malyshev, D.A.; Seo, Y.J.; Ordoukhanian, P.; Romesberg, F.E. PCR with an expanded genetic alphabet. J. Am. Chem. Soc. 2009, 131, 14620–14621. [Google Scholar] [CrossRef] [Green Version]
- Malyshev, D.A.; Pfaff, D.A.; Ippoliti, S.I.; Hwang, G.T.; Dwyer, T.J.; Romesberg, F.E. Solution structure, mechanism of replication, and optimization of an unnatural base pair. Chem. Eur. J. 2010, 16, 12650–12659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirao, I.; Mitsui, T.; Kimoto, M.; Yokoyama, S. An efficient unnatural base pair for PCR amplification. J. Am. Chem. Soc. 2007, 129, 15549–15555. [Google Scholar] [CrossRef] [PubMed]
- Morohashi, N.; Kimoto, M.; Sato, A.; Kawai, R.; Hirao, I. Site-specific incorporation of functional components into RNA by an unnatural base pair transcription system. Molecules 2012, 17, 2855–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimoto, M.; Hirao, I. Site-specific incorporation of extra components into RNA by transcription using unnatural base pair systems. Methods Mol. Biol. 2010, 634, 355–369. [Google Scholar] [PubMed]
- Hirao, I.; Kimoto, M.; Mitsui, T.; Fujiwara, T.; Kawai, R.; Sato, A.; Harada, Y.; Yokoyama, S. An unnatural hydrophobic base pair system: Site-specific incorporation of nucleotide analogs into DNA and RNA. Nat. Methods. 2006, 3, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, M.; Meyer, A.J.; Hirao, I.; Ellington, A.D. Genetic alphabet expansion transcription generating functional RNA molecules containing a five-letter alphabet including modified unnatural and natural base nucleotides by thermostable T7 RNA polymerase variants. Chem. Commun. 2017, 53, 12309–12312. [Google Scholar] [CrossRef]
- Cheetham, G.M.; Steitz, T.A. Insights into transcription: Structure and function of single-subunit DNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 2000, 10, 117–123. [Google Scholar] [CrossRef]
- Sousa, R. Structural and mechanistic relationships between nucleic acid polymerases. Trends Biochem. Sci. 1996, 21, 186–190. [Google Scholar] [CrossRef]
- Gudima, S. Deoxyribonucleotide-containing RNAs: A novel class of templates for HIV-1 reverse transcriptase. Nucleic Acids Res. 1997, 25, 4614–4618. [Google Scholar] [CrossRef] [Green Version]
- Rusakova, E.E.; Tunitskaya, V.L.; Memelova, L.V.; Kochetkova, S.V.; Kostyuk, D.A.; Kochetkov, S. N. Mutant T7 RNA polymerase is capable of catalyzing DNA primer extension reaction. FEBS Lett. 1998, 423, 189–192. [Google Scholar] [CrossRef] [Green Version]
- Chelliserrykattil, J.; Ellington, A.D. Evolution of a T7 RNA polymerase variant that transcribes 2′-O-methyl RNA. Nat. Biotechnol. 2004, 22, 1155–1160. [Google Scholar] [CrossRef]
- Frydman, J.; Erdjument-Bromage, H.; Tempst, P.; Hartl, F.U. Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase. Nat. Struct. Biol. 1999, 6, 697–705. [Google Scholar] [CrossRef]
- England, C.G.; Ehlerding, E.B.; Cai, W. NanoLuc: A small luciferase is brightening up the field of bioluminescence. Bioconjugate Chem. 2016, 27, 1175–1187. [Google Scholar] [CrossRef]
- Guillerez, J.; Lopez, P.; Proux, F.; Launay, H.; Dreyfus, M. A mutation in T7 RNA polymerase that facilitates promoter clearance. Proc. Natl. Acad. Sci. USA 2005, 102, 5958–5963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, A.J.; Garry, D.J.; Hall, B.; Byrom, M.M.; McDonald, H.G.; Yang, X.; Yin, Y.W.; Ellington, A.D. Transcription yield of fully 2′-modified RNA can be increased by the addition of thermostabilizing mutations to T7 RNA polymerase mutants. Nucleic Acids Res. 2015, 43, 7480–7488. [Google Scholar] [CrossRef] [Green Version]
- Rusakova, E.E.; Sugiyama, A.; Nishiya, Y.; Kawakami, B. RNA Polymerase Mutants with Increased Thermostability. U.S. Patent 7,507,567B2, 24 March 2009. [Google Scholar]
- Boulain, J.C.; Dassa, J.; Mesta, L.; Savatier, A.; Costa, N.; Muller, B.; L’hostis, G.; Stura, E.; Troesch, A.; Ducancel, F. Mutants with higher stability and specific activity from a single thermosensitive variant of T7 RNA polymerase. Protein Eng. Des. Sel. 2013, 26, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temiakov, D.; Patlan, V.; Anikin, M.; McAllister, W.T.; Yokoyama, S.; Vassylyev, D.G. Structural basis for substrate selection by T7 RNA Polymerase. Cell 2004, 116, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef]
- Maciejewska, A.M.; Lichota, K.D.; Kusmierek, J.T. Neighbouring bases in template influence base-pairing of isoguanine. Biochem. J. 2003, 369, 611–618. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | Transcription Activity | References |
|---|---|---|
| P266L | Increased thermal stability and promoter clearance | [53] |
| N433Q | Increased thermal stability | [54,55] |
| G542V | Involved in interactions with the 2′-hydroxyl moiety of ribonucleotides | [50] |
| R627S | Increased thermal stability, interactions with phosphate groups of NTPs, incorporation of ddNTP | [54,55] |
| S633P | Increased thermal stability | [54,55,56] |
| H772A | [56] | |
| F849I | Increased thermal stability | [54,55,56] |
| F849A | Increased thermal stability | [54,55,56] |
| F849Y | Increased thermal stability | [54,55,56] |
| FAL: P266L/Y639F/H784A | Increased thermal stability, can accept 2′-O-methyl triphosphates | [31,54,57] |
| Components | Final Concentration |
|---|---|
| TNT® Wheat Germ Extract (Promega, Madison, WI, USA) | 50% |
| TNT buffer (Promega, Madison, WI, USA) 25× | 1× |
| Amino Acid mix (Promega, Madison, WI, USA) 1 mM | 0.02 mM |
| RiboLock RNase Inhibitor (Thermo Fisher Scientific, Waltham, MA, USA) 40 U/µL | 0.8 U/µL |
| Nluc PCR product 10 ng/µL | 1 ng/µL |
| SP6 RNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA) 200 U/µL | 16 U/µL |
| T7 Polymerase PCR product | 10 ng/µL |
| Nano-Glo® (Promega, Madison, WI, USA) | 1% |
| Primer | Sequence |
|---|---|
| Forward primer (T7 RNAP) | 5′-ACG CCA AGC TAT TTA GGT GAC ACT ATA GAA T-3′ |
| Reverse primer (T7 RNAP) | 5′-TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTG GGC GAA TTG GCC AAG TCG GC-3′ |
| Forward primer (Nluc) | 5′-CCA GTG CCA AGC TTA ATA CGA CTC ACT ATA G-3′ |
| Reverse primer (NLuc) | 5′-TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTA AAC AGC TAT GAC CAT GAT T-3′ |
| T7_HindIII_F | 5′-TTT AAG CTT GCT TTT GAC ACA ACT GTG TTT ACT TGC AAT CCC CCA AAA CAG ACA CCA TGG GAT CTC ATC ATC ATC ATC ATC ACT CTG CTG GTG AAA ACC TTT ACT TCC AGG GTG TGG GAT CCA ACA CGA TTA ACA TCG CTA AGA A-3′ |
| T7_XhoI_R | 5′-AAA ACT CGA GTT ACG CGA ACG CGA AGT CC-3′ |
| Forward primer (T7 RNAP) | 5′-ACG CCA AGC TAT TTA GGT GAC ACT ATA GAA T-3′ |
| Reverse primer (T7 RNAP) | 5′-TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTG GGC GAA TTG GCC AAG TCG GC-3′ |
| Forward primer (Nluc) | 5′-CCA GTG CCA AGC TTA ATA CGA CTC ACT ATA G-3′ |
| Reverse primer (NLuc) | 5′-TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTA AAC AGC TAT GAC CAT GAT T-3′ |
| Reverse primer (V29-NaM2) | 5′-TG AAA CAA ACT GGA C-NaM-C ACC TCC CTG TTC AA-3′ |
| Forward primer (V29-iC2) | 5′-TT GAA CAG GGA GGT G-iC-G TCC AGT TTG TTT CAG AAT CTC-3′ |
| Reverse primer (V29-iG2) | 5′-GAG ATT CTG AAA CAA ACT GGA C-iG-C ACC TCC CTG TTC AA-3′ |
| Forward primer (V29-iG2) | 5′-TT GAA CAG GGA GGT G-iG-G TCC AGT TTG TTT CAG AAT CTC-3′ |
| Reverse primer (V29-iC2) | 5′-GAG ATT CTG AAA CAA ACT GGA C-iC-C ACC TCC CTG TTC AA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egorova, T.; Shuvalova, E.; Mukba, S.; Shuvalov, A.; Kolosov, P.; Alkalaeva, E. Method for Rapid Analysis of Mutant RNA Polymerase Activity on Templates Containing Unnatural Nucleotides. Int. J. Mol. Sci. 2021, 22, 5186. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105186
Egorova T, Shuvalova E, Mukba S, Shuvalov A, Kolosov P, Alkalaeva E. Method for Rapid Analysis of Mutant RNA Polymerase Activity on Templates Containing Unnatural Nucleotides. International Journal of Molecular Sciences. 2021; 22(10):5186. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105186
Chicago/Turabian StyleEgorova, Tatiana, Ekaterina Shuvalova, Sabina Mukba, Alexey Shuvalov, Peter Kolosov, and Elena Alkalaeva. 2021. "Method for Rapid Analysis of Mutant RNA Polymerase Activity on Templates Containing Unnatural Nucleotides" International Journal of Molecular Sciences 22, no. 10: 5186. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105186