
Neuroprotective Strategies in Aneurysmal Subarachnoid Hemorrhage (aSAH)

 and
and
Abstract
:1. Introduction
2. Role of Neuroinflammation in aSAH
2.1. Activation of the Immune System
2.2. Acute Events Following aSAH
2.3. Subacute/Chronic Events Following aSAH
2.4. Microglia
2.5. Endocannabinoids
2.6. Metalloproteinases
2.7. High Mobility Group Box 1
2.8. Autophagy and NF-κB-Mediated Inflammation
2.9. Meningeal Lymphatics
2.10. Novel Therapies Targeting Neuroinflammation
3. Role of Thromboinflammation in aSAH
3.1. Microthrombus Formation
3.2. The von Willebrand Factor (vWF) and ADAMTS-13
3.3. The Contact-Kinin System and Activated FXII
3.4. Platelet Receptors
3.5. Potential Neuroprotective Approaches
4. Role of Metabolism in aSAH
4.1. Metabolism with Regard to Early Brain Injury
4.2. Metabolism with Regard to Delayed Brain Injury
4.3. Potential Neuroprotective Approaches Targeting Metabolism
5. Role of Cerebral Vasospasm in aSAH
5.1. Pathophysiology of Cerebral Vasospasm
5.2. Potential Neuroprotective Approaches Targeting Cerebral Vasospasm and Hypoperfusion
5.2.1. Magnesium Sulfate
5.2.2. Hypercapnia
5.2.3. Hypothermia
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| aSAH | Aneurysmal subarachnoid hemorrhage |
| CVS | Cerebral vasospasm |
| DCI | Delayed cerebral ischemia |
| BBB | Blood-brainbarrier |
| CSF | Cerebrospinal fluid |
| TLR4 | Toll-like receptor 4 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| CNS | Central nervous system |
| IL | Interleukin |
| TNFα | Tumor necrosis factor alpha |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| eCBs | Endocannabinoids |
| AEA | Anandamide |
| 2-AG | 2-arachidonoyl-glycerol |
| ECS | Endocannabinoid system |
| CBRs | Cannabinoid receptors |
| CB1R | Cannabinoid type 1 receptor |
| CB2R | Cannabinoid type 2 receptor |
| MMPs | Metalloproteinases |
| HMGB1 | High mobility group box 1 |
| TBI | Traumatic brain injury |
| EBI | Early brain injury |
| DAMP | Damage-associated molecular pattern |
| RAGE | Receptor for advanced glycation end products |
| JAK2 | Janus-Kinase 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| COX-2 | Cyclooxygenase-2 |
| PGE-2 | Prostaglandin E2 |
| IL-1Ra | IL-1 receptor antagonist |
| vWF | von Willebrand Factor |
| ADAMTS-13 | A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 |
| tPA | Tissue plasminogen activator+ |
| GP | Platelet glycoprotein |
| ICH | Intracerebral hemorrhage |
| ICP | Intracranial pressure |
| CPP | Cerebral perfusion pressure |
| CBF | Cerebral blood flow |
| MABP | Mean artery blood pressure |
| ATP | Adenosine triphosphate |
| LPR | Lactate-to-pyruvate ratio |
| CMD | Cerebral microdialysis |
| CMRO2 | Cerebral metabolic rate for oxygen |
| PET | Positron emission tomography |
| ptiO2 | Tissue oxygenation |
| PDH | Pyruvate dehydrogenase |
| DCA | Dichloroacetate |
| ALCAR | Acetyl-L-carnitine |
| EGCG | Epigallocatechin-3-gallate |
| PaCO2 | Arterial partial pressure of carbon dioxide |
References
- Hackenberg, K.A.M.; Hanggi, D.; Etminan, N. Unruptured intracranial aneurysms. Stroke J. Cereb. Circ. 2018, 49, 2268–2275. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2014, 10, 44–58. [Google Scholar] [CrossRef]
- Schneider, U.C.; Xu, R.; Vajkoczy, P. Inflammatory events following subarachnoid hemorrhage (SAH). Curr. Neuropharmacol. 2018, 16, 1385–1395. [Google Scholar] [CrossRef]
- Vergouwen, M.D.; Vermeulen, M.; van Gijn, J.; Rinkel, G.J.; Wijdicks, E.F.; Muizelaar, J.P.; Mendelow, A.D.; Juvela, S.; Yonas, H.; Terbrugge, K.G.; et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: Proposal of a multidisciplinary research group. Stroke J. Cereb. Circ. 2010, 41, 2391–2395. [Google Scholar] [CrossRef] [Green Version]
- Jaja, B.N.R.; Saposnik, G.; Lingsma, H.F.; Macdonald, E.; Thorpe, K.E.; Mamdani, M.; Steyerberg, E.W.; Molyneux, A.; Manoel, A.L.O.; Schatlo, B.; et al. Development and validation of outcome prediction models for aneurysmal subarachnoid haemorrhage: The SAHIT multinational cohort study. BMJ 2018, 360, 5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraghty, J.R.; Davis, J.L.; Testai, F.D. Neuroinflammation and microvascular dysfunction after experimental subarachnoid hemorrhage: Emerging components of early brain injury related to outcome. Neurocritical. Care 2019, 31, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Sarrafzadeh, A.; Haux, D.; Sakowitz, O.; Benndorf, G.; Herzog, H.; Kuechler, I.; Unterberg, A. Acute focal neurological deficits in aneurysmal subarachnoid hemorrhage: Relation of clinical course, CT findings, and metabolite abnormalities monitored with bedside microdialysis. Stroke J. Cereb. Circ. 2003, 34, 1382–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermaier, T.; Jauss, A.; Eriskat, J.; Kunze, E.; Roosen, K. The temporal profile of cerebral blood flow and tissue metabolites indicates sustained metabolic depression after experimental subarachnoid hemorrhage in rats. Neurosurgery 2011, 68, 223–229, discussion 229–230. [Google Scholar] [CrossRef]
- Lilla, N.; Fullgraf, H.; Stetter, C.; Kohler, S.; Ernestus, R.I.; Westermaier, T. First description of reduced pyruvate dehydrogenase enzyme activity following subarachnoid hemorrhage (SAH). Front. Neurosci. 2017, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J. Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: A review. Acta Neurochir. Suppl. 2013, 115, 233–238. [Google Scholar]
- Sela, S.; Shurtz-Swirski, R.; Awad, J.; Shapiro, G.; Nasser, L.; Shasha, S.M.; Kristal, B. The involvement of peripheral polymorphonuclear leukocytes in the oxidative stress and inflammation among cigarette smokers. Isr. Med. Assoc. J. 2002, 4, 1015–1019. [Google Scholar]
- Zeyu, Z.; Yuanjian, F.; Cameron, L.; Sheng, C. The role of immune inflammation in aneurysmal subarachnoid hemorrhage. Exp. Neurol. 2021, 336, 113535. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J.; Fu, X.; Siu, A.; Rasmussen, P.A.; Hazen, S.L.; Ransohoff, R.M. CSF neutrophils are implicated in the development of vasospasm in subarachnoid hemorrhage. Neurocrit. Care 2010, 12, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucke-Wold, B.P.; Logsdon, A.F.; Manoranjan, B.; Turner, R.C.; McConnell, E.; Vates, G.E.; Huber, J.D.; Rosen, C.L.; Simard, J.M. Aneurysmal subarachnoid hemorrhage and neuroinflammation: A comprehensive review. Int. J. Mol. Sci. 2016, 17, 497. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.C.; Schiffler, J.; Hakiy, N.; Horn, P.; Vajkoczy, P. Functional analysis of Pro-inflammatory properties within the cerebrospinal fluid after subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflamm. 2012, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Broadwell, R.D. Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 1995, 2, 241–248. [Google Scholar] [CrossRef]
- Bulters, D.; Gaastra, B.; Zolnourian, A.; Alexander, S.; Ren, D.; Blackburn, S.L.; Borsody, M.; Dore, S.; Galea, J.; Iihara, K.; et al. Haemoglobin scavenging in intracranial bleeding: Biology and clinical implications. Nat. Rev. Neurol. 2018, 14, 416–432. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Marton, L.S.; Andrus, P.K.; Hall, E.D.; Johns, L.; Sajdak, M. Time course of production of hydroxyl free radical after subarachnoid hemorrhage in dogs. Life Sci. 2004, 75, 979–989. [Google Scholar] [CrossRef]
- Gallia, G.L.; Tamargo, R.J. Leukocyte-endothelial cell interactions in chronic vasospasm after subarachnoid hemorrhage. Neurol. Res. 2006, 28, 750–758. [Google Scholar] [CrossRef]
- Dietrich, H.H.; Dacey, R.G., Jr. Molecular keys to the problems of cerebral vasospasm. Neurosurgery 2000, 46, 517–530. [Google Scholar] [CrossRef]
- Klein, R.S.; Garber, C.; Howard, N. Infectious immunity in the central nervous system and brain function. Nat. Immunol. 2017, 18, 132–141. [Google Scholar] [CrossRef]
- Mathiesen, T.; Andersson, B.; Loftenius, A.; von Holst, H. Increased interleukin-6 levels in cerebrospinal fluid following subarachnoid hemorrhage. J. Neurosurg. 1993, 78, 562–567. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, T.; Okuda, Y.; Kaito, N.; Abe, T. Cytokine production in cerebrospinal fluid after subarachnoid haemorrhage. Neurol. Res. 1995, 17, 106–108. [Google Scholar] [CrossRef]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graça-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [Green Version]
- Crowley, R.W.; Medel, R.; Kassell, N.F.; Dumont, A.S. New insights into the causes and therapy of cerebral vasospasm following subarachnoid hemorrhage. Drug. Discov. Today 2008, 13, 254–260. [Google Scholar] [CrossRef]
- Pradilla, G.; Chaichana, K.L.; Hoang, S.; Huang, J.; Tamargo, R.J. Inflammation and cerebral vasospasm after subarachnoid hemorrhage. Neurosurg. Clin. N. Am. 2010, 21, 365–379. [Google Scholar] [CrossRef]
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Heinz, R.; Brandenburg, S.; Nieminen-Kelhä, M.; Kremenetskaia, I.; Boehm-Sturm, P.; Vajkoczy, P.; Schneider, U.C. Microglia as target for anti-inflammatory approaches to prevent secondary brain injury after subarachnoid hemorrhage (SAH). J. Neuroinflamm. 2021, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.C.; Davids, A.M.; Brandenburg, S.; Müller, A.; Elke, A.; Magrini, S.; Atangana, E.; Turkowski, K.; Finger, T.; Gutenberg, A.; et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol. 2015, 130, 215–231. [Google Scholar] [CrossRef]
- Patricio, F.; Morales-Andrade, A.A.; Patricio-Martinez, A.; Limon, I.D. Cannabidiol as a therapeutic target: Evidence of its neuroprotective and neuromodulatory function in parkinson’s disease. Front. Pharm. 2020, 11, 595635. [Google Scholar] [CrossRef] [PubMed]
- Benyo, Z.; Ruisanchez, E.; Leszl-Ishiguro, M.; Sandor, P.; Pacher, P. Endocannabinoids in cerebrovascular regulation. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H785–H801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S.S.; Hayes, J.P.; Fiore, N.T.; Moalem-Taylor, G. The cannabinoid system and microglia in health and disease. Neuropharmacology 2021, 190, 108555. [Google Scholar] [CrossRef]
- Zhang, X.; Ares, W.J.; Taussky, P.; Ducruet, A.F.; Grandhi, R. Role of matrix metalloproteinases in the pathogenesis of intracranial aneurysms. Neurosurg. Focus 2019, 47, E4. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, S.R.; Hafez, A.; Rezai Jahromi, B.; Kinfe, T.M.; Lamprecht, A.; Niemela, M.; Muhammad, S. Role of damage associated molecular pattern molecules (DAMPs) in aneurysmal subarachnoid hemorrhage (aSAH). Int. J. Mol. Sci. 2018, 19, 2035. [Google Scholar] [CrossRef] [Green Version]
- Balanca, B.; Desmurs, L.; Grelier, J.; Perret-Liaudet, A.; Lukaszewicz, A.C. DAMPs and RAGE pathophysiology at the acute phase of brain injury: An overview. Int. J. Mol. Sci. 2021, 22, 2439. [Google Scholar] [CrossRef]
- Aucott, H.; Lundberg, J.; Salo, H.; Klevenvall, L.; Damberg, P.; Ottosson, L.; Andersson, U.; Holmin, S.; Erlandsson Harris, H. Neuroinflammation in response to intracerebral injections of different HMGB1 redox isoforms. J. Innate Immun. 2018, 10, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.K.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Shaikh, M.F. HMGB1-Mediated neuroinflammatory responses in brain injuries: Potential mechanisms and therapeutic opportunities. Int. J. Mol. Sci. 2020, 21, 4609. [Google Scholar] [CrossRef]
- Sun, Q.; Wu, W.; Hu, Y.C.; Li, H.; Zhang, D.; Li, S.; Li, W.; Li, W.D.; Ma, B.; Zhu, J.H.; et al. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflamm. 2014, 11, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, J.Y.; Pang, H.G.; Huang, T.Q.; Song, J.N.; Li, D.D.; Zhao, Y.L.; Ma, X.D. AG490 ameliorates early brain injury via inhibition of JAK2/STAT3-mediated regulation of HMGB1 in subarachnoid hemorrhage. Exp. Med. 2018, 15, 1330–1338. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.J.; Xu, J.; Ma, B.Y.; Chen, G.; Gu, P.Y.; Wei, D.; Hu, W.X. Effect of glycyrrhizin on traumatic brain injury in rats and its mechanism. Chin. J. Traumatol. 2014, 17, 1–7. [Google Scholar]
- Chaudhry, S.R.; Guresir, A.; Stoffel-Wagner, B.; Fimmers, R.; Kinfe, T.M.; Dietrich, D.; Lamprecht, A.; Vatter, H.; Guresir, E.; Muhammad, S. Systemic high-mobility group box-1: A novel predictive biomarker for cerebral vasospasm in aneurysmal subarachnoid hemorrhage. Crit. Care Med. 2018, 46, e1023–e1028. [Google Scholar] [CrossRef]
- Pawlowska, E.; Szczepanska, J.; Wisniewski, K.; Tokarz, P.; Jaskolski, D.J.; Blasiak, J. NF-kappaB-mediated inflammation in the pathogenesis of intracranial aneurysm and subarachnoid hemorrhage. does autophagy play a role? Int. J. Mol. Sci. 2018, 19, 1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frosen, J.; Cebral, J.; Robertson, A.M.; Aoki, T. Flow-induced, inflammation-mediated arterial wall remodeling in the formation and progression of intracranial aneurysms. Neurosurg. Focus 2019, 47, E21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Aoki, T.; Frosen, J.; Fukuda, M.; Bando, K.; Shioi, G.; Tsuji, K.; Ollikainen, E.; Nozaki, K.; Laakkonen, J.; Narumiya, S. Prostaglandin E2-EP2-NF-kappaB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci. Signal. 2017, 10, eaah6037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cai, H.; Wang, Z.; Li, J.; Wang, K.; Yu, Z.; Chen, G. Induction of autophagy by cystatin C: A potential mechanism for prevention of cerebral vasospasm after experimental subarachnoid hemorrhage. Eur. J. Med. Res. 2013, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Li, J.; Chen, J.; Hu, Q.; Gu, C.; Lin, W.; Chen, G. Endoplasmic reticulum stress is associated with neuroprotection against apoptosis via autophagy activation in a rat model of subarachnoid hemorrhage. Neurosci. Lett. 2014, 563, 160–165. [Google Scholar] [CrossRef]
- Luo, C.; Yao, X.; Li, J.; He, B.; Liu, Q.; Ren, H.; Liang, F.; Li, M.; Lin, H.; Peng, J.; et al. Paravascular pathways contribute to vasculitis and neuroinflammation after subarachnoid hemorrhage independently of glymphatic control. Cell Death Dis. 2016, 7, e2160. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Kostrikov, S.; Mehta, R.I.; Nedergaard, M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin. Sci. 2017, 131, 2257–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Mesquita, S.; Louveau, A.; Vaccari, A.; Smirnov, I.; Cornelison, R.C.; Kingsmore, K.M.; Contarino, C.; Onengut-Gumuscu, S.; Farber, E.; Raper, D.; et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 2018, 560, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018, 21, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, L.; Xu, H.; Xing, L.; Zhuang, Z.; Zheng, Y.; Li, X.; Wang, C.; Chen, S.; Guo, Z.; et al. Meningeal lymphatics clear erythrocytes that arise from subarachnoid hemorrhage. Nat. Commun. 2020, 11, 3159. [Google Scholar] [CrossRef] [PubMed]
- Vadokas, G.; Koehler, S.; Weiland, J.; Lilla, N.; Stetter, C.; Westermaier, T. Early antiinflammatory therapy attenuates brain damage after sah in rats. Transl. Neurosci. 2019, 10, 104–111. [Google Scholar] [CrossRef]
- Gomis, P.; Graftieaux, J.P.; Sercombe, R.; Hettler, D.; Scherpereel, B.; Rousseaux, P. Randomized, double-blind, placebo-controlled, pilot trial of high-dose methylprednisolone in aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2010, 112, 681–688. [Google Scholar] [CrossRef]
- Feigin, V.L.; Anderson, N.; Rinkel, G.J.; Algra, A.; van Gijn, J.; Bennett, D.A. Corticosteroids for aneurysmal subarachnoid haemorrhage and primary intracerebral haemorrhage. Cochrane Database Syst. Rev. 2005, 3, Cd004583. [Google Scholar] [CrossRef]
- Belayev, L.; Saul, I.; Huh, P.W.; Finotti, N.; Zhao, W.; Busto, R.; Ginsberg, M.D. Neuroprotective effect of high-dose albumin therapy against global ischemic brain injury in rats. Brain Res. 1999, 845, 107–111. [Google Scholar] [CrossRef]
- Liu, Y.; Belayev, L.; Zhao, W.; Busto, R.; Belayev, A.; Ginsberg, M.D. Neuroprotective effect of treatment with human albumin in permanent focal cerebral ischemia: Histopathology and cortical perfusion studies. Eur. J. Pharm. 2001, 428, 193–201. [Google Scholar] [CrossRef]
- Suarez, J.I.; Martin, R.H.; Calvillo, E.; Dillon, C.; Bershad, E.M.; Macdonald, R.L.; Wong, J.; Harbaugh, R. The Albumin in Subarachnoid Hemorrhage (ALISAH) multicenter pilot clinical trial: Safety and neurologic outcomes. Stroke 2012, 43, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Hopkins, S.J.; Hulme, S.; Galea, J.P.; Hoadley, M.; Vail, A.; Hutchinson, P.J.; Grainger, S.; Rothwell, N.J.; King, A.T.; et al. The effect of intravenous interleukin-1 receptor antagonist on inflammatory mediators in cerebrospinal fluid after subarachnoid haemorrhage: A phase II randomised controlled trial. J. Neuroinflamm. 2014, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galea, J.; Ogungbenro, K.; Hulme, S.; Patel, H.; Scarth, S.; Hoadley, M.; Illingworth, K.; McMahon, C.J.; Tzerakis, N.; King, A.T.; et al. Reduction of inflammation after administration of interleukin-1 receptor antagonist following aneurysmal subarachnoid hemorrhage: Results of the Subcutaneous Interleukin-1Ra in SAH (SCIL-SAH) study. J. Neurosurg. 2018, 128, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, S.R.; Kahlert, U.D.; Kinfe, T.M.; Lamprecht, A.; Niemela, M.; Hanggi, D.; Muhammad, S. Elevated systemic IL-10 levels indicate immunodepression leading to nosocomial infections after aneurysmal subarachnoid hemorrhage (SAH) in patients. Int. J. Mol. Sci. 2020, 21, 1569. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, S.R.; Kinfe, T.M.; Lamprecht, A.; Niemela, M.; Dobreva, G.; Hanggi, D.; Muhammad, S. Elevated level of cerebrospinal fluid and systemic chemokine CCL5 is a predictive biomarker of clinical outcome after aneurysmal subarachnoid hemorrhage (aSAH). Cytokine 2020, 133, 155142. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Sherchan, P.; Krafft, P.R.; Rolland, W.B.; Soejima, Y.; Zhang, J.H. Cannabinoid type 2 receptor stimulation attenuates brain edema by reducing cerebral leukocyte infiltration following subarachnoid hemorrhage in rats. J. Neurol. Sci. 2014, 342, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Sherchan, P.; Soejima, Y.; Hasegawa, Y.; Flores, J.; Doycheva, D.; Zhang, J.H. Cannabinoid receptor type 2 agonist attenuates apoptosis by activation of phosphorylated CREB-Bcl-2 pathway after subarachnoid hemorrhage in rats. Exp. Neurol. 2014, 261, 396–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, M.; Sherchan, P.; Soejima, Y.; Doycheva, D.; Zhao, D.; Zhang, J.H. Cannabinoid receptor Type 2 agonist attenuates acute neurogenic pulmonary edema by preventing neutrophil migration after subarachnoid hemorrhage in rats. Acta Neurochir. Suppl. 2016, 121, 135–139. [Google Scholar]
- Behrouz, R.; Birnbaum, L.; Grandhi, R.; Johnson, J.; Misra, V.; Palacio, S.; Seifi, A.; Topel, C.; Garvin, R.; Caron, J.L. Cannabis use and outcomes in patients with aneurysmal subarachnoid hemorrhage. Stroke J. Cereb. Circ. 2016, 47, 1371–1373. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira Manoel, A.L.; Macdonald, R.L. Neuroinflammation as a target for intervention in subarachnoid hemorrhage. Front. Neurol. 2018, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.; Li, C.; Hu, Q.; Wang, Y.; Sun, J.; Gao, F.; Yang, M.; Sun, B.; Mao, L. Low-Dose IL-2 treatment affords protection against subarachnoid hemorrhage injury by expanding peripheral regulatory T cells. ACS Chem. Neurosci. 2021, 12, 430–440. [Google Scholar] [CrossRef]
- Han, M.; Cao, Y.; Guo, X.; Chu, X.; Li, T.; Xue, H.; Xin, D.; Yuan, L.; Ke, H.; Li, G.; et al. Mesenchymal stem cell-derived extracellular vesicles promote microglial M2 polarization after subarachnoid hemorrhage in rats and involve the AMPK/NF-kappaB signaling pathway. Biomed. Pharm. 2021, 133, 111048. [Google Scholar] [CrossRef]
- Li, P.; Li, X.; Deng, P.; Wang, D.; Bai, X.; Li, Y.; Luo, C.; Belguise, K.; Wang, X.; Wei, X.; et al. Activation of adenosine A3 receptor reduces early brain injury by alleviating neuroinflammation after subarachnoid hemorrhage in elderly rats. Aging (Albany Ny) 2020, 13, 694–713. [Google Scholar] [CrossRef]
- Gao, Y.Y.; Tao, T.; Wu, D.; Zhuang, Z.; Lu, Y.; Wu, L.Y.; Liu, G.J.; Zhou, Y.; Zhang, D.D.; Wang, H.; et al. MFG-E8 attenuates inflammation in subarachnoid hemorrhage by driving microglial M2 polarization. Exp. Neurol. 2021, 336, 113532. [Google Scholar] [CrossRef]
- Kremer, B.; Coburn, M.; Weinandy, A.; Nolte, K.; Clusmann, H.; Veldeman, M.; Hollig, A. Argon treatment after experimental subarachnoid hemorrhage: Evaluation of microglial activation and neuronal survival as a subanalysis of a randomized controlled animal trial. Med. Gas. Res. 2020, 10, 103–109. [Google Scholar] [CrossRef]
- Luo, Y.; Fang, Y.; Kang, R.; Lenahan, C.; Gamdzyk, M.; Zhang, Z.; Okada, T.; Tang, J.; Chen, S.; Zhang, J.H. Inhibition of EZH2 (Enhancer of Zeste Homolog 2) Attenuates Neuroinflammation via H3k27me3/SOCS3/TRAF6/NF-kappaB (Trimethylation of Histone 3 Lysine 27/Suppressor of Cytokine Signaling 3/Tumor Necrosis Factor Receptor Family 6/Nuclear Factor-kappaB) in a Rat Model of Subarachnoid Hemorrhage. Stroke 2020, 51, 3320–3331. [Google Scholar]
- Chen, T.; Zhu, J.; Wang, Y.H. RNF216 mediates neuronal injury following experimental subarachnoid hemorrhage through the Arc/Arg3.1-AMPAR pathway. Faseb J. 2020, 34, 15080–15092. [Google Scholar] [CrossRef]
- Xu, P.; Hong, Y.; Xie, Y.; Yuan, K.; Li, J.; Sun, R.; Zhang, X.; Shi, X.; Li, R.; Wu, J.; et al. TREM-1 Exacerbates Neuroinflammatory Injury via NLRP3 Inflammasome-Mediated Pyroptosis in Experimental Subarachnoid Hemorrhage. Transl. Stroke Res. 2020. [Google Scholar] [CrossRef]
- Liu, G.J.; Tao, T.; Wang, H.; Zhou, Y.; Gao, X.; Gao, Y.Y.; Hang, C.H.; Li, W. Functions of resolvin D1-ALX/FPR2 receptor interaction in the hemoglobin-induced microglial inflammatory response and neuronal injury. J. Neuroinflamm. 2020, 17, 239. [Google Scholar] [CrossRef]
- Duan, H.; Li, L.; Shen, S.; Ma, Y.; Yin, X.; Liu, Z.; Yuan, C.; Wang, Y.; Zhang, J. Hydrogen Sulfide Reduces Cognitive Impairment in Rats After Subarachnoid Hemorrhage by Ameliorating Neuroinflammation Mediated by the TLR4/NF-kappaB Pathway in Microglia. Front. Cell Neurosci. 2020, 14, 210. [Google Scholar] [CrossRef]
- Liao, L.S.; Zhang, M.W.; Gu, Y.J.; Sun, X.C. Targeting CCL20 inhibits subarachnoid hemorrhage-related neuroinflammation in mice. Aging (Albany Ny) 2020, 12, 14849–14862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, X.; Peng, J.; Xie, Y.; Du, F.; Guo, K.; Feng, Y.; Zhang, L.; Chen, L.; Jiang, Y. TSPO ligand Ro5–4864 modulates microglia/macrophages polarization after subarachnoid hemorrhage in mice. Neurosci. Lett. 2020, 729, 134977. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Wu, X.; Feng, D.; Zhang, Y.; Chen, X.; Ma, C.; Shen, H.; Li, X.; Li, H.; Zhang, J.; et al. Cerebral cavernous malformation 3 relieves subarachnoid hemorrhage-induced neuroinflammation in rats through inhibiting NF-kB signaling pathway. Brain Res. Bull. 2020, 160, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.; Wu, D.; Liang, T.; Pan, P.; Yuan, G.; Li, X.; Li, H.; Shen, H.; Wang, Z.; Chen, G. Systemic exosomal miR-193b-3p delivery attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage in mice. J. Neuroinflamm. 2020, 17, 74. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhuang, Z.; Lu, Y.; Tao, T.; Zhou, Y.; Liu, G.; Wang, H.; Zhang, D.; Wu, L.; Dai, H.; et al. Curcumin Mitigates Neuro-Inflammation by Modulating Microglia Polarization Through Inhibiting TLR4 Axis Signaling Pathway Following Experimental Subarachnoid Hemorrhage. Front. Neurosci. 2019, 13, 1223. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Liu, G.J.; Shi, X.; Zhou, Y.; Lu, Y.; Gao, Y.Y.; Zhang, X.S.; Wang, H.; Wu, L.Y.; Chen, C.L.; et al. DHEA Attenuates Microglial Activation via Induction of JMJD3 in Experimental Subarachnoid Haemorrhage. J. Neuroinflamm. 2019, 16, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Li, T.; Gao, L.; Zheng, J.; Yan, J.; Zhang, J.; Shao, A. Apelin-13/APJ system attenuates early brain injury via suppression of endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation and oxidative stress in a AMPK-dependent manner after subarachnoid hemorrhage in rats. J. Neuroinflamm. 2019, 16, 247. [Google Scholar] [CrossRef]
- Xu, W.; Mo, J.; Ocak, U.; Travis, Z.D.; Enkhjargal, B.; Zhang, T.; Wu, P.; Peng, J.; Li, T.; Zuo, Y.; et al. Activation of Melanocortin 1 Receptor Attenuates Early Brain Injury in a Rat Model of Subarachnoid Hemorrhage viathe Suppression of Neuroinflammation through AMPK/TBK1/NF-kappaB Pathway in Rats. Neurotherapeutics 2020, 17, 294–308. [Google Scholar] [CrossRef]
- Okada, T.; Lei, L.; Nishikawa, H.; Nakano, F.; Nakatsuka, Y.; Suzuki, H. TAK-242, Toll-Like Receptor 4 Antagonist, Attenuates Brain Edema in Subarachnoid Hemorrhage Mice. Acta Neurochir. Suppl. 2020, 127, 77–81. [Google Scholar]
- Zuo, Y.; Huang, L.; Enkhjargal, B.; Xu, W.; Umut, O.; Travis, Z.D.; Zhang, G.; Tang, J.; Liu, F.; Zhang, J.H. Activation of retinoid X receptor by bexarotene attenuates neuroinflammation via PPARgamma/SIRT6/FoxO3a pathway after subarachnoid hemorrhage in rats. J. Neuroinflamm. 2019, 16, 47. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Venkat, P.; Chopp, M.; Zhang, Q.; Yan, T.; Chen, J. RP001 hydrochloride improves neurological outcome after subarachnoid hemorrhage. J. Neurol. Sci. 2019, 399, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, R.; Yin, J.; Guo, S.; Chen, Y.; Fan, H.; Li, G.; Li, Z.; Li, X.; Zhang, X.; et al. Mesenchymal stem cells alleviate the early brain injury of subarachnoid hemorrhage partly by suppression of Notch1-dependent neuroinflammation: Involvement of Botch. J. Neuroinflamm. 2019, 16, 8. [Google Scholar] [CrossRef]
- Liu, F.Y.; Cai, J.; Wang, C.; Ruan, W.; Guan, G.P.; Pan, H.Z.; Li, J.R.; Qian, C.; Chen, J.S.; Wang, L.; et al. Fluoxetine attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage: A possible role for the regulation of TLR4/MyD88/NF-kappaB signaling pathway. J. Neuroinflamm. 2018, 15, 347. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Peng, J.; Matei, N.; Yang, P.; Kuai, L.; Wu, Y.; Chen, L.; Vitek, M.P.; Li, F.; Sun, X.; et al. Apolipoprotein E Exerts a Whole-Brain Protective Property by Promoting M1? Microglia Quiescence After Experimental Subarachnoid Hemorrhage in Mice. Transl Stroke Res. 2018, 9, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, W.; Yin, J.; Chen, Y.; Guo, S.; Fan, H.; Li, X.; Zhang, X.; He, X.; Duan, C. TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J. Neuroinflamm. 2018, 15, 231. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, W.H.; Xu, L.B.; Shao, M.F.; Chen, Z.P.; Zhu, G.C.; Hou, Q. Resident Microglia Activate before Peripheral Monocyte Infiltration and p75NTR Blockade Reduces Microglial Activation and Early Brain Injury after Subarachnoid Hemorrhage. ACS Chem. Neurosci. 2019, 10, 412–423. [Google Scholar] [CrossRef]
- Qu, J.; Zhao, H.; Li, Q.; Pan, P.; Ma, K.; Liu, X.; Feng, H.; Chen, Y. MST1 Suppression Reduces Early Brain Injury by Inhibiting the NF-kappaB/MMP-9 Pathway after Subarachnoid Hemorrhage in Mice. Behav. Neurol. 2018, 2018, 6470957. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Enkhjargal, B.; Huang, L.; Zhang, T.; Sun, C.; Xie, Z.; Wu, P.; Mo, J.; Tang, J.; Xie, Z.; et al. Aggf1 attenuates neuroinflammation and BBB disruption via PI3K/Akt/NF-kappaB pathway after subarachnoid hemorrhage in rats. J. Neuroinflamm. 2018, 15, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Kawakita, F.; Nishikawa, H.; Nakano, F.; Liu, L.; Suzuki, H. Selective Toll-Like Receptor 4 Antagonists Prevent Acute Blood-Brain Barrier Disruption After Subarachnoid Hemorrhage in Mice. Mol. Neurobiol. 2019, 56, 976–985. [Google Scholar] [CrossRef]
- Tu, L.; Yang, X.L.; Zhang, Q.; Wang, Q.; Tian, T.; Liu, D.; Qu, X.; Tian, J.Y. Bexarotene attenuates early brain injury via inhibiting micoglia activation through PPARgamma after experimental subarachnoid hemorrhage. Neurol. Res. 2018, 40, 702–708. [Google Scholar]
- Yin, J.; Li, R.; Liu, W.; Chen, Y.; Zhang, X.; Li, X.; He, X.; Duan, C. Neuroprotective Effect of Protein Phosphatase 2A/Tristetraprolin Following Subarachnoid Hemorrhage in Rats. Front. Neurosci. 2018, 12, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Jin, J.; Fan, L.; Xu, H.; He, P.; Li, J.; Chen, T.; Ruan, W.; Chen, G. Rolipram Attenuates Early Brain Injury Following Experimental Subarachnoid Hemorrhage in Rats: Possibly via Regulating the SIRT1/NF-kappaB Pathway. Neurochem. Res. 2018, 43, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Huang, L.; Enkhjargal, B.; Reis, C.; Wan, W.; Tang, J.; Cheng, Y.; Zhang, J.H. Recombinant Netrin-1 binding UNC5B receptor attenuates neuroinflammation and brain injury via PPARgamma/NFkappaB signaling pathway after subarachnoid hemorrhage in rats. Brain Behav. Immun. 2018, 69, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Li, J.R.; Xu, H.Z.; Nie, S.; Peng, Y.C.; Fan, L.F.; Wang, Z.J.; Wu, C.; Yan, F.; Chen, J.Y.; Gu, C.; et al. Fluoxetine-enhanced autophagy ameliorates early brain injury via inhibition of NLRP3 inflammasome activation following subrachnoid hemorrhage in rats. J. Neuroinflamm. 2017, 14, 186. [Google Scholar] [CrossRef]
- Xu, H.; Li, J.; Wang, Z.; Feng, M.; Shen, Y.; Cao, S.; Li, T.; Peng, Y.; Fan, L.; Chen, J.; et al. Methylene blue attenuates neuroinflammation after subarachnoid hemorrhage in rats through the Akt/GSK-3beta/MEF2D signaling pathway. Brain Behav. Immun. 2017, 65, 125–139. [Google Scholar] [CrossRef]
- Xu, J.; Xu, Z.; Yan, A. Prostaglandin E2 EP4 Receptor Activation Attenuates Neuroinflammation and Early Brain Injury Induced by Subarachnoid Hemorrhage in Rats. Neurochem. Res. 2017, 42, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Hao, G.; Dong, Y.; Huo, R.; Wen, K.; Zhang, Y.; Liang, G. Rutin Inhibits Neuroinflammation and Provides Neuroprotection in an Experimental Rat Model of Subarachnoid Hemorrhage, Possibly Through Suppressing the RAGE-NF-kappaB Inflammatory Signaling Pathway. Neurochem. Res. 2016, 41, 1496–1504. [Google Scholar] [CrossRef]
- Guo, Z.; Hu, Q.; Xu, L.; Guo, Z.N.; Ou, Y.; He, Y.; Yin, C.; Sun, X.; Tang, J.; Zhang, J.H. Lipoxin A4 Reduces Inflammation Through Formyl Peptide Receptor 2/p38 MAPK Signaling Pathway in Subarachnoid Hemorrhage Rats. Stroke 2016, 47, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Vergouwen, M.D.; Vermeulen, M.; Coert, B.A.; Stroes, E.S.; Roos, Y.B. Microthrombosis after aneurysmal subarachnoid hemorrhage: An additional explanation for delayed cerebral ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 1761–1770. [Google Scholar] [CrossRef]
- Sehba, F.A.; Hou, J.; Pluta, R.M.; Zhang, J.H. The importance of early brain injury after subarachnoid hemorrhage. Prog. Neurobiol. 2012, 97, 14–37. [Google Scholar]
- McBride, D.W.; Blackburn, S.L.; Peeyush, K.T.; Matsumura, K.; Zhang, J.H. The Role of Thromboinflammation in Delayed Cerebral Ischemia after Subarachnoid Hemorrhage. Front. Neurol. 2017, 8, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, A.; Ammassam Veettil, R.; Hong, S.H.; Matsumura, K.; Kumar, T.P.; Yan, Y.; Blackburn, S.L.; Ballester, L.Y.; Marrelli, S.P.; McCullough, L.D.; et al. Microthrombi Correlates With Infarction and Delayed Neurological Deficits After Subarachnoid Hemorrhage in Mice. Stroke 2020, 51, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Frontera, J.A.; Aledort, L.; Gordon, E.; Egorova, N.; Moyle, H.; Patel, A.; Bederson, J.B.; Sehba, F. Early platelet activation, inflammation and acute brain injury after a subarachnoid hemorrhage: A pilot study. J. Thromb. Haemost. 2012, 10, 711–713. [Google Scholar] [CrossRef]
- Friedrich, B.; Muller, F.; Feiler, S.; Scholler, K.; Plesnila, N. Experimental subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: An in-vivo microscopy study. J. Cereb. Blood Flow Metab. 2012, 32, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siler, D.A.; Gonzalez, J.A.; Wang, R.K.; Cetas, J.S.; Alkayed, N.J. Intracisternal administration of tissue plasminogen activator improves cerebrospinal fluid flow and cortical perfusion after subarachnoid hemorrhage in mice. Transl. Stroke Res. 2014, 5, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabri, M.; Ai, J.; Lakovic, K.; D’Abbondanza, J.; Ilodigwe, D.; Macdonald, R.L. Mechanisms of microthrombi formation after experimental subarachnoid hemorrhage. Neuroscience 2012, 224, 26–37. [Google Scholar] [CrossRef]
- Andereggen, L.; Neuschmelting, V.; von Gunten, M.; Widmer, H.R.; Fandino, J.; Marbacher, S. The role of microclot formation in an acute subarachnoid hemorrhage model in the rabbit. Biomed. Res. Int. 2014, 2014, 161702. [Google Scholar] [CrossRef]
- Stein, S.C.; Browne, K.D.; Chen, X.H.; Smith, D.H.; Graham, D.I. Thromboembolism and delayed cerebral ischemia after subarachnoid hemorrhage: An autopsy study. Neurosurgery 2006, 59, 781–787, discussion 787–8. [Google Scholar] [CrossRef]
- Boluijt, J.; Meijers, J.C.; Rinkel, G.J.; Vergouwen, M.D. Hemostasis and fibrinolysis in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage: A systematic review. J. Cereb. Blood Flow Metab. 2015, 35, 724–733. [Google Scholar] [CrossRef]
- Chauhan, A.K.; Kisucka, J.; Brill, A.; Walsh, M.T.; Scheiflinger, F.; Wagner, D.D. ADAMTS13: A new link between thrombosis and inflammation. J. Exp. Med. 2008, 205, 2065–2074. [Google Scholar] [CrossRef]
- Stoll, G.; Nieswandt, B. Thrombo-inflammation in acute ischaemic stroke-implications for treatment. Nat. Rev. Neurol. 2019, 15, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Muroi, C.; Fujioka, M.; Mishima, K.; Irie, K.; Fujimura, Y.; Nakano, T.; Fandino, J.; Keller, E.; Iwasaki, K.; Fujiwara, M. Effect of ADAMTS-13 on cerebrovascular microthrombosis and neuronal injury after experimental subarachnoid hemorrhage. J. Thromb. Haemost. 2014, 12, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergouwen, M.D.; Knaup, V.L.; Roelofs, J.J.; de Boer, O.J.; Meijers, J.C. Effect of recombinant ADAMTS-13 on microthrombosis and brain injury after experimental subarachnoid hemorrhage. J. Thromb. Haemost. 2014, 12, 943–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, H.; Wang, Y.; Ai, J.; Brathwaite, S.; Ni, H.; Macdonald, R.L.; Hol, E.M.; Meijers, J.C.M.; Vergouwen, M.D.I. Role of von Willebrand factor and ADAMTS-13 in early brain injury after experimental subarachnoid hemorrhage. J. Thromb. Haemost. 2018, 16, 1413–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergouwen, M.D.; Bakhtiari, K.; van Geloven, N.; Vermeulen, M.; Roos, Y.B.; Meijers, J.C. Reduced ADAMTS13 activity in delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2009, 29, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Putzer, A.S.; Worthmann, H.; Grosse, G.M.; Goetz, F.; Martens-Lobenhoffer, J.; Dirks, M.; Kielstein, J.T.; Lichtinghagen, R.; Budde, U.; Bode-Boger, S.M.; et al. ADAMTS13 activity is associated with early neurological improvement in acute ischemic stroke patients treated with intravenous thrombolysis. J. Thromb. Thrombolysis 2020, 49, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.H.; Macdonald, R.L.; Keller, E.; Unruptured Intracranial, A.; Investigators, S.C.P. Biospecimens and Molecular and Cellular Biomarkers in Aneurysmal Subarachnoid Hemorrhage Studies: Common Data Elements and Standard Reporting Recommendations. Neurocrit Care 2019, 30 (Suppl. 1), 46–59. [Google Scholar] [CrossRef]
- Jabbarli, R.; Pierscianek, D.; Darkwah Oppong, M.; Sato, T.; Dammann, P.; Wrede, K.H.; Kaier, K.; Kohrmann, M.; Forsting, M.; Kleinschnitz, C.; et al. Laboratory biomarkers of delayed cerebral ischemia after subarachnoid hemorrhage: A systematic review. Neurosurg Rev. 2020, 43, 825–833. [Google Scholar] [CrossRef]
- Albert-Weissenberger, C.; Siren, A.L.; Kleinschnitz, C. Ischemic stroke and traumatic brain injury: The role of the kallikrein-kinin system. Prog. Neurobiol. 2013, 101–102, 65–82. [Google Scholar] [CrossRef]
- De Meyer, S.F.; Denorme, F.; Langhauser, F.; Geuss, E.; Fluri, F.; Kleinschnitz, C. Thromboinflammation in Stroke Brain Damage. Stroke 2016, 47, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Kleinschnitz, C.; Stoll, G.; Bendszus, M.; Schuh, K.; Pauer, H.U.; Burfeind, P.; Renne, C.; Gailani, D.; Nieswandt, B.; Renne, T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J. Exp. Med. 2006, 203, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagedorn, I.; Schmidbauer, S.; Pleines, I.; Kleinschnitz, C.; Kronthaler, U.; Stoll, G.; Dickneite, G.; Nieswandt, B. Factor XIIa inhibitor recombinant human albumin Infestin-4 abolishes occlusive arterial thrombus formation without affecting bleeding. Circulation 2010, 121, 1510–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heydenreich, N.; Nolte, M.W.; Gob, E.; Langhauser, F.; Hofmeister, M.; Kraft, P.; Albert-Weissenberger, C.; Brede, M.; Varallyay, C.; Gobel, K.; et al. C1-inhibitor protects from brain ischemia-reperfusion injury by combined antiinflammatory and antithrombotic mechanisms. Stroke 2012, 43, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Albert-Weissenberger, C.; Mencl, S.; Schuhmann, M.K.; Salur, I.; Gob, E.; Langhauser, F.; Hopp, S.; Hennig, N.; Meuth, S.G.; Nolte, M.W.; et al. C1-Inhibitor protects from focal brain trauma in a cortical cryolesion mice model by reducing thrombo-inflammation. Front. Cell Neurosci. 2014, 8, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopp, S.; Albert-Weissenberger, C.; Mencl, S.; Bieber, M.; Schuhmann, M.K.; Stetter, C.; Nieswandt, B.; Schmidt, P.M.; Monoranu, C.M.; Alafuzoff, I.; et al. Targeting coagulation factor XII as a novel therapeutic option in brain trauma. Ann. Neurol. 2016, 79, 970–982. [Google Scholar] [CrossRef] [Green Version]
- Hopp, S.; Nolte, M.W.; Stetter, C.; Kleinschnitz, C.; Siren, A.L.; Albert-Weissenberger, C. Alleviation of secondary brain injury, posttraumatic inflammation, and brain edema formation by inhibition of factor XIIa. J. Neuroinflamm. 2017, 14, 39. [Google Scholar] [CrossRef] [Green Version]
- Gobel, K.; Pankratz, S.; Asaridou, C.M.; Herrmann, A.M.; Bittner, S.; Merker, M.; Ruck, T.; Glumm, S.; Langhauser, F.; Kraft, P.; et al. Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells. Nat. Commun. 2016, 7, 11626. [Google Scholar] [CrossRef] [Green Version]
- Revenko, A.S.; Gao, D.; Crosby, J.R.; Bhattacharjee, G.; Zhao, C.; May, C.; Gailani, D.; Monia, B.P.; MacLeod, A.R. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood 2011, 118, 5302–5311. [Google Scholar] [CrossRef] [Green Version]
- Simao, F.; Ustunkaya, T.; Clermont, A.C.; Feener, E.P. Plasma kallikrein mediates brain hemorrhage and edema caused by tissue plasminogen activator therapy in mice after stroke. Blood 2017, 129, 2280–2290. [Google Scholar] [CrossRef] [PubMed]
- Albert-Weissenberger, C.; Hopp, S.; Nieswandt, B.; Siren, A.L.; Kleinschnitz, C.; Stetter, C. How is the formation of microthrombi after traumatic brain injury linked to inflammation? J. Neuroimmunol. 2019, 326, 9–13. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Pozgajova, M.; Pham, M.; Bendszus, M.; Nieswandt, B.; Stoll, G. Targeting platelets in acute experimental stroke: Impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation 2007, 115, 2323–2330. [Google Scholar] [CrossRef] [Green Version]
- Kraft, P.; Schuhmann, M.K.; Fluri, F.; Lorenz, K.; Zernecke, A.; Stoll, G.; Nieswandt, B.; Kleinschnitz, C. Efficacy and Safety of Platelet Glycoprotein Receptor Blockade in Aged and Comorbid Mice With Acute Experimental Stroke. Stroke 2015, 46, 3502–3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuhmann, M.K.; Guthmann, J.; Stoll, G.; Nieswandt, B.; Kraft, P.; Kleinschnitz, C. Blocking of platelet glycoprotein receptor Ib reduces “thrombo-inflammation” in mice with acute ischemic stroke. J. Neuroinflamm. 2017, 14, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuhmann, M.K.; Stoll, G.; Bieber, M.; Vogtle, T.; Hofmann, S.; Klaus, V.; Kraft, P.; Seyhan, M.; Kollikowski, A.M.; Papp, L.; et al. CD84 Links T Cell and Platelet Activity in Cerebral Thrombo-Inflammation in Acute Stroke. Circ. Res. 2020, 127, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Iwabuchi, T.; Tanaka, T.; Kanayama, S.; Ottomo, M.; Hatanaka, M.; Aihara, H. Prevention of cerebral vasospasm with OKY-046 an imidazole derivative and a thromboxane synthetase inhibitor. A preliminary co-operative clinical study. Acta Neurochir. (Wien.) 1985, 77, 133–141. [Google Scholar] [CrossRef]
- Tokiyoshi, K.; Ohnishi, T.; Nii, Y. Efficacy and toxicity of thromboxane synthetase inhibitor for cerebral vasospasm after subarachnoid hemorrhage. Surg. Neurol. 1991, 36, 112–118. [Google Scholar] [CrossRef]
- Hirashima, Y.; Endo, S.; Kato, R.; Takaku, A. Prevention of cerebrovasospasm following subarachnoid hemorrhage in rabbits by the platelet-activating factor antagonist, E5880. J. Neurosurg. 1996, 84, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Hirashima, Y.; Endo, S.; Nukui, H.; Kobayashi, N.; Takaku, A. Effect of a platelet-activating factor receptor antagonist, E5880, on cerebral vasospasm after aneurysmal subarachnoid hemorrhage--open clinical trial to investigate efficacy and safety. Neurol. Med. Chir. (Tokyo) 2001, 41, 165–175, discussion 175–6. [Google Scholar] [CrossRef] [Green Version]
- Lagier, D.; Tonon, D.; Garrigue, P.; Guillet, B.; Giacomino, L.; Martin, J.C.; Alessi, M.C.; Bruder, N.; Velly, L.J. Thromboxane-prostaglandin receptor antagonist, terutroban, prevents neurovascular events after subarachnoid haemorrhage: A nanoSPECT study in rats. Crit Care 2019, 23, 42. [Google Scholar] [CrossRef]
- Shi, L.; Xu, L.; Shi, L.; Brandon, D.; Chen, S.; Zhang, J. Intraventricular Recombinant Tissue Plasminogen Activator in Treatment of Aneurysmal Intraventricular Hemorrhage: A Meta-Analysis. Curr. Drug Targets 2017, 18, 1399–1407. [Google Scholar] [CrossRef]
- Vergouwen, M.D.; Meijers, J.C.; Geskus, R.B.; Coert, B.A.; Horn, J.; Stroes, E.S.; van der Poll, T.; Vermeulen, M.; Roos, Y.B. Biologic effects of simvastatin in patients with aneurysmal subarachnoid hemorrhage: A double-blind, placebo-controlled randomized trial. J. Cereb. Blood Flow Metab. 2009, 29, 1444–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhou, D.; Guo, J.; Ren, Z.; Zhou, L.; Wang, S.; Zhang, Y.; Xu, B.; Zhao, K.; Wang, R.; et al. Fasudil Aneurysmal Subarachnoid Hemorrhage Study, G. Efficacy and safety of fasudil in patients with subarachnoid hemorrhage: Final results of a randomized trial of fasudil versus nimodipine. Neurol. Med. Chir. (Tokyo) 2011, 51, 679–683. [Google Scholar] [CrossRef] [Green Version]
- James, R.F.; Kramer, D.R.; Aljuboori, Z.S.; Parikh, G.; Adams, S.W.; Eaton, J.C.; Abou Al-Shaar, H.; Badjatia, N.; Mack, W.J.; Simard, J.M. Novel Treatments in Neuroprotection for Aneurysmal Subarachnoid Hemorrhage. Curr. Treat. Options Neurol. 2016, 18, 38. [Google Scholar] [CrossRef]
- Li, L.; Lou, X.; Zhang, K.; Yu, F.; Zhao, Y.; Jiang, P. Hydrochloride fasudil attenuates brain injury in ICH rats. Transl. Neurosci. 2020, 11, 75–86. [Google Scholar] [CrossRef]
- Vagnozzi, R.; Signoretti, S.; Cristofori, L.; Alessandrini, F.; Floris, R.; Isgro, E.; Ria, A.; Marziali, S.; Zoccatelli, G.; Tavazzi, B.; et al. Assessment of metabolic brain damage and recovery following mild traumatic brain injury: A multicentre, proton magnetic resonance spectroscopic study in concussed patients. Brain 2010, 133, 3232–3242. [Google Scholar] [CrossRef]
- Bederson, J.B.; Germano, I.M.; Guarino, L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke 1995, 26, 1086–1091. [Google Scholar] [CrossRef]
- Oddo, M.; Levine, J.M.; Frangos, S.; Maloney-Wilensky, E.; Carrera, E.; Daniel, R.T.; Levivier, M.; Magistretti, P.J.; LeRoux, P.D. Brain lactate metabolism in humans with subarachnoid hemorrhage. Stroke 2012, 43, 1418–1421. [Google Scholar] [CrossRef] [Green Version]
- Sehba, F.A.; Bederson, J.B. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol. Res. 2006, 28, 381–398. [Google Scholar] [CrossRef]
- Sarrafzadeh, A.S.; Sakowitz, O.W.; Lanksch, W.R.; Unterberg, A.W. Time course of various interstitial metabolites following subarachnoid hemorrhage studied by on-line microdialysis. Acta Neurochir. Suppl. 2001, 77, 145–147. [Google Scholar]
- Cesarini, K.G.; Enblad, P.; Ronne-Engstrom, E.; Marklund, N.; Salci, K.; Nilsson, P.; Hardemark, H.G.; Hillered, L.; Persson, L. Early cerebral hyperglycolysis after subarachnoid haemorrhage correlates with favourable outcome. Acta Neurochir. 2002, 144, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Sarrafzadeh, A.; Haux, D.; Plotkin, M.; Ludemann, L.; Amthauer, H.; Unterberg, A. Bedside microdialysis reflects dysfunction of cerebral energy metabolism in patients with aneurysmal subarachnoid hemorrhage as confirmed by 15 O-H2 O-PET and 18 F-FDG-PET. J. Neuroradiol. 2005, 32, 348–351. [Google Scholar] [CrossRef]
- Carpenter, D.A.; Grubb, R.L., Jr.; Tempel, L.W.; Powers, W.J. Cerebral oxygen metabolism after aneurysmal subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 1991, 11, 837–844. [Google Scholar] [CrossRef] [Green Version]
- Westermaier, T.; Jauss, A.; Eriskat, J.; Kunze, E.; Roosen, K. Time-course of cerebral perfusion and tissue oxygenation in the first 6 h after experimental subarachnoid hemorrhage in rats. J. Cereb. Blood Flow Metab. 2009, 29, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis. 2018, 9, 924–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S.H.; Lan, J.; Esposito, E.; Ning, M.; Balaj, L.; Ji, X.; Lo, E.H.; Hayakawa, K. Extracellular Mitochondria in Cerebrospinal Fluid and Neurological Recovery After Subarachnoid Hemorrhage. Stroke 2017, 48, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Youn, D.H.; Kim, B.J.; Kim, Y.; Jeon, J.P. Extracellular Mitochondrial Dysfunction in Cerebrospinal Fluid of Patients with Delayed Cerebral Ischemia after Aneurysmal Subarachnoid Hemorrhage. Neurocrit Care 2020, 33, 422–428. [Google Scholar] [CrossRef]
- Macdonald, R.L. Origins of the Concept of Vasospasm. Stroke 2016, 47, e11–e15. [Google Scholar] [CrossRef] [Green Version]
- Etminan, N.; Vergouwen, M.D.; Ilodigwe, D.; Macdonald, R.L. Effect of pharmaceutical treatment on vasospasm, delayed cerebral ischemia, and clinical outcome in patients with aneurysmal subarachnoid hemorrhage: A systematic review and meta-analysis. J. Cereb. Blood Flow Metab. 2011, 31, 1443–1451. [Google Scholar] [CrossRef] [Green Version]
- Rowland, M.J.; Hadjipavlou, G.; Kelly, M.; Westbrook, J.; Pattinson, K.T. Delayed cerebral ischaemia after subarachnoid haemorrhage: Looking beyond vasospasm. Br. J. Anaesth 2012, 109, 315–329. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, J.R.; Testai, F.D. Delayed Cerebral Ischemia after Subarachnoid Hemorrhage: Beyond Vasospasm and Towards a Multifactorial Pathophysiology. Curr. Atheroscler. Rep. 2017, 19, 50. [Google Scholar] [CrossRef]
- Helbok, R.; Kofler, M.; Schiefecker, A.J.; Gaasch, M.; Rass, V.; Pfausler, B.; Beer, R.; Schmutzhard, E. Clinical Use of Cerebral Microdialysis in Patients with Aneurysmal Subarachnoid Hemorrhage-State of the Art. Front. Neurol. 2017, 8, 565. [Google Scholar] [CrossRef] [Green Version]
- Samuelsson, C.; Hillered, L.; Enblad, P.; Ronne-Engstrom, E. Microdialysis patterns in subarachnoid hemorrhage patients with focus on ischemic events and brain interstitial glutamine levels. Acta Neurochir (Wien.) 2009, 151, 437–446, discussion 446. [Google Scholar] [CrossRef]
- Helbok, R.; Madineni, R.C.; Schmidt, M.J.; Kurtz, P.; Fernandez, L.; Ko, S.B.; Choi, A.; Stuart, M.R.; Connolly, E.S.; Lee, K.; et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit. Care 2011, 14, 162–167. [Google Scholar] [CrossRef]
- Patet, C.; Quintard, H.; Zerlauth, J.B.; Maibach, T.; Carteron, L.; Suys, T.; Bouzat, P.; Bervini, D.; Levivier, M.; Daniel, R.T.; et al. Bedside cerebral microdialysis monitoring of delayed cerebral hypoperfusion in comatose patients with poor grade aneurysmal subarachnoid haemorrhage. J. Neurol. Neurosurg. Psychiatry 2017, 88, 332–338. [Google Scholar] [CrossRef]
- Dorhout Mees, S.M.; Rinkel, G.J.; Feigin, V.L.; Algra, A.; van den Bergh, W.M.; Vermeulen, M.; van Gijn, J. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev. 2007, 2007, CD000277. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Henderson, G.N.; Yan, Z.; James, M.O. Clinical pharmacology and toxicology of dichloroacetate. Env. Health Perspect 1998, 106 (Suppl. 4), 989–994. [Google Scholar]
- Thibodeau, A.; Geng, X.; Previch, L.E.; Ding, Y. Pyruvate dehydrogenase complex in cerebral ischemia-reperfusion injury. Brain Circ. 2016, 2, 61–66. [Google Scholar]
- Wang, P.; Chen, M.; Yang, Z.; Yu, T.; Zhu, J.; Zhou, L.; Lin, J.; Fang, X.; Huang, Z.; Jiang, L.; et al. Activation of Pyruvate Dehydrogenase Activity by Dichloroacetate Improves Survival and Neurologic Outcomes After Cardiac Arrest in Rats. Shock 2018, 49, 704–711. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.H.; Park, J.B.; Suh, S.W. The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury. Cells 2019, 8, 405. [Google Scholar] [CrossRef] [Green Version]
- Jalal, F.Y.; Bohlke, M.; Maher, T.J. Acetyl-L-carnitine reduces the infarct size and striatal glutamate outflow following focal cerebral ischemia in rats. Ann. N. Y. Acad. Sci. 2010, 1199, 95–104. [Google Scholar] [CrossRef]
- Martin, E.; Rosenthal, R.E.; Fiskum, G. Pyruvate dehydrogenase complex: Metabolic link to ischemic brain injury and target of oxidative stress. J. Neurosci. Res. 2005, 79, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, R.E.; Bogaert, Y.E.; Fiskum, G. Delayed therapy of experimental global cerebral ischemia with acetyl-L-carnitine in dogs. Neurosci. Lett. 2005, 378, 82–87. [Google Scholar] [CrossRef]
- Zanelli, S.A.; Solenski, N.J.; Rosenthal, R.E.; Fiskum, G. Mechanisms of ischemic neuroprotection by acetyl-L-carnitine. Ann. N. Y. Acad. Sci. 2005, 1053, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, R.E.; Williams, R.; Bogaert, Y.E.; Getson, P.R.; Fiskum, G. Prevention of postischemic canine neurological injury through potentiation of brain energy metabolism by acetyl-L-carnitine. Stroke 1992, 23, 1312–1317. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Niu, H.; Wu, C.; Li, Y.; Wang, K.; Zhang, J.; Wang, Y.; Yang, S. The autophagy-lysosomal system in subarachnoid haemorrhage. J. Cell Mol. Med. 2016, 20, 1770–1778. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, L.; Zhang, H.; Diao, X.; Zhao, S.; Zhou, W. Reduction in Autophagy by (-)-Epigallocatechin-3-Gallate (EGCG): A Potential Mechanism of Prevention of Mitochondrial Dysfunction After Subarachnoid Hemorrhage. Mol. Neurobiol. 2017, 54, 392–405. [Google Scholar] [CrossRef]
- Daou, B.J.; Koduri, S.; Thompson, B.G.; Chaudhary, N.; Pandey, A.S. Clinical and experimental aspects of aneurysmal subarachnoid hemorrhage. Cns Neurosci. 2019, 25, 1096–1112. [Google Scholar] [CrossRef]
- Van Lieshout, J.H.; Dibué-Adjei, M.; Cornelius, J.F.; Slotty, P.J.; Schneider, T.; Restin, T.; Boogaarts, H.D.; Steiger, H.J.; Petridis, A.K.; Kamp, M.A. An introduction to the pathophysiology of aneurysmal subarachnoid hemorrhage. Neurosurg Rev. 2018, 41, 917–930. [Google Scholar] [CrossRef]
- Vatter, H.; Zimmermann, M.; Tesanovic, V.; Raabe, A.; Schilling, L.; Seifert, V. Cerebrovascular characterization of clazosentan, the first nonpeptide endothelin receptor antagonist clinically effective for the treatment of cerebral vasospasm. Part I: Inhibitory effect on endothelin(A) receptor-mediated contraction. J. Neurosurg. 2005, 102, 1101–1107. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Kassell, N.F.; Mayer, S.; Ruefenacht, D.; Schmiedek, P.; Weidauer, S.; Frey, A.; Roux, S.; Pasqualin, A.; CONSCIOUS-1 Investigators. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): Randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke 2008, 39, 3015–3021. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, R.L.; Higashida, R.T.; Keller, E.; Mayer, S.A.; Molyneux, A.; Raabe, A.; Vajkoczy, P.; Wanke, I.; Bach, D.; Frey, A.; et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: A randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 2011, 10, 618–625. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Higashida, R.T.; Keller, E.; Mayer, S.A.; Molyneux, A.; Raabe, A.; Vajkoczy, P.; Wanke, I.; Bach, D.; Frey, A.; et al. Randomized trial of clazosentan in patients with aneurysmal subarachnoid hemorrhage undergoing endovascular coiling. Stroke 2012, 43, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Bergh, W.M.; Algra, A.; van der Sprenkel, J.W.; Tulleken, C.A.; Rinkel, G.J. Hypomagnesemia after aneurysmal subarachnoid hemorrhage. Neurosurgery 2003, 52, 276–281, discussion 281–2. [Google Scholar] [CrossRef] [PubMed]
- Westermaier, T.; Stetter, C.; Kunze, E.; Willner, N.; Raslan, F.; Vince, G.H.; Ernestus, R.I. Magnesium treatment for neuroprotection in ischemic diseases of the brain. Exp. Transl. Stroke Med. 2013, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Bergh, W.M.; Algra, A.; van Kooten, F.; Dirven, C.M.; van Gijn, J.; Vermeulen, M.; Rinkel, G.J. Magnesium sulfate in aneurysmal subarachnoid hemorrhage: A randomized controlled trial. Stroke 2005, 36, 1011–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stippler, M.; Crago, E.; Levy, E.I.; Kerr, M.E.; Yonas, H.; Horowitz, M.B.; Kassam, A. Magnesium infusion for vasospasm prophylaxis after subarachnoid hemorrhage. J. Neurosurg. 2006, 105, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Kunze, E.; Lilla, N.; Stetter, C.; Ernestus, R.I.; Westermaier, T. Magnesium Protects in Episodes of Critical Perfusion after Aneurysmal SAH. Transl. Neurosci. 2018, 9, 99–105. [Google Scholar] [CrossRef]
- Westermaier, T.; Stetter, C.; Vince, G.H.; Pham, M.; Tejon, J.P.; Eriskat, J.; Kunze, E.; Matthies, C.; Ernestus, R.I.; Solymosi, L.; et al. Prophylactic intravenous magnesium sulfate for treatment of aneurysmal subarachnoid hemorrhage: A randomized, placebo-controlled, clinical study. Crit Care Med. 2010, 38, 1284–1290. [Google Scholar] [CrossRef]
- Dorhout Mees, S.M.; Algra, A.; Vandertop, W.P.; van Kooten, F.; Kuijsten, H.A.; Boiten, J.; van Oostenbrugge, R.J.; Al-Shahi Salman, R.; Lavados, P.M.; Rinkel, G.J.; et al. Magnesium for aneurysmal subarachnoid haemorrhage (MASH-2): A randomised placebo-controlled trial. Lancet 2012, 380, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.K.; Poon, W.S.; Chan, M.T.; Boet, R.; Gin, T.; Ng, S.C.; Zee, B.C. Intravenous magnesium sulphate for aneurysmal subarachnoid hemorrhage (IMASH): A randomized, double-blinded, placebo-controlled, multicenter phase III trial. Stroke 2010, 41, 921–926. [Google Scholar] [CrossRef]
- Pickard, J.D.; Murray, G.D.; Illingworth, R.; Shaw, M.D.; Teasdale, G.M.; Foy, P.M.; Humphrey, P.R.; Lang, D.A.; Nelson, R.; Richards, P.; et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989, 298, 636–642. [Google Scholar] [CrossRef] [Green Version]
- Amory, C.F.; Varelas, P.N. Magnesium and Hydrogen in Subarachnoid Hemorrhage: Is Neuroprotection Finally a Reality? Stroke 2021, 52, 28–30. [Google Scholar] [CrossRef]
- Takeuchi, S.; Kumagai, K.; Toyooka, T.; Otani, N.; Wada, K.; Mori, K. Intravenous Hydrogen Therapy With Intracisternal Magnesium Sulfate Infusion in Severe Aneurysmal Subarachnoid Hemorrhage. Stroke 2021, 52, 20–27. [Google Scholar] [CrossRef]
- Westermaier, T.; Stetter, C.; Kunze, E.; Willner, N.; Holzmeier, J.; Kilgenstein, C.; Lee, J.Y.; Ernestus, R.I.; Roewer, N.; Muellenbach, R.M. Controlled transient hypercapnia: A novel approach for the treatment of delayed cerebral ischemia after subarachnoid hemorrhage? J. Neurosurg. 2014, 121, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Westermaier, T.; Stetter, C.; Kunze, E.; Willner, N.; Holzmeier, J.; Weiland, J.; Koehler, S.; Lotz, C.; Kilgenstein, C.; Ernestus, R.I.; et al. Controlled Hypercapnia Enhances Cerebral Blood Flow and Brain Tissue Oxygenation After Aneurysmal Subarachnoid Hemorrhage: Results of a Phase 1 Study. Neurocrit. Care 2016, 25, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Petridis, A.K.; Doukas, A.; Kienke, S.; Maslehaty, H.; Mahvash, M.; Barth, H.; Mehdorn, H.M. The effect of lung-protective permissive hypercapnia in intracerebral pressure in patients with subarachnoid haemorrhage and ARDS. A retrospective study. Acta Neurochir. (Wien.) 2010, 152, 2143–2145. [Google Scholar] [CrossRef]
- Lilla, N.; Rinne, C.; Weiland, J.; Linsenmann, T.; Ernestus, R.I.; Westermaier, T. Early Transient Mild Hypothermia Attenuates Neurologic Deficits and Brain Damage After Experimental Subarachnoid Hemorrhage in Rats. World Neurosurg. 2018, 109, e88–e98. [Google Scholar] [CrossRef]
- Török, E.; Klopotowski, M.; Trabold, R.; Thal, S.C.; Plesnila, N.; Schöller, K. Mild hypothermia (33 degrees C) reduces intracranial hypertension and improves functional outcome after subarachnoid hemorrhage in rats. Neurosurgery 2009, 65, 352–359. [Google Scholar] [CrossRef]
- Ma, J.; Wang, Z.; Liu, C.; Shen, H.; Chen, Z.; Yin, J.; Zuo, G.; Duan, X.; Li, H.; Chen, G. Pramipexole-Induced Hypothermia Reduces Early Brain Injury via PI3K/AKT/GSK3β pathway in Subarachnoid Hemorrhage rats. Sci Rep. 2016, 6, 23817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seule, M.; Muroi, C.; Sikorski, C.; Hugelshofer, M.; Winkler, K.; Keller, E. Therapeutic hypothermia reduces middle cerebral artery flow velocity in patients with severe aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2014, 20, 255–262. [Google Scholar] [CrossRef] [Green Version]


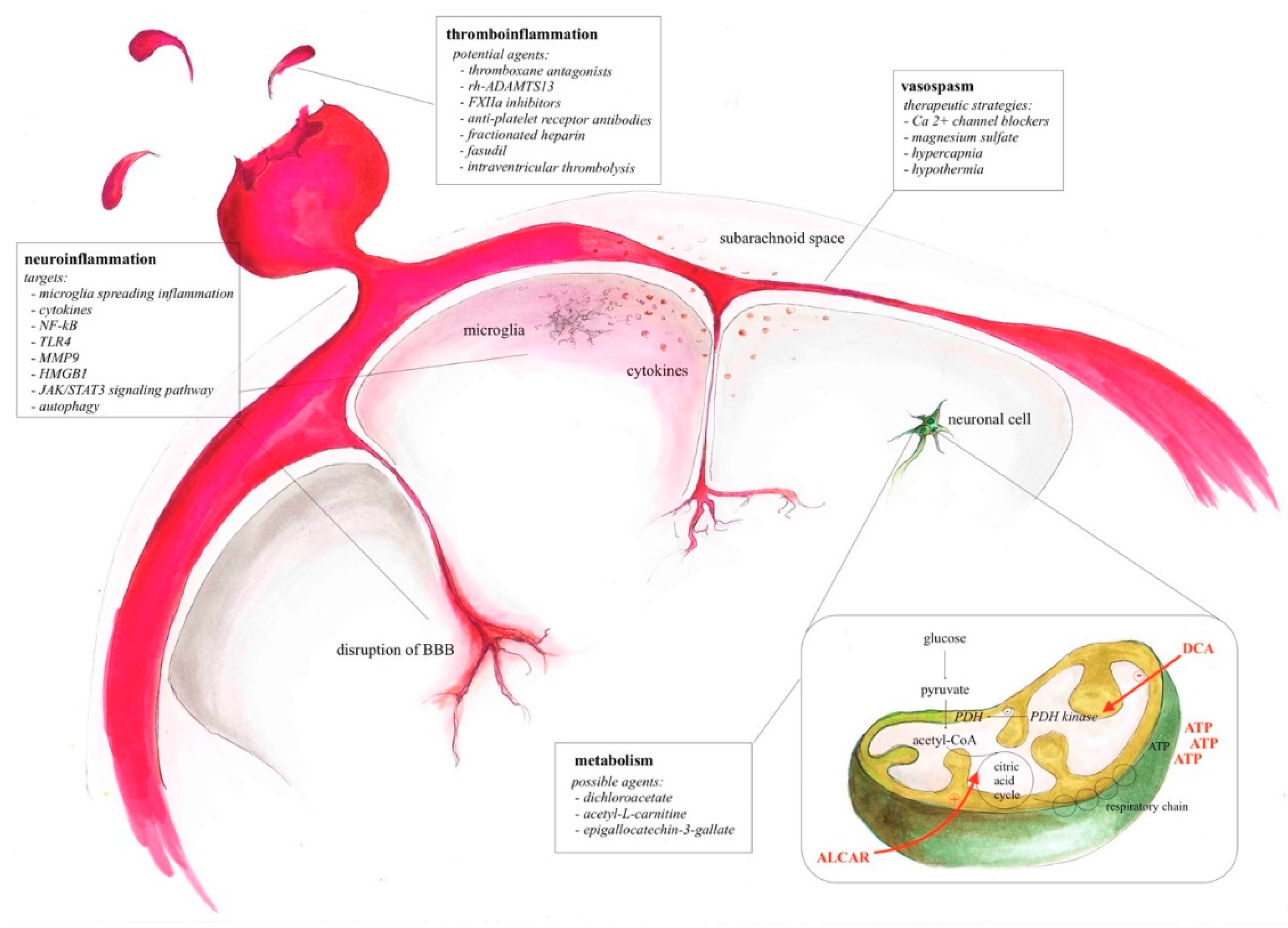{kind=link}
| Therapeutic Agent | Target | Model | Outcome Measures/Findings | Reference |
|---|---|---|---|---|
| LPS, PLX3397 | Microglia | Experimental aSAH in mice | Reduction in neuronal cell death | Heinz, R. et al., 2021 [29] |
| Interleukin-2 (IL-2) | Regulatory T-cells | Experimental aSAH in rats | Reduction in neuronal injury and proinflammatory factors, increase in neuronal functions | Dong et al., 2021 [71] |
| Mesenchymal stem cell-derived extracellular vesicles | Microglial M2 polarization | Experimental aSAH in rats | Reduction in inflammatory cytokines and inflammation, increase in neuroprotective effects | Han et al., 2021 [72] |
| Adenosine A3 receptor agonist | Microglial polarization | Experimental aSAH in rats | Increase in anti-inflammatory and neuroprotective effects | Li et al., 2020 [73] |
| Milk fat globule-epidural growth factor (MFG-EP) | Microglial polarization | Experimental aSAH in mice | Reduction in brain edema and proinflammatory factors, increase in neurological factors | Gao et al., 2021 [74] |
| Mixture of gas containing argon | Microglial inflammatory response | Experimental aSAH in rats | Reduction in early hippocampal neuronal damage | Kremer et al., 2020 [75] |
| EPZ6438 (specific EZH2 inhibitor) | EZH2 (enhancer of zeste homolog 2) | Experimental aSAH in rats | Reduction in attenuated neuroinflammatory brain injury | Luo et al., 2020 [76] |
| Oxyhemoglobin (OxyHb) | RNF26 (regulating TLRs) | Experimental aSAH in rats | Silence: reduction in neuronal injury and neurological dysfunction | Chen et al., 2020 [77] |
| LP-17 | TREM-1 myeloid cells | Experimental aSAH in mice | Amelioration of microglial pyroptosis | Xu et al., 2020 [78] |
| Resolvin D1 | Lipoxin A4 receptor/formyl peptide receptor 2 (ALX/FPR2) in microglia | Experimental aSAH in rats | Inhibiting H6 promoted microglial pro-inflammatory polarization, neuronal oxidant damage and death | Liu et al., 2020 [79] |
| Hydrogen sulfide (H2S) | TLR4/NF-κB pathway in microglia | Experimental aSAH in rats | Reduction in cognitive impairment and amelioration of neuroinflammation in microglia | Duan et al., 2020 [80] |
| Oxyhemoglobin (OxyHb) | CC chemokine ligand 20 (CCL20) | Experimental aSAH in mice | Reduction in apoptotic neurons | Liao et al., 2020 [81] |
| Translocator protein (TSPO) and TSPO ligand Ro5–4864 | Microglia/macrophages polarization | Experimental aSAH in mice | Improvement of neurological function, increase in expression of anti-inflammatory factors | Zhou et al., 2020 [82] |
| Oxyhemoglobin (OxyHb) | CCM3 overexpression and NF-κB signaling pathway | Experimental aSAH in rats | Reduction in cellular degeneration, neurocognitive impairment and inflammatory factors (TNF-a and IL-1β) | Peng et al., 2020 [83] |
| Modified exosomes (miR-193b-3p) | HDAC3, NF-κB | 1. aSAH patients and healthy controls to define profile 2. experimental aSAH in mice | Reduction in homological behavioral impairment, brain edema and BBB injury | Lai et al., 2020 [84] |
| Curcumin | M2 polarization through TLR4/MyD88/NF-κB signaling pathway | Experimental aSAH in tlr4−/− mice and wild type (WT) | Alleviation of neuroinflammation response, microglia phenotype shift and release of proinflammatory mediators | Gao et al., 2019 [85] |
| Dehydroepiandrosterone (DHEA) | Microglial activation | Experimental aSAH in C57BL/6 mice | Increase in neuroprotective effects, suppression of inflammation | Tao et al., 2019 [86] |
| Apelin-13 | Apelin receptor (APJ)/endoplasmic reticulum stress associated inflammation | Experimental aSAH in rats | Reduction in oxidative stress and neuroinflammation | Xu et al., 2019 [87] |
| BMS-470539 | Melanocortin 1 receptor (MC1R) | Experimental aSAH in rats | Suppression of microglial activation and neutrophil infiltration | Xu et al., 2020 [88] |
| TAK 242 (TLR4 antagonist) | Toll-like 4 receptor (TLR4) | Experimental aSAH in mice | Suppression of brain edema | Okada et al., 2020 [89] |
| Bexarotene | Retinoid X receptor | Experimental aSAH in rats | Decrease in neuroinflammation, improvement of neurological deficits | Zuo et al., 2019 [90] |
| RP 001 hydrochloride | S1P/S1PR pathway | Experimental aSAH in mice | Decrease in neuroinflammation, alleviation of neurological damage | Li et al., 2019 [91] |
| Bone marrow mesenchymal stem cells | Notch 1 signaling pathway | Experimental aSAH in rats | Amelioration of neurobehavioral impairments and BBB disruption | Liu et al., 2019 [92] |
| Fluoxetine | TLR4/MYD88/NF-κB pathway | Experimental aSAH in rats | Decrease in BBB disruption and brain edema, improvement of neurological function | Liu et al., 2018 [93] |
| Apolipoprotein E | Jak2/STAT3 signaling pathway | Experimental aSAH in mice | Decrease in oxidative stress and inflammation | Pang et al., 2018 [94] |
| TSG-6 | Microglial phenotype shift/SOCS3/STAT3 pathway | Experimental aSAH in rats | Amelioration of brain injury, decrease in proinflammatory mediators | Li et al., 2018 [95] |
| TAT-Pep5P | Resident microglia, p75 neurotrophin receptor (p75NTR) | Experimental aSAH in transgenic mice | Reduction in microglial activation, neuroinflammation and EBI | Xu et al., 2019 [96] |
| MST1 inhibitor XMU-MP-1 | MST1, NF-κB/MMP-9 pathway | Experimental aSAH in mice | Alleviation of neurological deficits, BBB, brain edema, neuroinflammation and white matter injury | Qu et al., 2018 [97] |
| rh-Aggf1 | PI3K/Akt/NF-κB pathway | Experimental aSAH in rats | Decrease in neuroinflammation and BBB disruption, improvement of neurological deficits | Zhu et al., 2018 [98] |
| IAXO-102 (TLR4 antagonist) | TLR4 | Experimental aSAH in C57BL/6 mice | Reduction in neurological impairments, brain edema, BBB disruption, increase in survival rates | Okada et al., 2019 [99] |
| Bexarotene | PPARγ | Experimental aSAH in C57BL/6 mice | Increase in neurological function, reduction in neuronal cell death and microglial activation | Tu et al., 2018 [100] |
| FTY720 (PP2A agonist) | Tristetraprolin (TTP), protein phosphatase 2A (PP2A) | Experimental aSAH in rats | Reduction in apoptosis, neuroinflammation and brain edema, increase in neurological function | Yin et al., 2018 [101] |
| Rolipram (specific phosphodiesterase-4 inhibitor) | SIRT1/NF-κB pathway | Experimental aSAH in rats | Reduction in brain edema, neurological dysfunction and neuronal cell death | Peng et al., 2018 [102] |
| Human Netrin-1 (rh-NTN-1) | UNC5B (receptor of NTN-1) | Experimental aSAH in rats | Increase in neurobehavioral function, reduction in brain edema and microglia activation | Xie et al., 2018 [103] |
| Fluoxetine, AC-YVAD-CMK (caspase-inhibitor) | NLRP3 inflammasome, caspase-1 | Experimental aSAH in rats | Increase in neurological function, reduction in brain edema and autophagy activation | Li et al., 2017 [104] |
| Methylene blue | Akt/GSK-3β/MEF2D pathway | Experimental aSAH in rats | Reduction in neurological dysfunction and brain edema | Xu et al., 2017 [105] |
| IL-1 receptor antagonist (IL-1Ra, anakinra) | Interleukin-1 (IL-1) | Randomized, open-label, clinical study in aSAH-patients | Difference in plasma IL-6, plasma pharmacokinetics for IL-1Ra, clinical outcome at 6 months | Galea et al., 2018 [63] |
| AE1–329 (EP4 selective agonist) | Prostanoid 4 receptor (EP4) | Experimental aSAH in rats | Reduction in neurological dysfunction, BBB damage, brain edema, reactivation of microglia, proinflammatory cytokines | Xu et al., 2017 [106] |
| Rutin | RAGE- NF-κB inflammatory signaling pathway | Experimental aSAH in rats | Increase in neurological function, reduction in BBB permeability, brain water content and neuronal cell death | Hao et al., 2016 [107] |
| Exogenous LXA4 (lipoxin A4) | Formyl peptide receptor 2 (FPR2), p38 MAPK | Experimental aSAH in rats | Increase in neurological functions, reduction in neutrophil infiltration and brain water content | Guo et al., 2016 [108] |
| Therapeutic Agent | Target | Model | Reference |
|---|---|---|---|
| Thromboxane antagonists, COX1-inhibitors, PAF antagonists | Platelet aggregation | Experimental and clinical aSAH | Lagier et al. [149], Suzuki et al. [145], Tokiyoshi et al. [146], Hirashima et al. [147,148] |
| intraventricular thrombolysis (rh tPA) | Clot clearance | Experimental and clinical aSAH | Shi et al. [150] |
| rh-ADAMTS13 | vWF-induced thrombosis and inflammation | Experimental and clinical aSAH | Muroi et al. [122], Vergouwen et al. [123,125], Wan et al. [124], Chauhan et al. [120] |
| FXIIa inhibitors (C1 inhibitor, rh infestin-4) | Contact kinin system (platelet aggregation and neuroinflammation) | Experimental stroke and TBI | Kleinschnitz et al. [131], Hagedorn et al. [132], Heydenreich et al. [133], Hopp et al. [135,136], Albert-Weissenberger et al. [134] |
| Anti-platelet receptor antibodies | Thrombosis, neuroinflammation, immune cells | Experimental stroke and TBI | Kleinschnitz et al. [141], Schuhmann et al. [143], Albert-Weissenberger et al. [140], Stoll and Nieswandt [121] |
| Fractionated heparin, glibenclamide, statins, anti-proinflammatory cytokine agents | Neuroinflammation | Experimental and clinical aSAH | James et al. [153], McBride et al. [111], Vergouwen et al. [151] |
| Fasudil (ROCK2 inhibitor) | Neuroinflammation | Experimental intracerebral hemorrhage (ICH) and clinical aSAH | McBride et al. [111], Li et al. [154], Zhao et al. [152] |
| Nimodipine | Vasospasms, thrombosis, leukocyte infiltration | Experimental and clinical aSAH | McBride et al. [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiland, J.; Beez, A.; Westermaier, T.; Kunze, E.; Sirén, A.-L.; Lilla, N. Neuroprotective Strategies in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2021, 22, 5442. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115442
Weiland J, Beez A, Westermaier T, Kunze E, Sirén A-L, Lilla N. Neuroprotective Strategies in Aneurysmal Subarachnoid Hemorrhage (aSAH). International Journal of Molecular Sciences. 2021; 22(11):5442. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115442
Chicago/Turabian StyleWeiland, Judith, Alexandra Beez, Thomas Westermaier, Ekkehard Kunze, Anna-Leena Sirén, and Nadine Lilla. 2021. "Neuroprotective Strategies in Aneurysmal Subarachnoid Hemorrhage (aSAH)" International Journal of Molecular Sciences 22, no. 11: 5442. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115442