
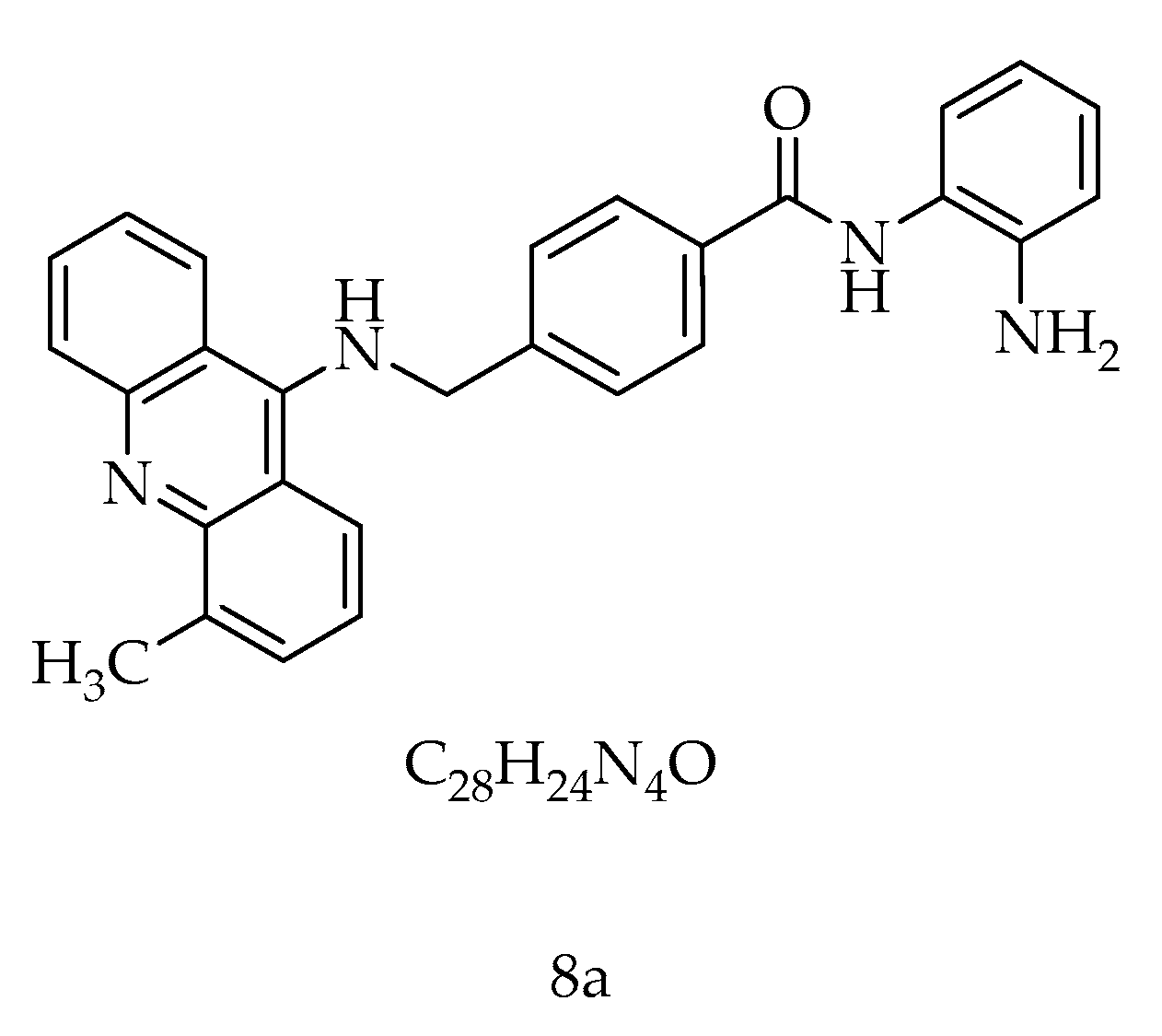
8a, a New Acridine Antiproliferative and Pro-Apoptotic Agent Targeting HDAC1/DNMT1

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
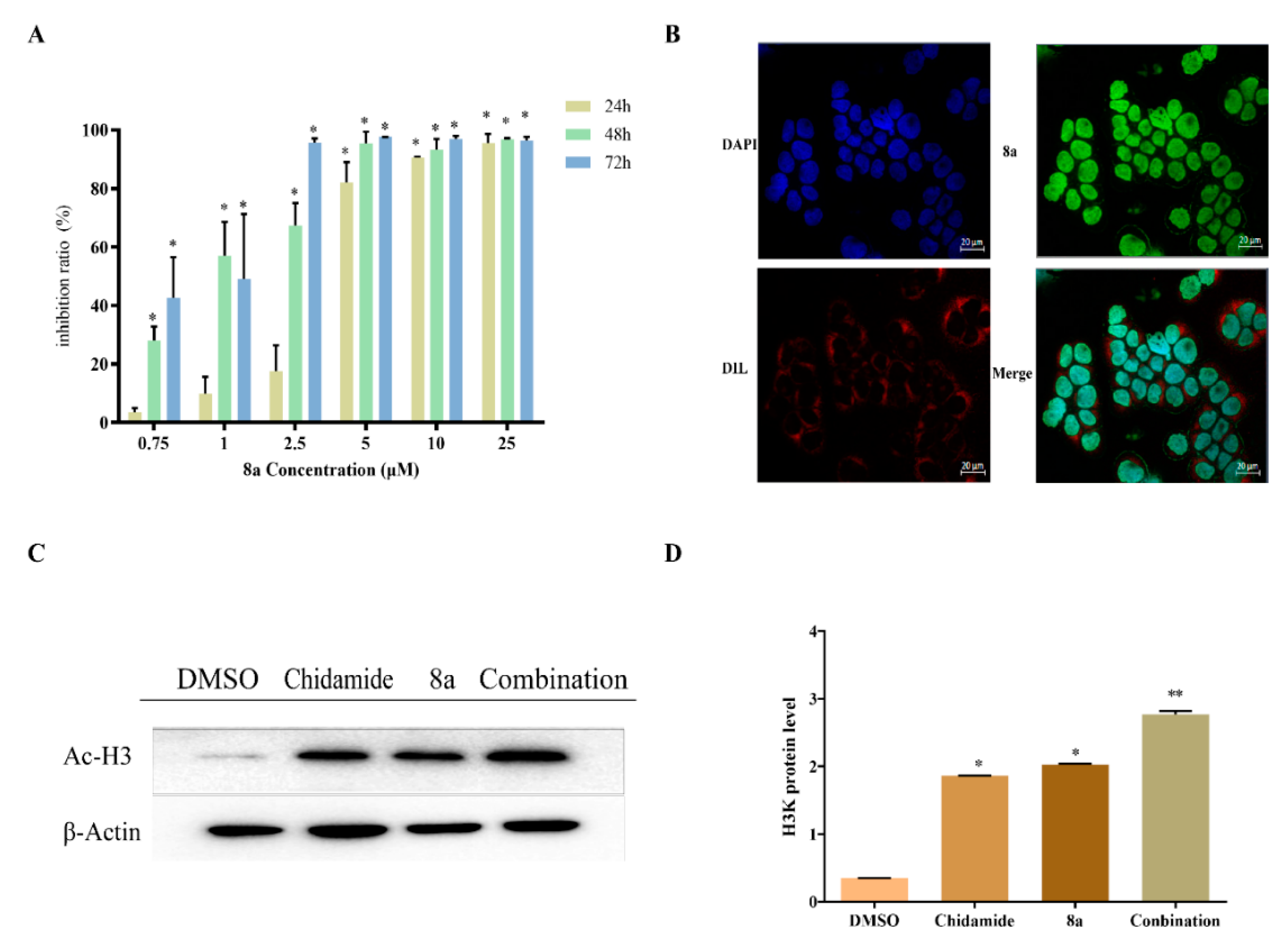
2.1. Antiproliferative Assay and Nuclear Localization
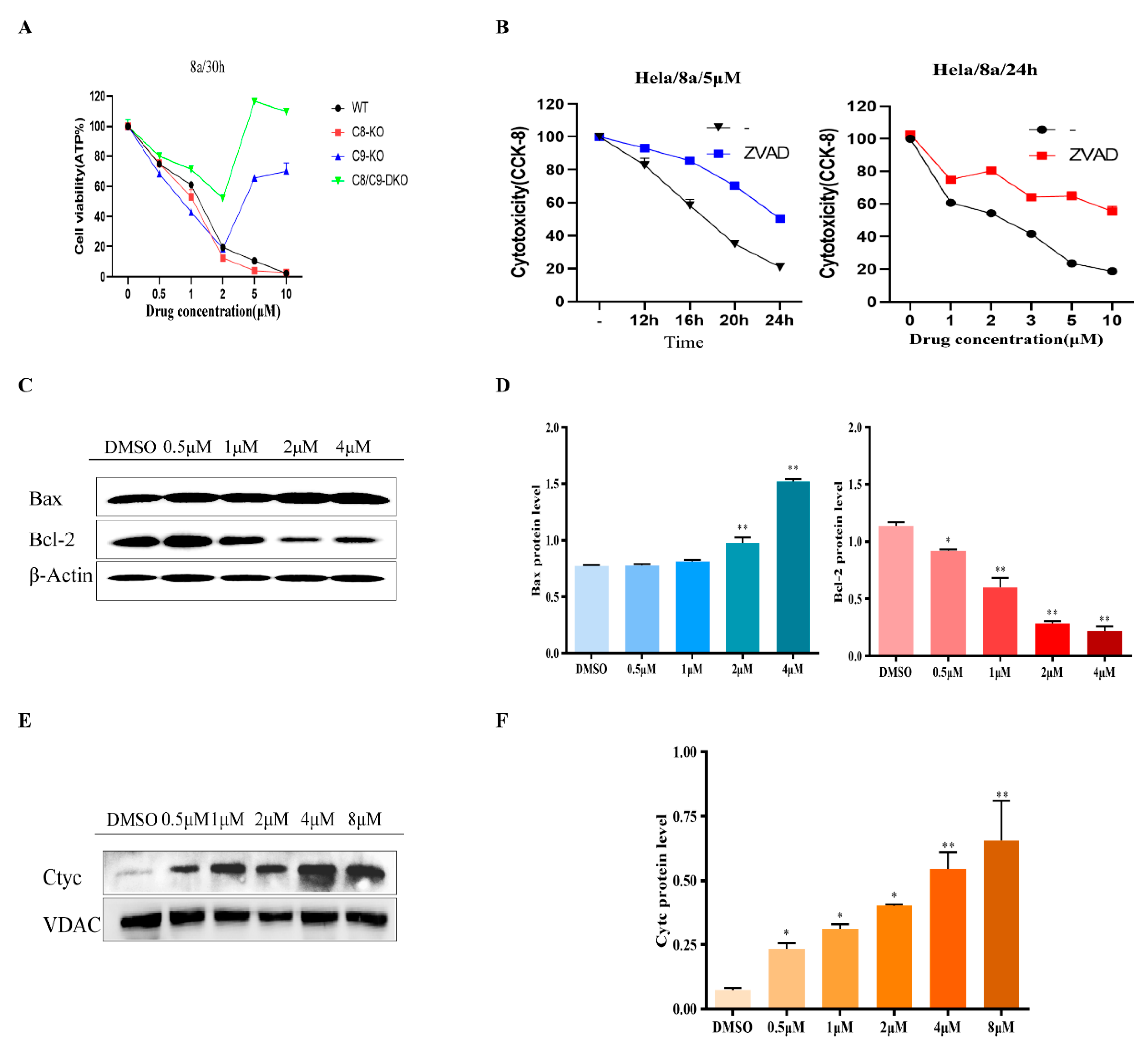
2.2. Inducing Apoptosis
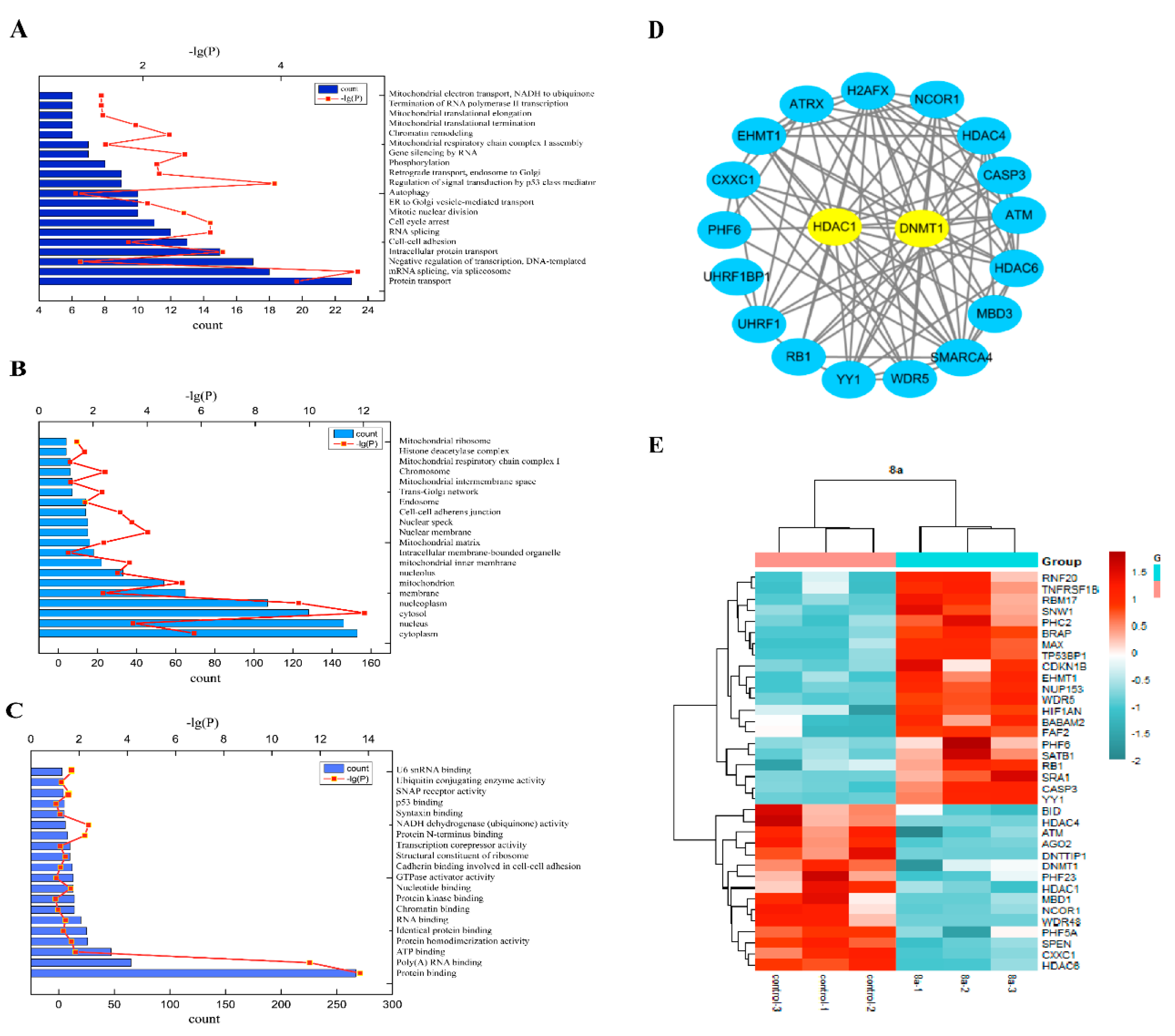
2.3. Proteomics Analysis
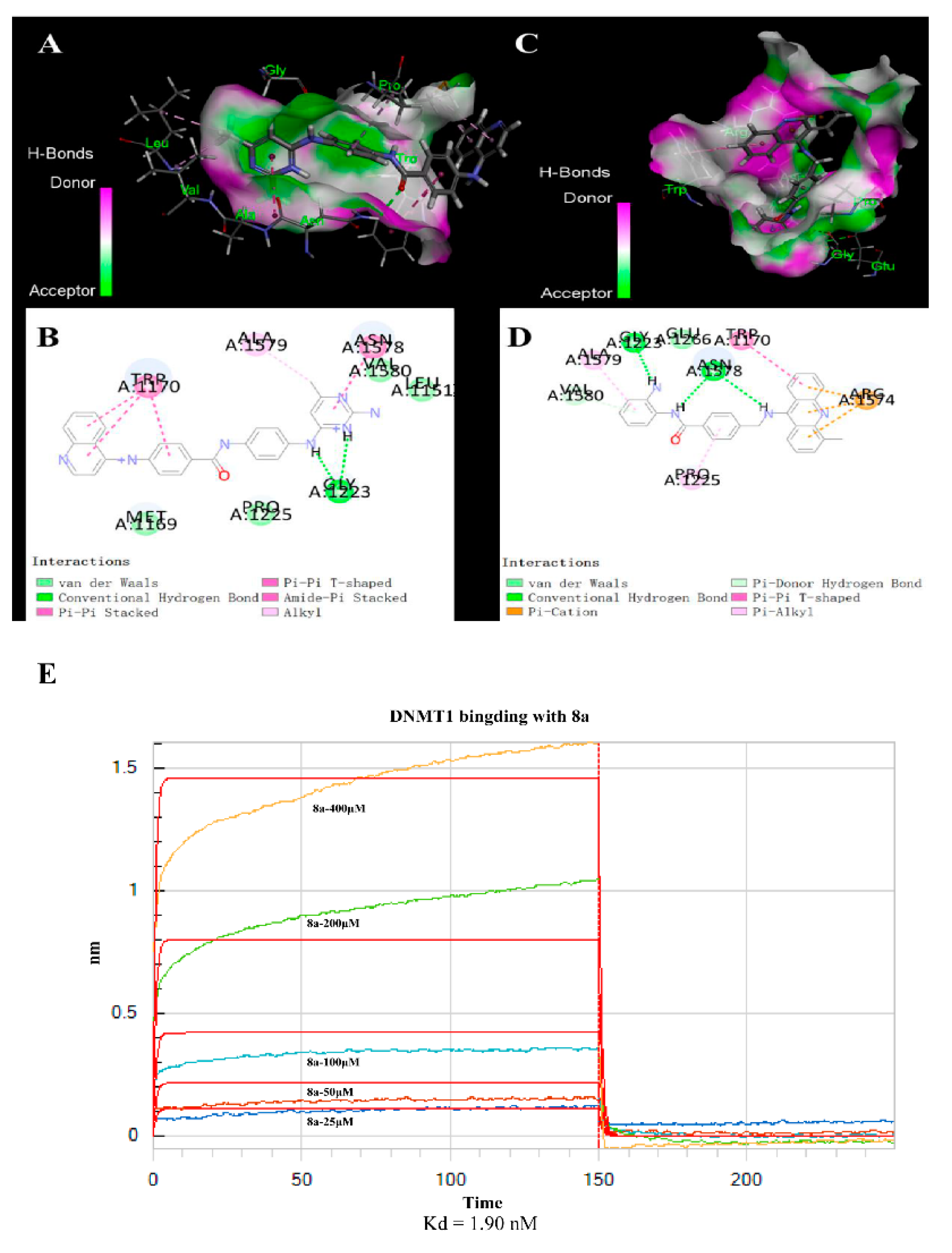
2.4. 8a-DNMT1 Molecular Docking and Interactions
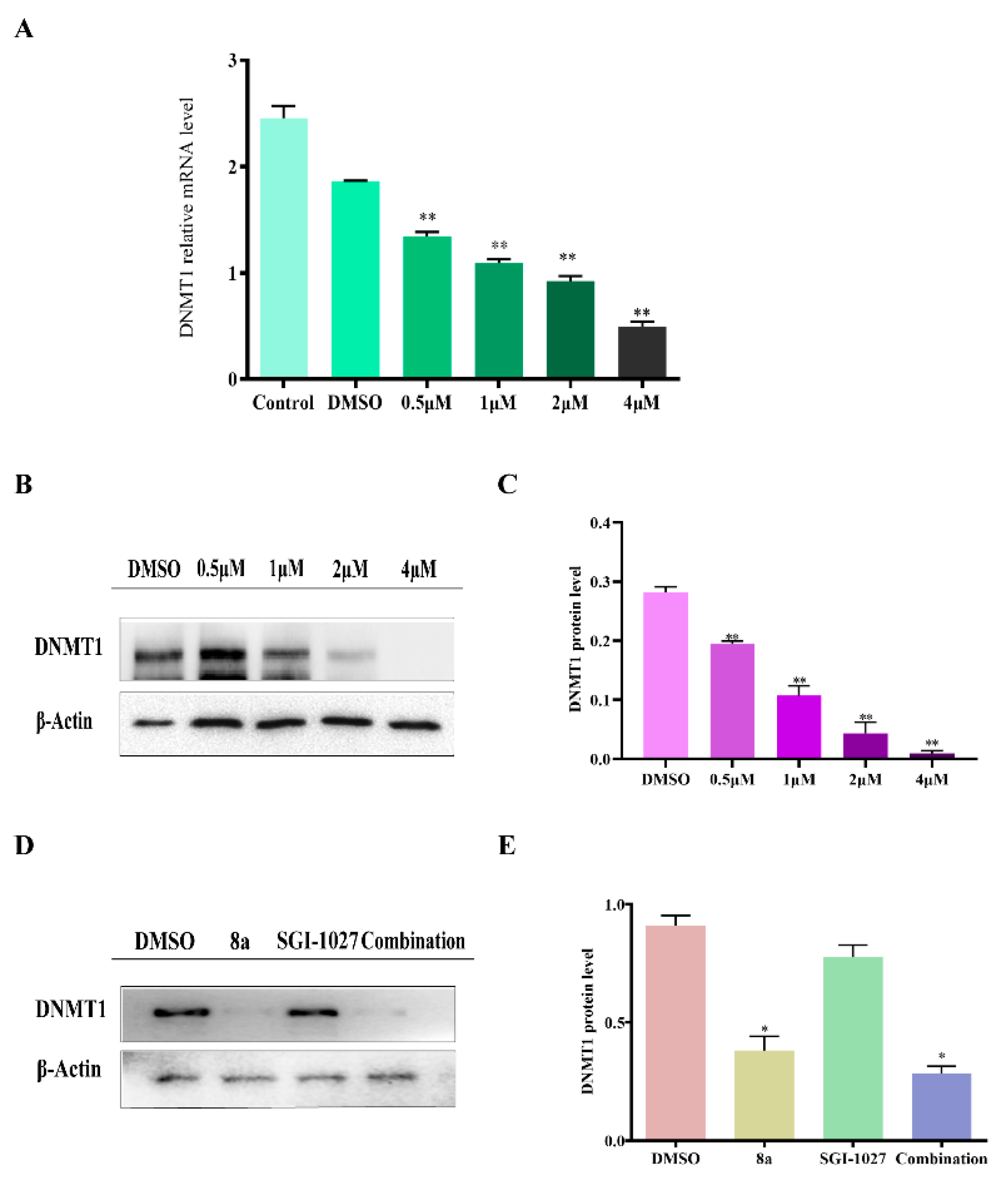
2.5. 8a Affected the Expression of DNMT1
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Viability Assay
4.3. Cell Co-Localization
4.4. Western Blot
4.5. Apoptosis Assay
4.6. ATP Assay Detection
4.7. Mitochondrial Potentiometric Membrane Detection
4.8. Reactive Oxygen Detection
4.9. Proteomics
4.10. Molecular Docking
4.11. Quantitative RT-PCR
4.12. Binding Specificity and Affinity Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peserico, A.; Simone, C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, D.; Li, C.; Jiang, J.; Huang, J.; Wang, J.; He, Z.; Zhang, J. Design, synthesis and biological evaluation of novel HDAC inhibitors with improved pharmacokinetic profile in breast cancer. Eur. J. Med. Chem. 2020, 205, 112648. [Google Scholar] [CrossRef]
- Matthews, G.M.; Mehdipour, P.; Cluse, L.A.; Falkenberg, K.J.; Wang, E.; Roth, M.; Santoro, F.; Vidacs, E.; Stanley, K.; House, C.M.; et al. Functional-genetic dissection of HDAC dependencies in mouse lymphoid and myeloid malignancies. Blood 2015, 126, 2392–2403. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Geng, M. Exploiting histone deacetylases for cancer therapy: From hematological malignancies to solid tumors. Sci. China Life Sci. 2017, 60, 94–97. [Google Scholar] [CrossRef] [Green Version]
- Carew, J.S.; Giles, F.J.; Nawrocki, S.T. Histone deacetylase inhibitors: Mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008, 269, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Siddiqui, H.; Solomon, D.A.; Gunawardena, R.W.; Wang, Y.; Knudsen, E.S. Histone deacetylation of RB-responsive promoters: Requisite for specific gene repression but dispensable for cell cycle inhibition. Mol. Cell Biol. 2003, 23, 7719–7731. [Google Scholar] [CrossRef] [Green Version]
- Herr, D.J.; Baarine, M.; Aune, S.E.; Li, X.; Ball, L.E.; Lemasters, J.J.; Beeson, C.C.; Chou, J.C.; Menick, D.R. HDAC1 localizes to the mitochondria of cardiac myocytes and contributes to early cardiac reperfusion injury. J. Mol. Cell. Cardiol. 2018, 114, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, S.; Mai, A.; Dyer, M.J.; Cohen, G.M. Inhibition of histone deacetylase class I but not class II is critical for the sensitization of leukemic cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2006, 66, 6785–6792. [Google Scholar] [CrossRef] [Green Version]
- Alhosin, M.; Omran, Z.; Zamzami, M.A.; Al-Malki, A.L.; Choudhry, H.; Mousli, M.; Bronner, C. Signalling pathways in UHRF1-dependent regulation of tumor suppressor genes in cancer. J. Exp. Clin. Cancer Res. 2016, 35, 174. [Google Scholar] [CrossRef] [Green Version]
- Sanaei, M.; Kavoosi, F. Histone Deacetylases and Histone Deacetylase Inhibitors: Molecular Mechanisms of Action in Various Cancers. Adv. Biomed. Res. 2019, 8, 63. [Google Scholar] [CrossRef]
- Hatzimichael, E.; Crook, T. Cancer epigenetics: New therapies and new challenges. J. Drug Deliv. 2013, 2013, 529312. [Google Scholar] [CrossRef] [Green Version]
- Abou Najem, S.; Khawaja, G.; Hodroj, M.H.; Babikian, P.; Rizk, S. Adjuvant epigenetic therapy of decitabine and suberoylanilide hydroxamic acid exerts anti-neoplastic effects in acute myeloid leukemia cells. Cells 2019, 8, 1480. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Sun, J.; Chen, W.; He, X.; Zhu, Y.; Dong, H.; Wang, H.; Li, Z.; Zhang, L.; Khaled, S.; et al. HDAC4 inhibition disrupts TET2 function in high-risk MDS and AML. Aging 2020, 12, 16759–16774. [Google Scholar] [CrossRef]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, S.; Özkan, T.; Özmen, F.; Baran, Y.; Sunguroğlu, A.; Kansu, E. The roles of epigenetic modifications of proapoptotic BID and BIM genes in imatinib-resistant chronic myeloid leukemia cells. Hematology 2013, 18, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS ONE 2014, 9, e87455. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. Overview of cancer epigenetics. Semin. Hematol. 2005, 42, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Nolan, L.; Johnson, P.W.; Ganesan, A.; Packham, G.; Crabb, S.J. Will histone deacetylase inhibitors require combination with other agents to fulfil their therapeutic potential? Br. J. Cancer 2008, 99, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Guerrant, W.; Patil, V.; Canzoneri, J.C.; Oyelere, A.K. Dual targeting of histone deacetylase and topoisomerase II with novel bifunctional inhibitors. J. Med. Chem. 2012, 55, 1465–1477. [Google Scholar] [CrossRef] [Green Version]
- Ting, L.Y.; Tan, Y.J.; Ein, O.C. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar]
- Zhang, B.; Wang, N.; Zhang, C.; Gao, C.; Zhang, W.; Chen, K.; Wu, W.; Chen, Y.; Tan, C.; Liu, F.; et al. Novel multi-substituted benzyl acridone derivatives as survivin inhibitors for hepatocellular carcinoma treatment. Eur. J. Med. Chem. 2017, 129, 337–348. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Q.; Liu, Z.; Wang, N.; Jin, H.; Liu, F.; Zhang, C.; He, S. Synthesis and anticancer research of N-(2-aminophenyl) benzamide acridine derivatives as dual topoisomerase I and isoform-selective HDAC inhibitors. ChemistrySelect 2020, 5, 8311–8318. [Google Scholar] [CrossRef]
- Neumann, H.; Hancock, S.M.; Buning, R.; Routh, A.; Chapman, L.; Somers, J.; Owen-Hughes, T.; van Noort, J.; Rhodes, D.; Chin, J.W. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 2009, 36, 153–163. [Google Scholar] [CrossRef]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Faraco, G.; Pancani, T.; Formentini, L.; Mascagni, P.; Fossati, G.; Leoni, F.; Moroni, F.; Chiarugi, A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol. Pharmacol. 2006, 70, 1876–1884. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, X.J.; Li, T.; Li, J.; Tang, Y.; Le, W. Valproic acid reduces neuritic plaque formation and improves learning deficits in APP(Swe) /PS1(A246E) transgenic mice via preventing the prenatal hypoxia-induced down-regulation of neprilysin. CNS Neurosci Ther. 2014, 20, 209–217. [Google Scholar] [CrossRef]
- Guo, X.; Ruan, H.; Li, X.; Qin, L.; Tao, Y.; Qi, X.; Gao, J.; Gan, L.; Duan, S.; Shen, W. Subcellular localization of class I histone deacetylases in the developing xenopus tectum. Front. Cell. Neurosci. 2015, 9, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garner, D.L.; Thomas, C.A. Organelle-specific probe JC-1 identifies membrane potential differences in the mitochondrial function of bovine sperm. Mol. Reprod. Dev. 1999, 53, 222–229. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, X.C.; Huang, Y.Y.; Ge, Y.P.; Sun, M.; Chen, W.L.; Liu, W.B.; Li, X.F. Carbonyl cyanide 3-chlorophenylhydrazone induced the imbalance of mitochondrial homeostasis in the liver of Megalobrama amblycephala: A dynamic study. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2021, 244, 109003. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Eot-Houllier, G.; Fulcrand, G.; Magnaghi-Jaulin, L.; Jaulin, C. Histone deacetylase inhibitors and genomic instability. Cancer Lett. 2009, 274, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.X.; Liu, R.; Wang, Q.; Li, B.S.; Xu, X.X.; Hu, M.; Chen, L.; Fu, Q.; Pu, D.M.; Hong, L. Realgar-induced apoptosis of cervical cancer cell line Siha via cytochrome c release and caspase-3 and caspase-9 activation. Chin. J. Integr. Med. 2012, 18, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Wieder, T.; Essmann, F.; Prokop, A.; Schmelz, K.; Schulze-Osthoff, K.; Beyaert, R.; Dörken, B.; Daniel, P.T. Activation of caspase-8 in drug-induced apoptosis of B-lymphoid cells is independent of CD95/Fas receptor-ligand interaction and occurs downstream of caspase-3. Blood 2001, 97, 1378–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajah, T.; Chow, S.C. The inhibition of human T cell proliferation by the caspase inhibitor z-VAD-FMK is mediated through oxidative stress. Toxicol. Appl. Pharmacol. 2014, 278, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.M.; Kuo, H.C.; Tseng, T.H.; Liu, J.Y.; Chu, C.Y. Berberine induces apoptosis through a mitochondria/caspases pathway in human hepatoma cells. Arch Toxicol. 2006, 80, 62–73. [Google Scholar] [CrossRef]
- García-Domínguez, P.; Dell’aversana, C.; Alvarez, R.; Altucci, L.; de Lera, A.R. Synthetic approaches to DNMT inhibitor SGI-1027 and effects on the U937 leukemia cell line. Bioorganic Med. Chem. Lett. 2013, 23, 1631–1635. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Ghoshal, K.; Denny, W.A.; Gamage, S.A.; Brooke, D.G.; Phiasivongsa, P.; Redkar, S.; Jacob, S.T. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009, 69, 4277–4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, C.; Xie, Y.K.; Chen, Y.J. Efflect of miR-34a-tangeted regulation of HDACI on apoptosis of AML cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2019, 27, 790–795. [Google Scholar] [CrossRef]
- New, M.; Olzscha, H.; La Thangue, N.B. HDAC inhibitor-based therapies: Can we interpret the code? Mol. Oncol. 2012, 6, 637–656. [Google Scholar] [CrossRef] [Green Version]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic regulators of the immune system: Impacts on cancer therapy and inflammatory diseases. Biomed. Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, M.; Kornbluth, S. Caspases and kinases in a death grip. Cell 2009, 138, 838–854. [Google Scholar] [CrossRef] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Dai, Y.; Grant, S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol. Cancer Ther. 2003, 2, 1273–1284. [Google Scholar]
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S.; et al. Combined inhibition of DNMT and HDAC blocks the tumorigenicity of cancer stem-like cells and attenuates mammary tumor growth. Cancer Res. 2016, 76, 3224–3235. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Zhao, R.; Ma, Y.; Wang, D.; Yan, C.; Zhou, D.; Yin, F.; Li, Z. The development of epigenetics and related inhibitors for targeted drug design in cancer therapy. Curr. Top. Med. Chem. 2018, 18, 2380–2394. [Google Scholar] [CrossRef]
- Pandey, M.; Sultana, S.; Gupta, K.P. Involvement of epigenetics and microRNA-29b in the urethane induced inception and establishment of mouse lung tumors. Exp. Mol. Pathol. 2014, 96, 61–70. [Google Scholar] [CrossRef]
- Kaur, J.; Daoud, A.; Eblen, S.T. Targeting chromatin remodeling for cancer therapy. Curr. Mol. Pharmacol. 2019, 12, 215–229. [Google Scholar] [CrossRef]
- Ghosh, R.; Gilda, J.E.; Gomes, A.V. The necessity of and strategies for improving confidence in the accuracy of western blots. Expert Rev. Proteom. 2014, 11, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Pi, W.S.; Cao, Z.Y.; Liu, J.M.; Peng, A.F.; Chen, W.Z.; Chen, J.W.; Huang, S.H.; Liu, Z.L. Potential molecular mechanisms of AURKB in the oncogenesis and progression of osteosarcoma cells: A Label-Free quantitative proteomics analysis. Technol. Cancer Res. Treat. 2018, 18. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Wang, Z.; Chen, X.; Qiu, H.; Gu, Y.; Wang, N.; Wang, T.; Wang, Z.; Ma, H.; Zhao, Y.; et al. 8a, a New Acridine Antiproliferative and Pro-Apoptotic Agent Targeting HDAC1/DNMT1. Int. J. Mol. Sci. 2021, 22, 5516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115516
Zhang Q, Wang Z, Chen X, Qiu H, Gu Y, Wang N, Wang T, Wang Z, Ma H, Zhao Y, et al. 8a, a New Acridine Antiproliferative and Pro-Apoptotic Agent Targeting HDAC1/DNMT1. International Journal of Molecular Sciences. 2021; 22(11):5516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115516
Chicago/Turabian StyleZhang, Qiting, Ziyan Wang, Xinyuan Chen, Haoxiang Qiu, Yifan Gu, Ning Wang, Tao Wang, Ze Wang, Huabin Ma, Yufen Zhao, and et al. 2021. "8a, a New Acridine Antiproliferative and Pro-Apoptotic Agent Targeting HDAC1/DNMT1" International Journal of Molecular Sciences 22, no. 11: 5516. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115516