
Exposure to the Methylselenol Precursor Dimethyldiselenide Induces a Reductive Endoplasmic Reticulum Stress in Saccharomyces cerevisiae


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Growth Inhibition by Methylselenol Precursors
2.2. Methylselenol Induces the Expression of Unfolded Protein Response (UPR) Targets
2.3. Selenomethionine Impairs Protein Folding in the Cytosol But Not in the Endoplasmc Reticulum
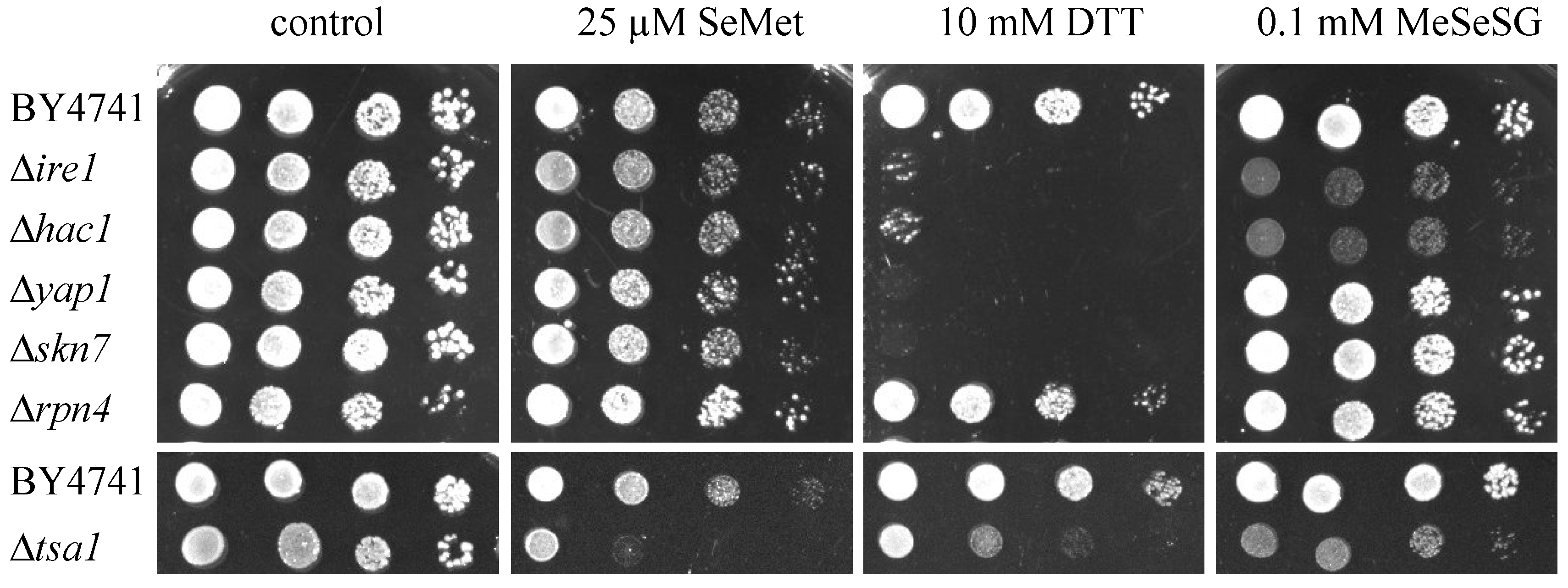
2.4. Null-Allele Strains of Genes Involved in the UPR Are Sensitive to Methylselenol but Not to Selenomethionine
2.5. Methylselenol Affects Oxidative Protein Folding in the ER
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Strains and Media
4.3. Growth Assays
4.4. Fluorescence Microscopy
4.5. Fluorescence Spectroscopy
4.6. Analysis of Cpy1p Maturation
4.7. In Vivo Oxidation State of Ero1p
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vinceti, M.; Filippini, T.; Del Giovane, C.; Dennert, G.; Zwahlen, M.; Brinkman, M.; Zeegers, M.P.A.; Horneber, M.; D’Amico, R.; Crespi, C.M. Selenium for preventing cancer. Cochrane Database Syst. Rev. 2018, 1, CD005195. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Boylan, M.; Selvam, A.; Spallholz, J.E.; Björnstedt, M. Redox-active selenium compounds: From toxicity and cell death to cancer treatment. Nutrients 2015, 7, 3536–3556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallenberg, M.; Misra, S.; Björnstedt, M. Selenium cytotoxicity in cancer. Basic Clin. Pharmacol. Toxicol. 2014, 114, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Lazard, M.; Dauplais, M.; Blanquet, S.; Plateau, P. Recent advances in the mechanism of selenoamino acids toxicity in eukaryotic cells. Biomol. Concepts 2017, 8, 93–104. [Google Scholar] [CrossRef]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, E.H. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar]
- Spallholz, J.E. On the nature of selenium toxicity and carcinostatic activity. Free. Radic. Biol. Med. 1994, 17, 45–64. [Google Scholar] [CrossRef]
- Jackson, I.M.; Combs, G.F. Selenium and anticarcinogenesis: Underlying mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.P.; Gandin, V. Selenium compounds as therapeutic agents in cancer. Biochim. Biophys. Acta (BBA) Gen. Subj. 2015, 1850, 1642–1660. [Google Scholar] [CrossRef]
- Drake, E.N. Cancer chemoprevention: Selenium as a prooxidant, not an antioxidant. Med. Hypotheses 2006, 67, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Spallholz, E.J.; Palace, V.P.; Reid, T.W. Methioninase and selenomethionine but not Semethylselenocysteine generate methylselenol and superoxide in an in vitro chemiluminescent assay: Implications for the nutritional carcinostatic activity of selenoamino acids. Biochem. Pharmacol. 2004, 67, 547–554. [Google Scholar] [CrossRef]
- Ganther, H.E. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: Complexities with thioredoxin reductase. Carcinogenesis 1999, 20, 1657–1666. [Google Scholar] [CrossRef] [Green Version]
- Zu, K.; Wu, Y.; Park, Y.M.; Ip, C. Peering down the kaleidoscope of thiol proteomics and unfolded protein response in studying the anticancer action of selenium. In Selenium: Its Molecular Biology and Role in Human Health; Hatfield, D.L., Berry, M.J., Gladyshev, V.N., Eds.; Springer: Boston, MA, USA, 2006; pp. 265–276. [Google Scholar]
- Park, E.M.; Choi, K.S.; Park, S.Y.; Kong, E.S.; Zu, E.K.; Wu, Y.; Zhang, H.; Ip, C.; Park, Y.M. A Display Thiol-Proteomics Approach to Characterize Global Redox Modification of Proteins by Selenium: Implications for the Anticancer Action of Selenium. Cancer Genom. Proteom. 2005, 2, 25–35. [Google Scholar]
- Cox, J.S.; Walter, P. A Novel Mechanism for Regulating Activity of a Transcription Factor That Controls the Unfolded Protein Response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, H.; Dong, Y.; Park, Y.-M.; Ip, C. Endoplasmic Reticulum Stress Signal Mediators Are Targets of Selenium Action. Cancer Res. 2005, 65, 9073–9079. [Google Scholar] [CrossRef] [Green Version]
- Zu, K.; Bihani, T.; Lin, A.; Park, Y.M.; Mori, K.; Ip, C. Enhanced selenium effect on growth arrest by BiP/GRP78 knockdown in p53-null human prostate cancer cells. Oncogene 2005, 25, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Shigemi, Z.; Manabe, K.; Hara, N.; Baba, Y.; Hosokawa, K.; Kagawa, H.; Watanabe, T.; Fujimuro, M. Methylseleninic acid and sodium selenite induce severe ER stress and subsequent apoptosis through UPR activation in PEL cells. Chem. Interact. 2017, 266, 28–37. [Google Scholar] [CrossRef]
- Goltyaev, M.V.; Mal’tseva, V.N.; Varlamova, E.G. Expression of ER-resident selenoproteins and activation of cancer cells apoptosis mechanisms under ER-stress conditions caused by methylseleninic acid. Gene 2020, 755, 144884. [Google Scholar] [CrossRef]
- Poerschke, R.L.; Franklin, M.R.; Moos, P.J. Modulation of redox status in human lung cell lines by organoselenocompounds: Selenazolidines, selenomethionine, and methylseleninic acid. Toxicol. Vitr. 2008, 22, 1761–1767. [Google Scholar] [CrossRef] [Green Version]
- Dauplais, M.; Bierla, K.; Maizeray, C.; Lestini, R.; Lobinski, R.; Plateau, P.; Szpunar, J.; Lazard, M. Methylselenol Produced In Vivo from Methylseleninic Acid or Dimethyl Diselenide Induces Toxic Protein Aggregation in Saccharomyces cerevisiae. Int. J. Mol. Sci. 2021, 22, 2241. [Google Scholar] [CrossRef]
- Boorstein, W.; Craig, E. Structure and regulation of the SSA4 HSP70 gene of Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 18912–18921. [Google Scholar] [CrossRef]
- Lee, J.; Godon, C.; Lagniel, G.; Spector, D.; Garin, J.; Labarre, J.; Toledano, M.B. Yap1 and Skn7 Control Two Specialized Oxidative Stress Response Regulons in Yeast. J. Biol. Chem. 1999, 274, 16040–16046. [Google Scholar] [CrossRef] [Green Version]
- Normington, K.; Kohno, K.; Kozutsumi, Y.; Gething, M.J.; Sambrook, J. S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 1989, 57, 1223–1236. [Google Scholar] [CrossRef]
- Lajoie, P.; Moir, R.D.; Willis, I.M.; Snapp, E.L. Kar2p availability defines distinct forms of endoplasmic reticulum stress in living cells. Mol. Biol. Cell 2012, 23, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Merksamer, P.I.; Trusina, A.; Papa, F.R. Real-Time Redox Measurements during Endoplasmic Reticulum Stress Reveal Interlinked Protein Folding Functions. Cell 2008, 135, 933–947. [Google Scholar] [CrossRef] [Green Version]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy Counterbalances Endoplasmic Reticulum Expansion during the Unfolded Protein Response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [Green Version]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Plateau, P.; Saveanu, C.; Lestini, R.; Dauplais, M.; Decourty, L.; Jacquier, A.; Blanquet, S.; Lazard, M. Exposure to selenomethionine causes selenocysteine misincorporation and protein aggregation in Saccharomyces cerevisiae. Sci. Rep. 2017, 7, 44761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rand, J.D.; Grant, C.M. The Thioredoxin System Protects Ribosomes against Stress-induced Aggregation. Mol. Biol. Cell 2006, 17, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two Enzymes in One: Two Yeast Peroxiredoxins Display Oxidative Stress-Dependent Switching from a Peroxidase to a Molecular Chaperone Function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef]
- Jämsä, E.; Simonen, M.; Makarow, M. Selective retention of secretory proteins in the yeast endoplasmic reticulum by treatment of cells with a reducing agent. Yeast 1994, 10, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Frand, A.R.; A Kaiser, C. Ero1p Oxidizes Protein Disulfide Isomerase in a Pathway for Disulfide Bond Formation in the Endoplasmic Reticulum. Mol. Cell 1999, 4, 469–477. [Google Scholar] [CrossRef]
- Kim, S.; Sideris, P.D.; Sevier, C.S.; Kaiser, C.A. Balanced Ero1 activation and inactivation establishes ER redox homeostasis. J. Cell Biol. 2012, 196, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Sevier, C.S.; Qu, H.; Heldman, N.; Gross, E.; Fass, D.; Kaiser, C.A. Modulation of Cellular Disulfide-Bond Formation and the ER Redox Environment by Feedback Regulation of Ero1. Cell 2007, 129, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyroche, G.; Saveanu, C.; Dauplais, M.; Lazard, M.; Beuneu, F.; Decourty, L.; Malabat, C.; Jacquier, A.; Blanquet, S.; Plateau, P. Sodium Selenide Toxicity Is Mediated by O2-Dependent DNA Breaks. PLoS ONE 2012, 7, e36343. [Google Scholar] [CrossRef] [Green Version]
- Oka, O.B.V.; Bulleid, N.J. Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 2425–2429. [Google Scholar] [CrossRef] [Green Version]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef] [Green Version]
- Toledano, M.B.; Kumar, C.; Le Moan, N.; Spector, D.; Tacnet, F. The system biology of thiol redox system in Escherichia coli and yeast: Differential functions in oxidative stress, iron metabolism and DNA synthesis. FEBS Lett. 2007, 581, 3598–3607. [Google Scholar] [CrossRef] [Green Version]
- Ponsero, A.J.; Igbaria, A.; Darch, M.A.; Miled, S.; Outten, C.E.; Winther, J.R.; Palais, G.; D’Autréaux, B.; Delaunay-Moisan, A.; Toledano, M.B. Endoplasmic Reticulum Transport of Glutathione by Sec61 Is Regulated by Ero1 and Bip. Mol. Cell 2017, 67, 962–973.e5. [Google Scholar] [CrossRef] [Green Version]
- Gromer, S.; Gross, J.H. Methylseleninate Is a Substrate Rather Than an Inhibitor of Mammalian Thioredoxin Reductase. J. Biol. Chem. 2002, 277, 9701–9706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radomska, D.; Czarnomysy, R.; Radomski, D.; Bielawski, K. Selenium Compounds as Novel Potential Anticancer Agents. Int. J. Mol. Sci. 2021, 22, 1009. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A.P. Organic selenium compounds as potential chemotherapeutic agents for improved cancer treatment. Free. Radic. Biol. Med. 2018, 127, 80–97. [Google Scholar] [CrossRef]
- Shore, G.C.; Papa, F.R.; Oakes, A.S. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Frand, A.R.; A Kaiser, C. The ERO1 Gene of Yeast Is Required for Oxidation of Protein Dithiols in the Endoplasmic Reticulum. Mol. Cell 1998, 1, 161–170. [Google Scholar] [CrossRef]
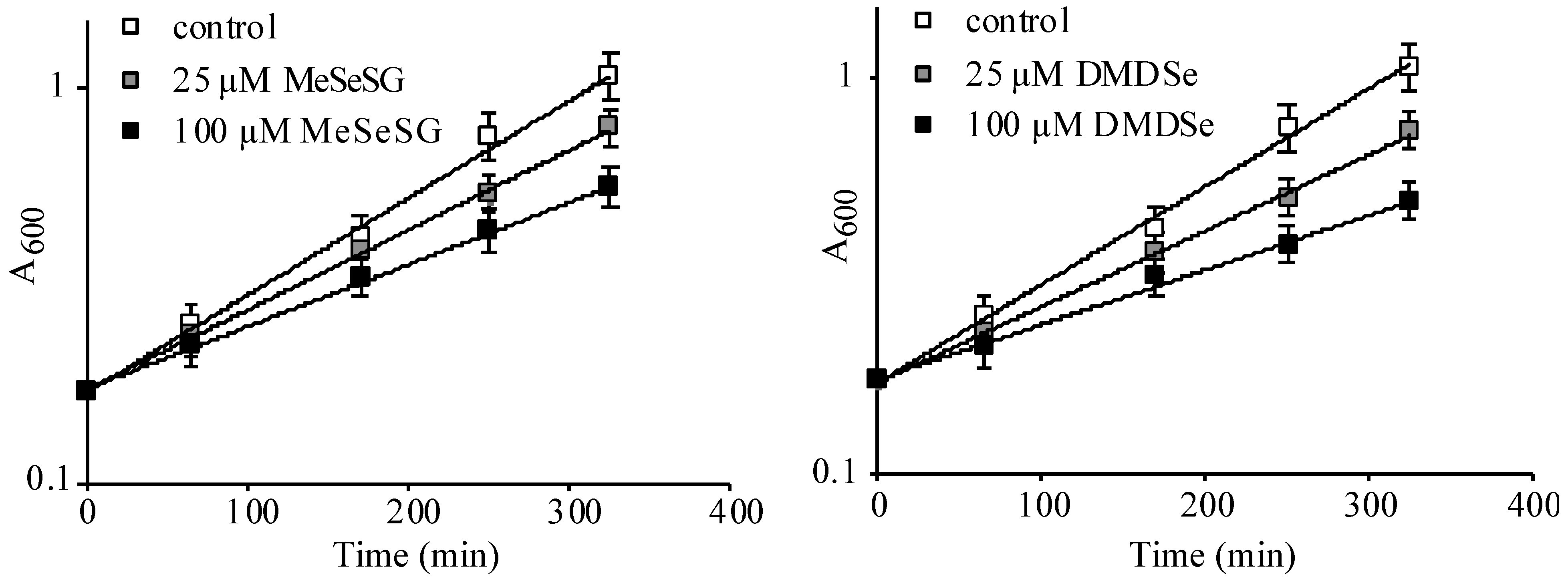
 ), 25 (
), 25 (  ) or 100 µM (
) or 100 µM (  ) of methylselenoglutathione (MeSeSG) (left panel) or dimethyldiselenide (DMDSe) (right panel) were added and cell growth was monitored by measuring the OD600 at various times during 6 h. Data were plotted on a semi-log scale as a function of time. Doubling times of 119 ± 14 (control), 147 ± 16 (25 µM MeSeSG), 192 ± 19 (100 µM MeSeSG), 157 ± 16 (25 µM DMDSe) and 223 ± 24 (100 µM DMDSe) min were calculated by using regression analysis. The results are the mean and range of two experiments.
), 25 ( ) or 100 µM ( ) of methylselenoglutathione (MeSeSG) (left panel) or dimethyldiselenide (DMDSe) (right panel) were added and cell growth was monitored by measuring the OD600 at various times during 6 h. Data were plotted on a semi-log scale as a function of time. Doubling times of 119 ± 14 (control), 147 ± 16 (25 µM MeSeSG), 192 ± 19 (100 µM MeSeSG), 157 ± 16 (25 µM DMDSe) and 223 ± 24 (100 µM DMDSe) min were calculated by using regression analysis. The results are the mean and range of two experiments.
) of methylselenoglutathione (MeSeSG) (left panel) or dimethyldiselenide (DMDSe) (right panel) were added and cell growth was monitored by measuring the OD600 at various times during 6 h. Data were plotted on a semi-log scale as a function of time. Doubling times of 119 ± 14 (control), 147 ± 16 (25 µM MeSeSG), 192 ± 19 (100 µM MeSeSG), 157 ± 16 (25 µM DMDSe) and 223 ± 24 (100 µM DMDSe) min were calculated by using regression analysis. The results are the mean and range of two experiments.
), 25 ( ) or 100 µM ( ) of methylselenoglutathione (MeSeSG) (left panel) or dimethyldiselenide (DMDSe) (right panel) were added and cell growth was monitored by measuring the OD600 at various times during 6 h. Data were plotted on a semi-log scale as a function of time. Doubling times of 119 ± 14 (control), 147 ± 16 (25 µM MeSeSG), 192 ± 19 (100 µM MeSeSG), 157 ± 16 (25 µM DMDSe) and 223 ± 24 (100 µM DMDSe) min were calculated by using regression analysis. The results are the mean and range of two experiments. ) or in ∆ire1 (
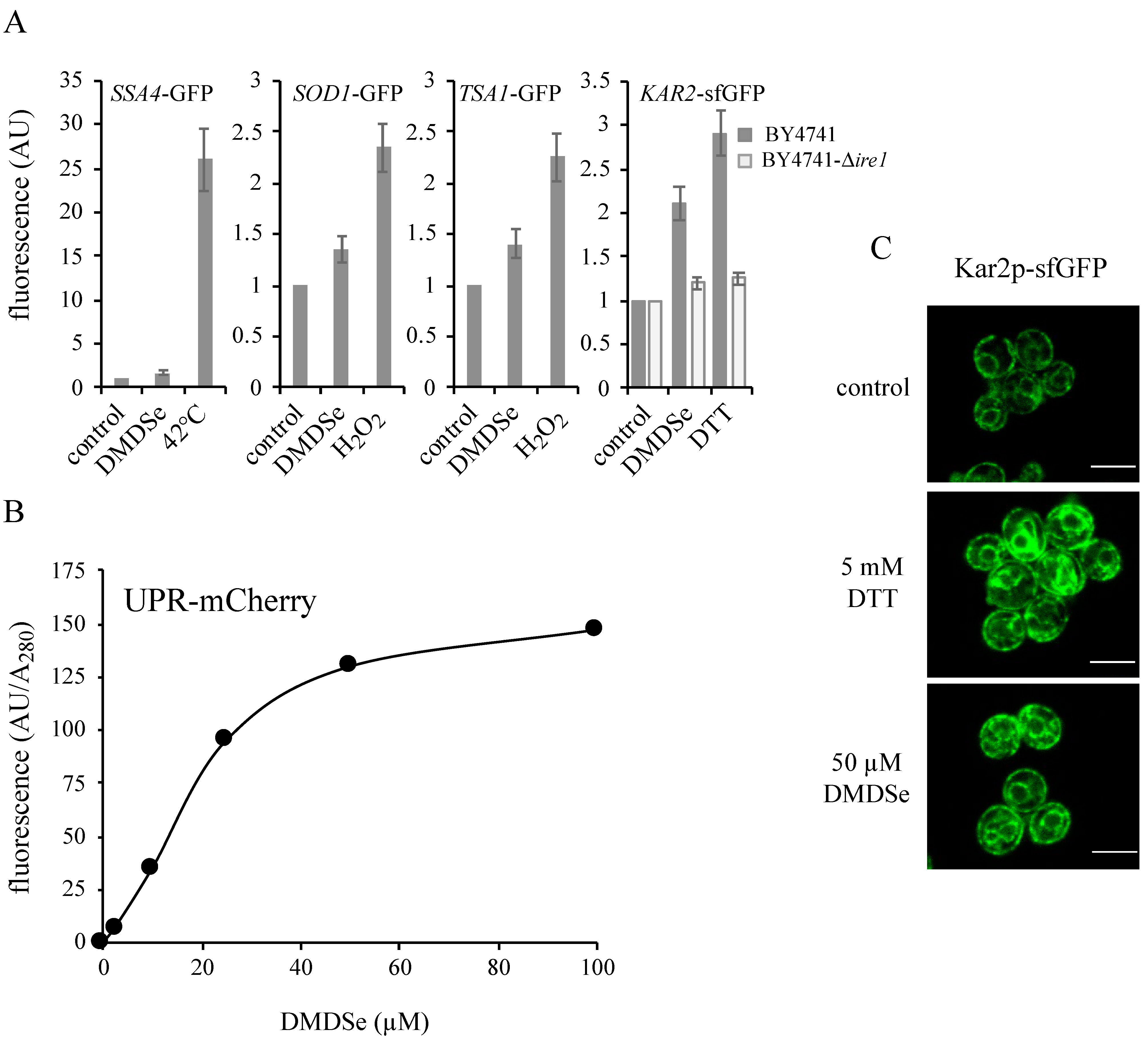
) or in ∆ire1 (  ) cell extracts. The fluorescence in whole cell extracts was recorded at 508 nm and normalized to the optical density of the extracts at 280 nm. The fluorescence intensity in the absence of toxic (control) was set as 1. The results are the mean ± S.D. of at least 3 experiments. (B) BY4741 cells expressing the fluorescent protein UPR-mCherry were grown in SC without uracil supplemented with 100 µM methionine. After incubation for 2 h with the indicated concentrations of DMDSe, the fluorescence in whole cell extracts was recorded at 610 nm. The autofluorescence of an extract from BY4741 cells in the absence of stress was subtracted from the results. (C) Kar2p-sfGFP localization was monitored by confocal fluorescence microscopy in living cells grown in SC + 100 µM methionine after 75 min of exposure to 50 µM DMDSe or 5 mM DTT. Bar equals 5 µm.
) or in ∆ire1 ( ) cell extracts. The fluorescence in whole cell extracts was recorded at 508 nm and normalized to the optical density of the extracts at 280 nm. The fluorescence intensity in the absence of toxic (control) was set as 1. The results are the mean ± S.D. of at least 3 experiments. (B) BY4741 cells expressing the fluorescent protein UPR-mCherry were grown in SC without uracil supplemented with 100 µM methionine. After incubation for 2 h with the indicated concentrations of DMDSe, the fluorescence in whole cell extracts was recorded at 610 nm. The autofluorescence of an extract from BY4741 cells in the absence of stress was subtracted from the results. (C) Kar2p-sfGFP localization was monitored by confocal fluorescence microscopy in living cells grown in SC + 100 µM methionine after 75 min of exposure to 50 µM DMDSe or 5 mM DTT. Bar equals 5 µm.
) cell extracts. The fluorescence in whole cell extracts was recorded at 508 nm and normalized to the optical density of the extracts at 280 nm. The fluorescence intensity in the absence of toxic (control) was set as 1. The results are the mean ± S.D. of at least 3 experiments. (B) BY4741 cells expressing the fluorescent protein UPR-mCherry were grown in SC without uracil supplemented with 100 µM methionine. After incubation for 2 h with the indicated concentrations of DMDSe, the fluorescence in whole cell extracts was recorded at 610 nm. The autofluorescence of an extract from BY4741 cells in the absence of stress was subtracted from the results. (C) Kar2p-sfGFP localization was monitored by confocal fluorescence microscopy in living cells grown in SC + 100 µM methionine after 75 min of exposure to 50 µM DMDSe or 5 mM DTT. Bar equals 5 µm.
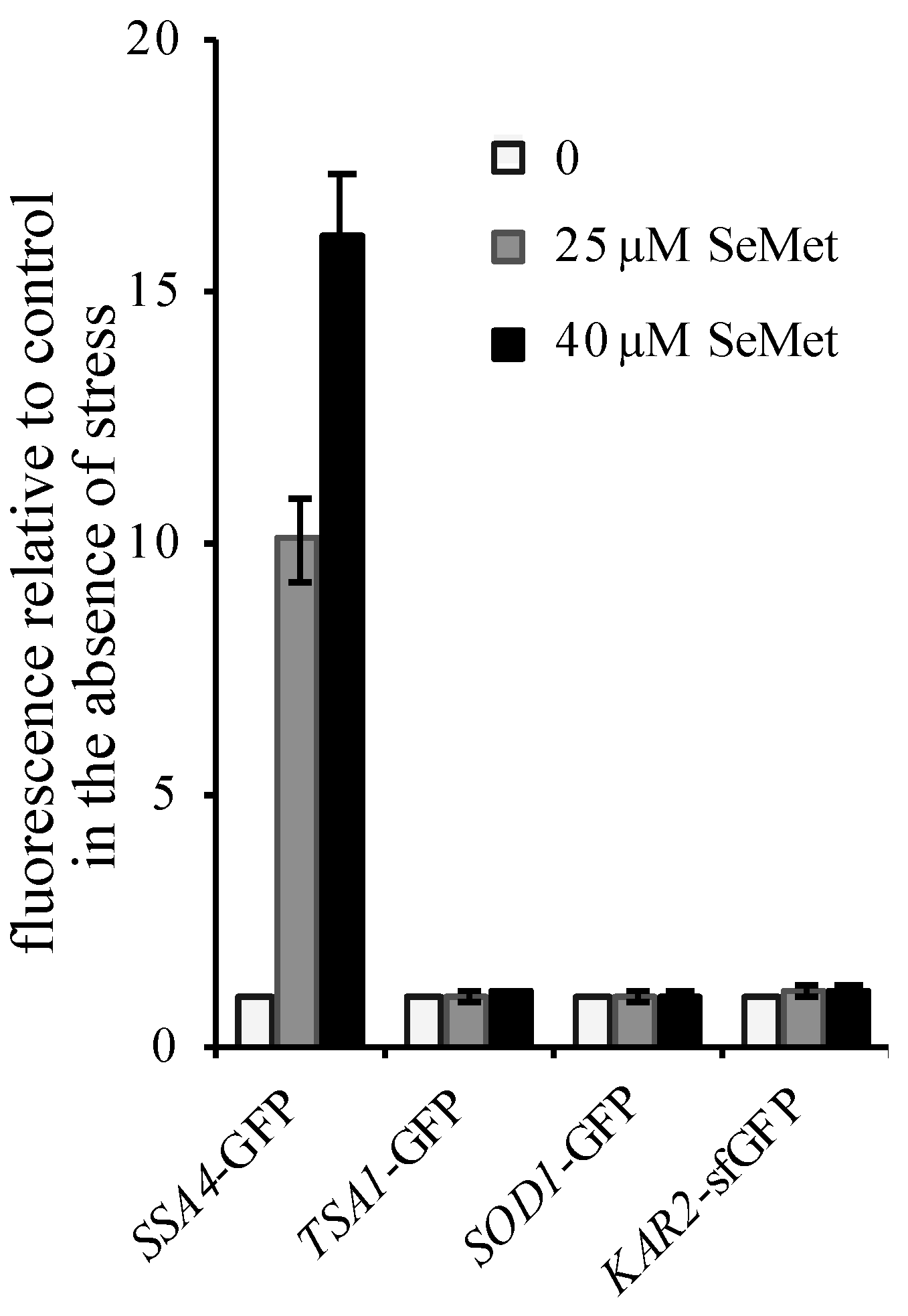
) or in ∆ire1 ( ) cell extracts. The fluorescence in whole cell extracts was recorded at 508 nm and normalized to the optical density of the extracts at 280 nm. The fluorescence intensity in the absence of toxic (control) was set as 1. The results are the mean ± S.D. of at least 3 experiments. (B) BY4741 cells expressing the fluorescent protein UPR-mCherry were grown in SC without uracil supplemented with 100 µM methionine. After incubation for 2 h with the indicated concentrations of DMDSe, the fluorescence in whole cell extracts was recorded at 610 nm. The autofluorescence of an extract from BY4741 cells in the absence of stress was subtracted from the results. (C) Kar2p-sfGFP localization was monitored by confocal fluorescence microscopy in living cells grown in SC + 100 µM methionine after 75 min of exposure to 50 µM DMDSe or 5 mM DTT. Bar equals 5 µm. ), 25 ( ) or 40 µM ( ) SeMet. The fluorescence in whole cell extracts was recorded at 508 nm and normalized to the optical density of the extracts at 280 nm. The fluorescence intensity in the absence of toxic was set as 1. The results are the mean ± S.D. of at least 3 experiments.
), 25 ( ) or 40 µM ( ) SeMet. The fluorescence in whole cell extracts was recorded at 508 nm and normalized to the optical density of the extracts at 280 nm. The fluorescence intensity in the absence of toxic was set as 1. The results are the mean ± S.D. of at least 3 experiments.
), 25 ( ) or 40 µM ( ) SeMet. The fluorescence in whole cell extracts was recorded at 508 nm and normalized to the optical density of the extracts at 280 nm. The fluorescence intensity in the absence of toxic was set as 1. The results are the mean ± S.D. of at least 3 experiments.
), 25 ( ) or 40 µM ( ) SeMet. The fluorescence in whole cell extracts was recorded at 508 nm and normalized to the optical density of the extracts at 280 nm. The fluorescence intensity in the absence of toxic was set as 1. The results are the mean ± S.D. of at least 3 experiments.


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dauplais, M.; Mahou, P.; Plateau, P.; Lazard, M. Exposure to the Methylselenol Precursor Dimethyldiselenide Induces a Reductive Endoplasmic Reticulum Stress in Saccharomyces cerevisiae. Int. J. Mol. Sci. 2021, 22, 5467. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115467
Dauplais M, Mahou P, Plateau P, Lazard M. Exposure to the Methylselenol Precursor Dimethyldiselenide Induces a Reductive Endoplasmic Reticulum Stress in Saccharomyces cerevisiae. International Journal of Molecular Sciences. 2021; 22(11):5467. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115467
Chicago/Turabian StyleDauplais, Marc, Pierre Mahou, Pierre Plateau, and Myriam Lazard. 2021. "Exposure to the Methylselenol Precursor Dimethyldiselenide Induces a Reductive Endoplasmic Reticulum Stress in Saccharomyces cerevisiae" International Journal of Molecular Sciences 22, no. 11: 5467. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115467