
Effects of Atypical Antipsychotics, Clozapine, Quetiapine and Brexpiprazole on Astroglial Transmission Associated with Connexin43


Abstract
:1. Introduction
2. Results
2.1. Effects of Mood-Stabilizing Antipsychotics on Astroglial l-Glutamate Release
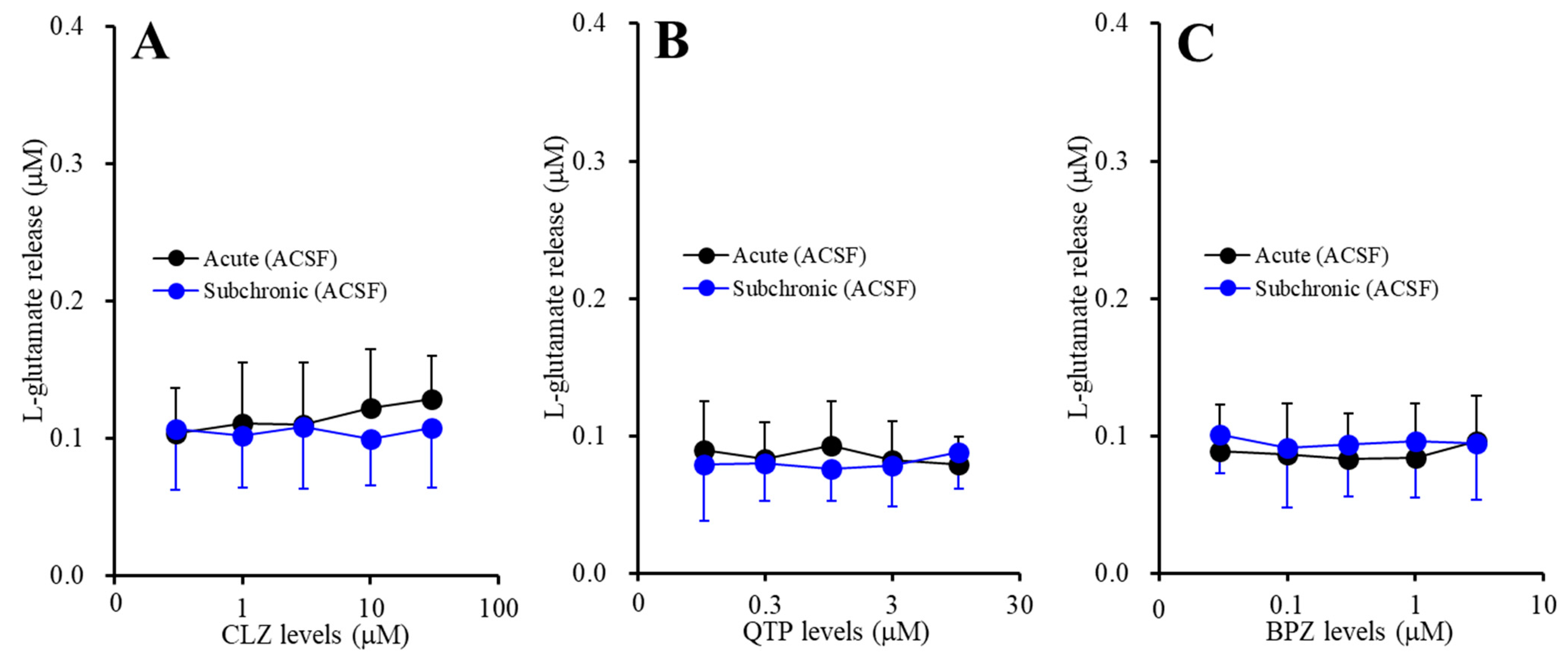
2.1.1. Concentration-Dependent Effects of Acute and Subchronic Administration of Mood-Stabilizing Antipsychotics on Astroglial l-glutamate Release during Resting Stage (Study-1)
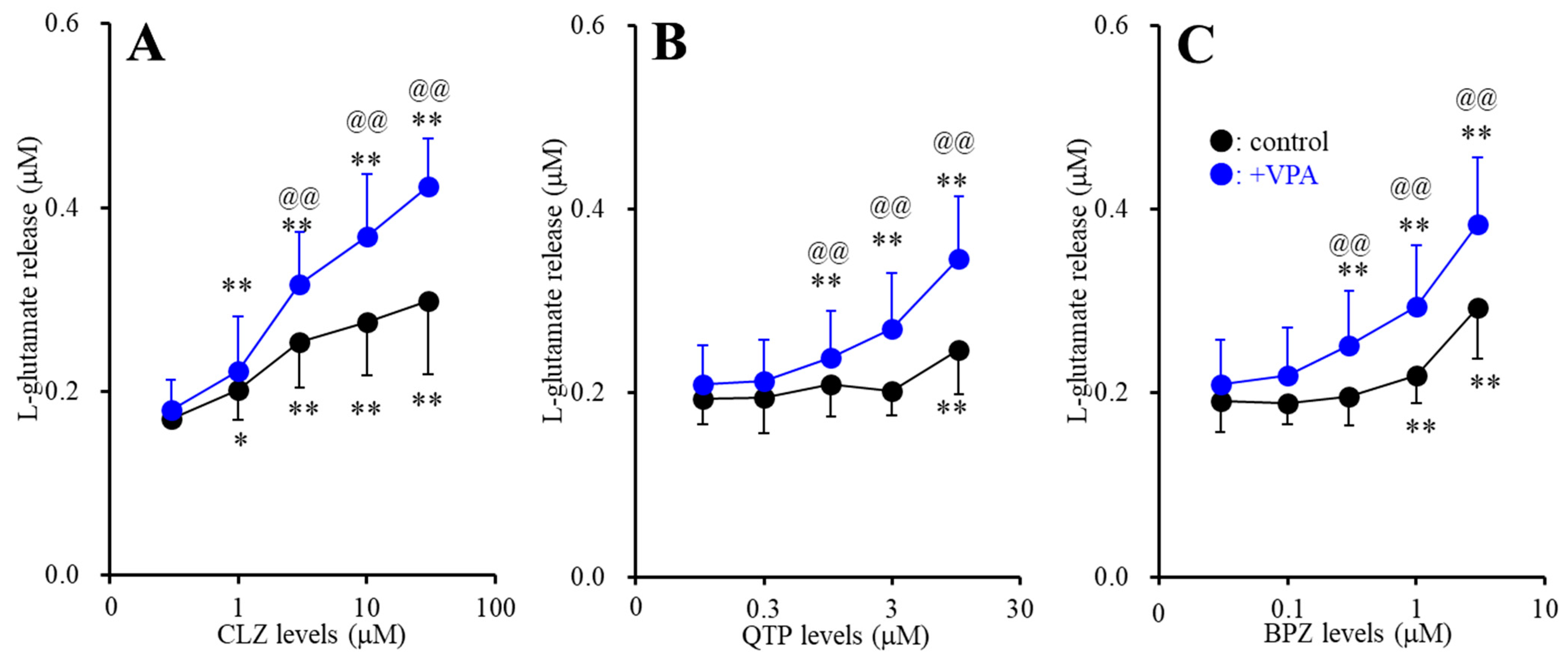
2.1.2. Interaction between Subchronic Administration of Therapeutic-Relevant Concentration of Valproate (VPA) and Acute Administration of Antipsychotics on l-Glutamate Release through Activated Hemichannel (Study-2)
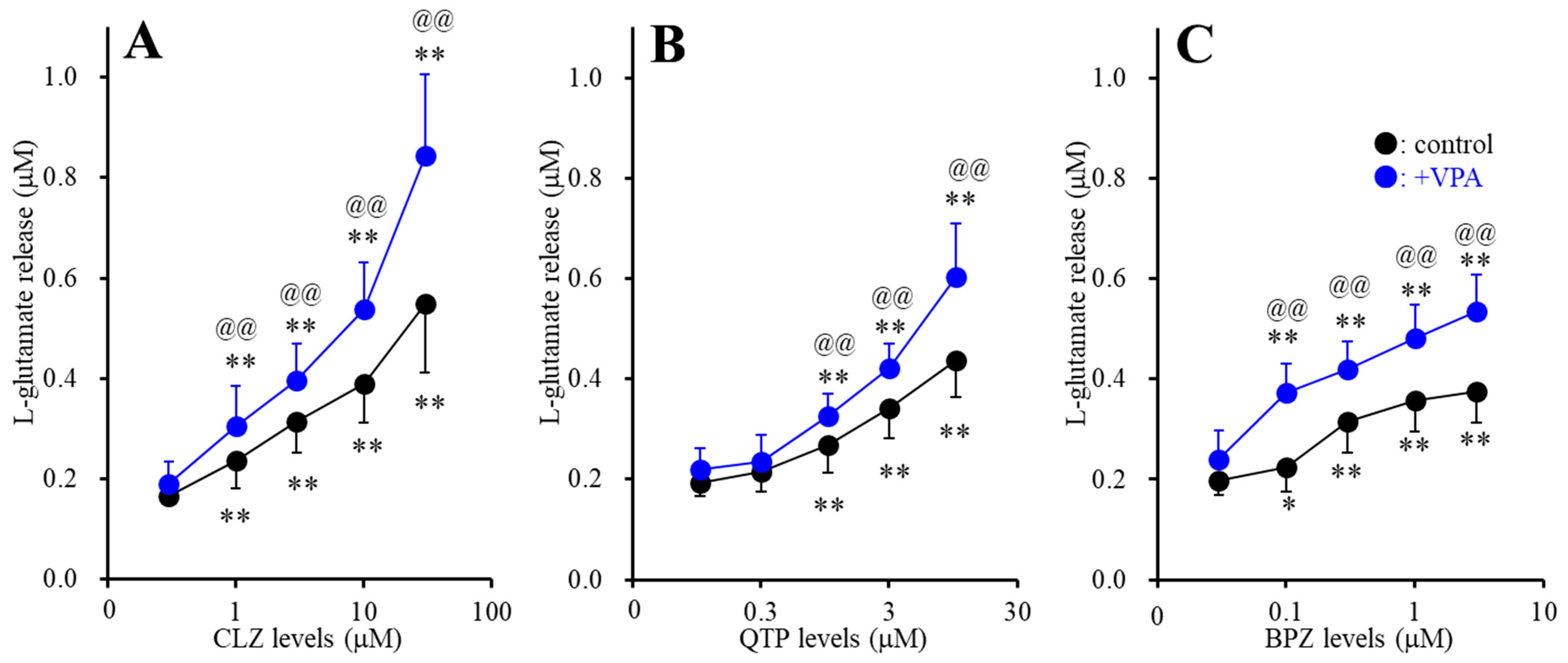
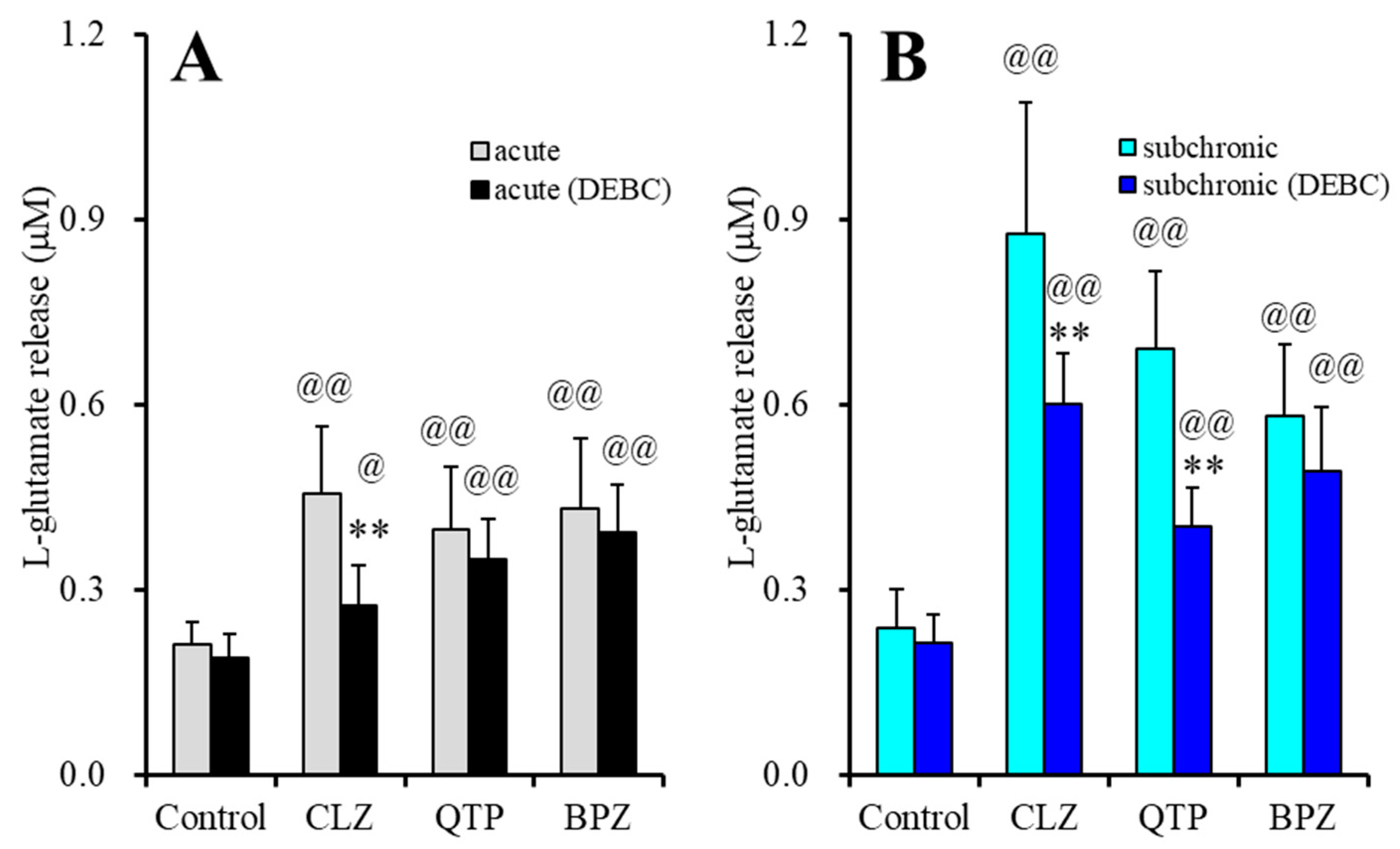
2.1.3. Interaction between Subchronic Administrations of Therapeutic-Relevant Concentration of VPA and Antipsychotics on l-Glutamate Release through Activated Hemichannel (Study-3)
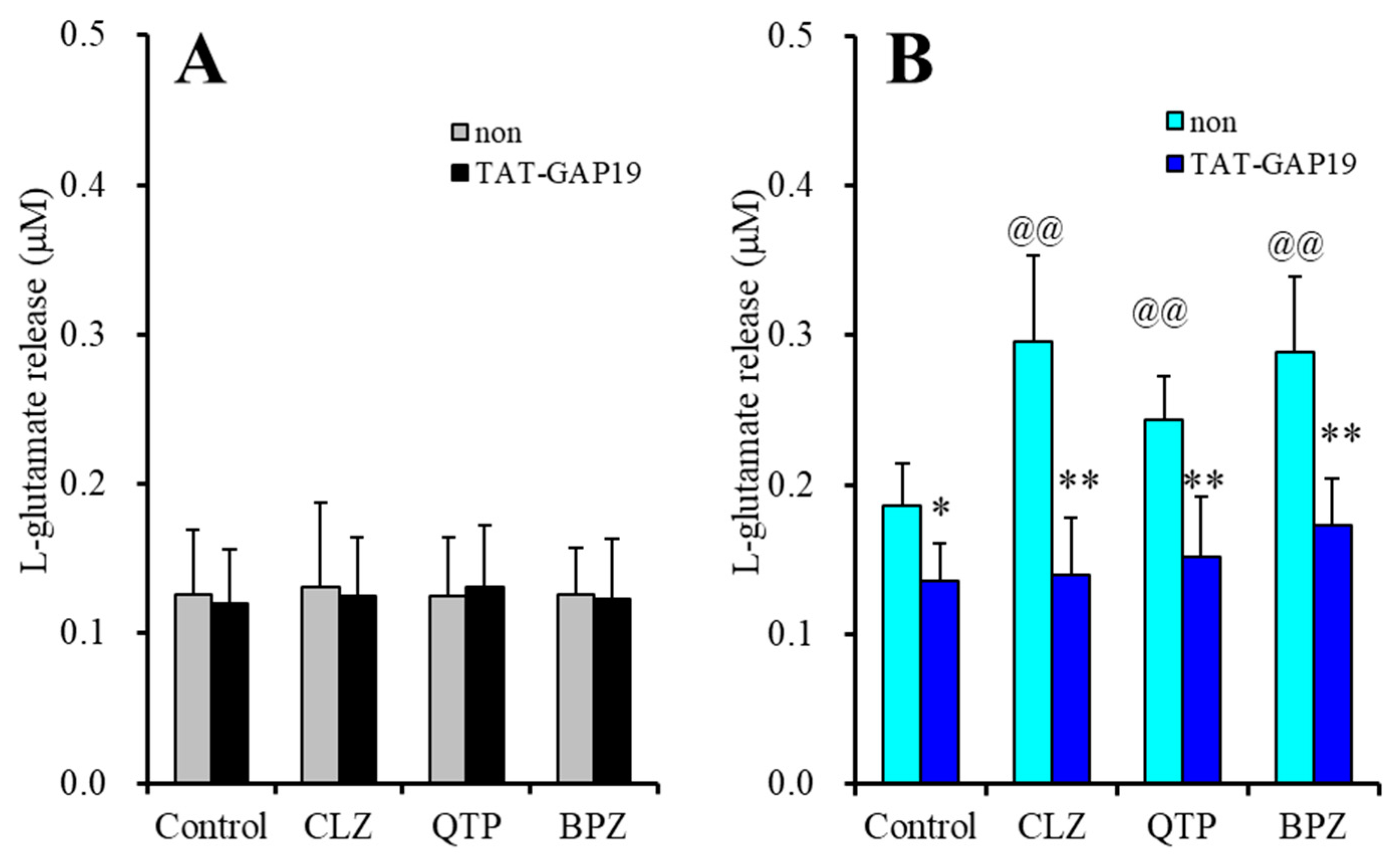
2.1.4. Effects of Cx43 Inhibitor on Astroglial l-glutamate Release through Activated Hemichannel Enhanced by Acute Administration of Antipsychotics (Study-4)
2.1.5. Effects of Acute Administration of Protein Kinase B (Akt) Inhibitor on Astroglial l-glutamate Release through Activated Hemichannel Enhanced by Acute and Subchronic Administration of Antipsychotics, after the Subchronic Administration of Therapeutic-Relevant Concentration of VPA (Study-5)
2.2. Effects of Mood-Stabilizing Antipsychotics on Expression of Cx43 Protein in the Astroglial Plasma Membrane Fraction
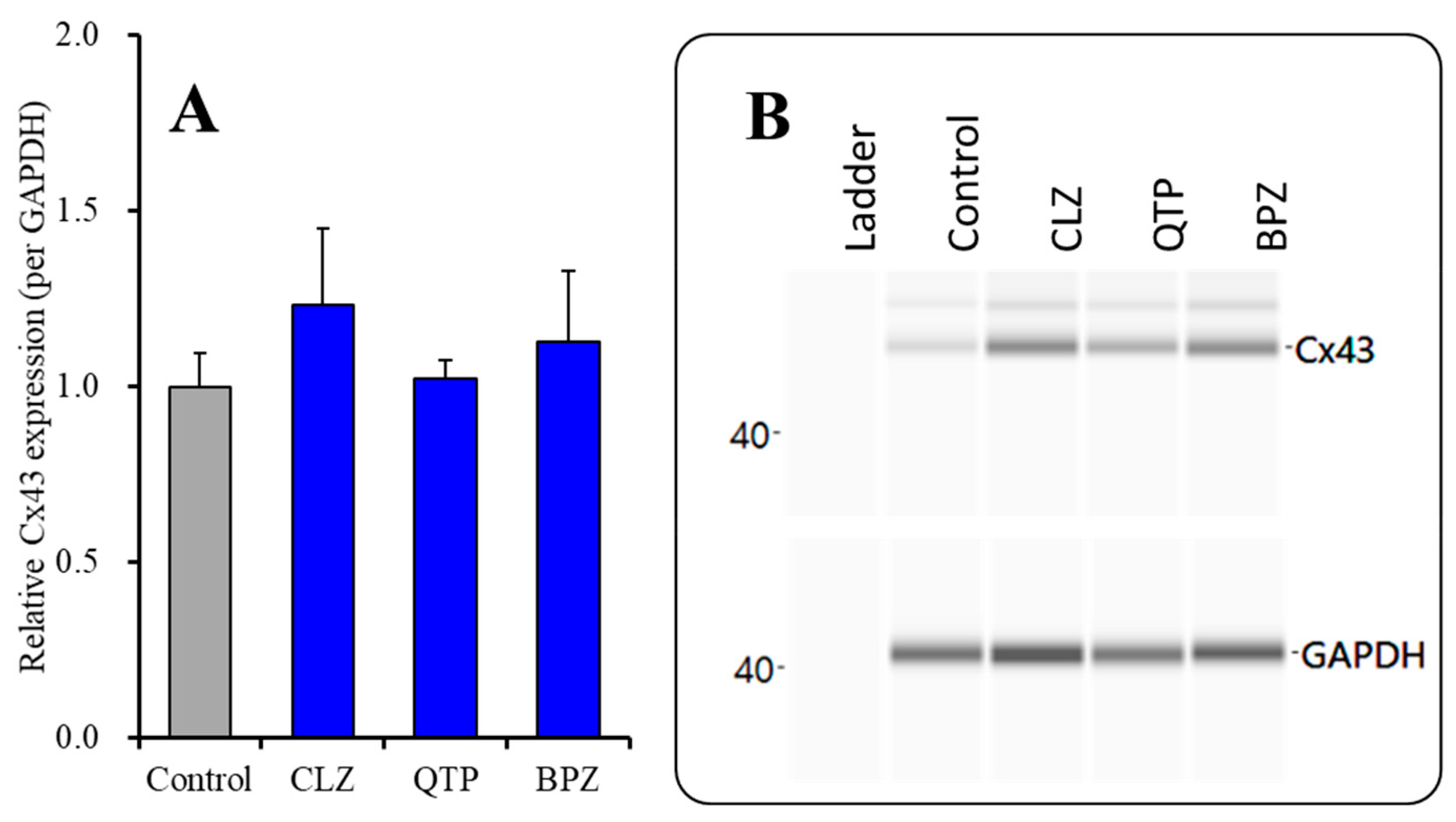
2.2.1. Effects of Subchronic Administration of Therapeutic-Relevant Concentrations of Antipsychotics alone on Cx43 Expression in the Plasma Membrane Fraction
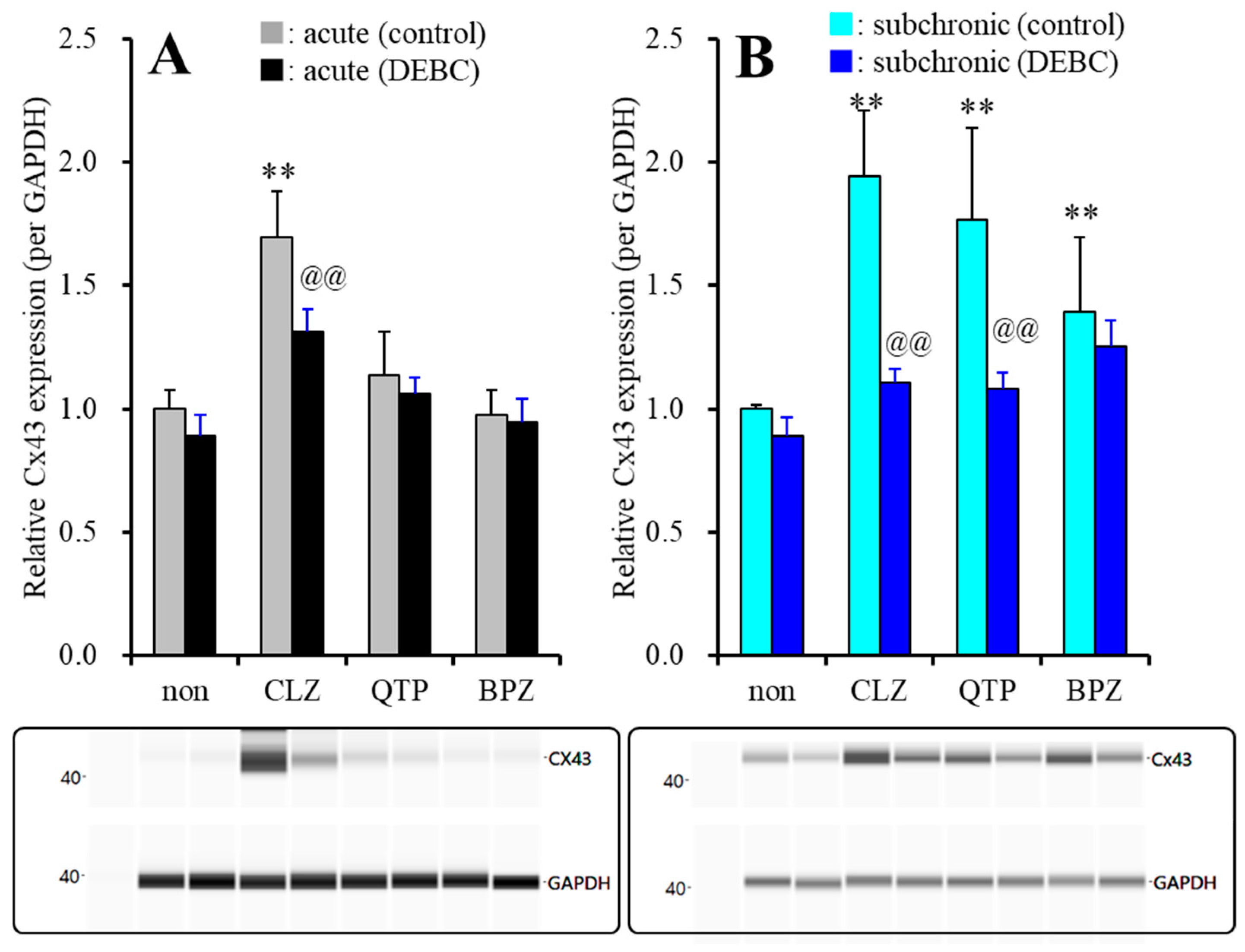
2.2.2. Effects of Acute and Subchronic Administrations of Therapeutic-Relevant Concentration of Antipsychotics on Cx43 Expression in the Astroglial Plasma Membrane, after Subchronic Administration of Therapeutic-Relevant Concentration of VPA
3. Discussion
3.1. Mechanisms of Mood-Stabilizing Antipsychotics, CLZ, QTP and BPZ, on Astroglial l-Glutamate Release through Hemichannel
3.2. Mechanisms of Clinical Action of Mood-Stabilizing Antipsychotics Associated with Cx43
4. Materials and Methods
4.1. Chemical Agents
4.2. Preparation of Primary Astrocyte Culture
4.3. Ultra-High-Performance Liquid Chromatography (UHPLC)
4.4. Capillary Immunoblotting Analysis
4.5. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Okubo, R.; Hasegawa, T.; Fukuyama, K.; Shiroyama, T.; Okada, M. Current limitations and candidate potential of 5-ht7 receptor antagonism in psychiatric pharmacotherapy. Front. Psychiatry 2021, 12, 623684. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Oka, T.; Nakamoto, M.; Fukuyama, K.; Shiroyama, T. Astroglial connexin43 as a potential target for a mood stabiliser. Int. J. Mol. Sci. 2020, 22, 339. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Murata, M. A working hypothesis regarding identical pathomechanisms between clinical efficacy and adverse reaction of clozapine via the activation of connexin43. Int. J. Mol. Sci. 2020, 21, 7019. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Li, Z.; Kaneda, Y.; Ichikawa, J. Serotonin receptors: Their key role in drugs to treat schizophrenia. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2003, 27, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Massey, B.W. The role of serotonin receptors in the action of atypical antipsychotic drugs. Curr. Opin. Pharmacol. 2011, 11, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kawano, Y.; Fukuyama, K.; Motomura, E.; Shiroyama, T. Candidate strategies for development of a rapid-acting antidepressant class that does not result in neuropsychiatric adverse effects: Prevention of ketamine-induced neuropsychiatric adverse reactions. Int. J. Mol. Sci. 2020, 21, 7951. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Matsumoto, R.; Yamamoto, Y.; Fukuyama, K. Effects of subchronic administrations of vortioxetine, lurasidone, and escitalopram on thalamocortical glutamatergic transmission associated with serotonin 5-ht7 receptor. Int. J. Mol. Sci 2021, 22, 1351. [Google Scholar] [CrossRef]
- Fukuyama, K.; Ueda, Y.; Okada, M. Effects of carbamazepine, lacosamide and zonisamide on gliotransmitter release associated with activated astroglial hemichannels. Pharmaceuticals 2020, 13, 117. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of astroglial connexin is involved in concentration-dependent double-edged sword clinical action of clozapine. Cells 2020, 9, 414. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Okada, M. Age-dependent and sleep/seizure-induced pathomechanisms of autosomal dominant sleep-related hypermotor epilepsy. Int. J. Mol. Sci. 2020, 21, 8142. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Ruri, O.; Okada, M. Upregulated connexin 43 induced by loss-of-functional s284l-mutant alpha4 subunit of nicotinic ach receptor contributes to pathomechanisms of autosomal dominant sleep-related hypermotor epilepsy. Pharmaceuticals 2020, 13, 58. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Fukuzawa, M.; Okada, M. Upregulated and hyperactivated thalamic connexin 43 plays important roles in pathomechanisms of cognitive impairment and seizure of autosomal dominant sleep-related hypermotor epilepsy with s284l-mutant α4 subunit of nicotinic ach receptor. Pharmaceuticals 2020, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Carbamazepine attenuates astroglial l-glutamate release induced by pro-inflammatory cytokines via chronically activation of adenosine a2a receptor. Int. J. Mol. Sci. 2019, 20, 3727. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by nmda receptor antagonism via activation of system xc−. Pharmacol. Res. Perspect. 2019, 7, e00457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Hasegawa, T.; Suzuki, D.; Motomura, E.; Okada, M. Amantadine combines astroglial system xc− activation with glutamate/nmda receptor inhibition. Biomolecules 2019, 9, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine normalizes a glutamatergic transmission abnormality induced by an impaired nmda receptor in the thalamocortical pathway via the activation of a group iii metabotropic glutamate receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, K.; Okada, M. Effects of levetiracetam on astroglial release of kynurenine-pathway metabolites. Br. J. Pharmacol. 2018, 175, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/glutamate antiporter and aripiprazole compensate nmda antagonist-induced dysfunction of thalamocortical l-glutamatergic transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, K.; Tanahashi, S.; Hoshikawa, M.; Shinagawa, R.; Okada, M. Zonisamide regulates basal ganglia transmission via astroglial kynurenine pathway. Neuropharmacology 2014, 76, 137–145. [Google Scholar] [CrossRef]
- Yamamura, S.; Hoshikawa, M.; Dai, K.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. Ono-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharmacol. 2013, 168, 1088–1100. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial d-serine and l-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M. Can rodent models elucidate pathomechanisms of genetic epilepsy? Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Jeanson, T.; Pondaven, A.; Ezan, P.; Mouthon, F.; Charveriat, M.; Giaume, C. Antidepressants impact connexin 43 channel functions in astrocytes. Front. Cell. Neurosci. 2015, 9, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Gangoso, E.; Yi, C.; Jeanson, T.; Kandelman, S.; Mantz, J.; Giaume, C. General anesthetics have differential inhibitory effects on gap junction channels and hemichannels in astrocytes and neurons. Glia 2016, 64, 524–536. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with s284l-mutant alpha4 subunit of nicotinic ach receptor. Br. J. Pharmacol. 2020, 177, 2143–2162. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-D.; Liu, Y.; Yuan, Y.-H.; Li, J.; Chen, N.-H. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology 2012, 37, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Moraga-Amaro, R.; Diaz-Galarce, R.; Rojas, S.; Maturana, C.J.; Stehberg, J.; Saez, J.C. Restraint stress increases hemichannel activity in hippocampal glial cells and neurons. Front. Cell. Neurosci. 2015, 9, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfeld, T.J.; Kloth, A.D.; Hsueh, B.; Runkle, M.B.; Kane, G.A.; Wang, S.S.; Gould, E. Gap junctions in the ventral hippocampal-medial prefrontal pathway are involved in anxiety regulation. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 15679–15688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathomechanism of nocturnal paroxysmal dystonia in autosomal dominant sleep-related hypermotor epilepsy with s284l-mutant α4 subunit of nicotinic ach receptor. Biomed. Pharmacother. 2020, 126, 110070. [Google Scholar] [CrossRef]
- Bernard, R.; Kerman, I.A.; Thompson, R.C.; Jones, E.G.; Bunney, W.E.; Barchas, J.D.; Schatzberg, A.F.; Myers, R.M.; Akil, H.; Watson, S.J. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol. Psychiatry 2011, 16, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Ernst, C.; Nagy, C.; Kim, S.; Yang, J.P.; Deng, X.; Hellstrom, I.C.; Choi, K.H.; Gershenfeld, H.; Meaney, M.J.; Turecki, G. Dysfunction of astrocyte connexins 30 and 43 in dorsal lateral prefrontal cortex of suicide completers. Biol. Psychiatry 2011, 70, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.; Torres-Platas, S.G.; Mechawar, N.; Turecki, G. Repression of astrocytic connexins in cortical and subcortical brain regions and prefrontal enrichment of h3k9me3 in depression and suicide. Int. J. Neuropsychopharmacol. 2017, 20, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel-Hidalgo, J.J.; Wilson, B.A.; Hussain, S.; Meshram, A.; Rajkowska, G.; Stockmeier, C.A. Reduced connexin 43 immunolabeling in the orbitofrontal cortex in alcohol dependence and depression. J. Psychiatr. Res. 2014, 55, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Lurasidone inhibits nmda antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-ht7 receptor blockade. Br. J. Pharmacol. 2019, 176, 4002–4018. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone sub-chronically activates serotonergic transmission via desensitization of 5-ht1a and 5-ht7 receptors in dorsal raphe nucleus. Pharmaceuticals 2019, 12, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Nakano, T.; Ueda, Y. Pharmacological discrimination of effects of mk801 on thalamocortical, mesothalamic, and mesocortical transmissions. Biomolecules 2019, 9, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel-Hidalgo, J.J.; Moulana, M.; Deloach, P.H.; Rajkowska, G. Chronic unpredictable stress reduces immunostaining for connexins 43 and 30 and myelin basic protein in the rat prelimbic and orbitofrontal cortices. Chronic Stress 2018, 2, 2470547018814186. [Google Scholar] [CrossRef]
- Jin, C.; Wang, Z.Z.; Zhou, H.; Lou, Y.X.; Chen, J.; Zuo, W.; Tian, M.T.; Wang, Z.Q.; Du, G.H.; Kawahata, I.; et al. Ginsenoside rg1-induced antidepressant effects involve the protection of astrocyte gap junctions within the prefrontal cortex. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2017, 75, 183–191. [Google Scholar] [CrossRef]
- Lou, Y.X.; Wang, Z.Z.; Xia, C.Y.; Mou, Z.; Ren, Q.; Liu, D.D.; Zhang, X.; Chen, N.H. The protective effect of ginsenoside rg1 on depression may benefit from the gap junction function in hippocampal astrocytes. Eur. J. Pharmacol. 2020, 882, 173309. [Google Scholar] [CrossRef] [PubMed]
- Quesseveur, G.; Portal, B.; Basile, J.A.; Ezan, P.; Mathou, A.; Halley, H.; Leloup, C.; Fioramonti, X.; Deglon, N.; Giaume, C.; et al. Attenuated levels of hippocampal connexin 43 and its phosphorylation correlate with antidepressant- and anxiolytic-like activities in mice. Front. Cell. Neurosci. 2015, 9, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel-Hidalgo, J.J.; Carter, K.; Deloach, P.H.; Sanders, L.; Pang, Y. Glucocorticoid-induced reductions of myelination and connexin 43 in mixed central nervous system cell cultures are prevented by mifepristone. Neuroscience 2019, 411, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D.; Reutiman, T.J.; Pandian, T.; Braun, N.N.; Haug, K. Chronic psychotropic drug treatment causes differential expression of connexin 43 and gfap in frontal cortex of rats. Schizophr. Res. 2008, 104, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Morioka, N.; Suekama, K.; Zhang, F.F.; Kajitani, N.; Hisaoka-Nakashima, K.; Takebayashi, M.; Nakata, Y. Amitriptyline up-regulates connexin43-gap junction in rat cultured cortical astrocytes via activation of the p38 and c-fos/ap-1 signalling pathway. Br. J. Pharmacol. 2014, 171, 2854–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkowska, G.; Miguel-Hidalgo, J.J.; Wei, J.; Dilley, G.; Pittman, S.D.; Meltzer, H.Y.; Overholser, J.C.; Roth, B.L.; Stockmeier, C.A. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 1999, 45, 1085–1098. [Google Scholar] [CrossRef]
- Ongur, D.; Drevets, W.C.; Price, J.L. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. USA 1998, 95, 13290–13295. [Google Scholar] [CrossRef] [Green Version]
- Cotter, D.; Mackay, D.; Chana, G.; Beasley, C.; Landau, S.; Everall, I.P. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 2002, 12, 386–394. [Google Scholar] [CrossRef]
- Bowley, M.P.; Drevets, W.C.; Ongur, D.; Price, J.L. Low glial numbers in the amygdala in major depressive disorder. Biol. Psychiatry 2002, 52, 404–412. [Google Scholar] [CrossRef]
- Chana, G.; Landau, S.; Beasley, C.; Everall, I.P.; Cotter, D. Two-dimensional assessment of cytoarchitecture in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia: Evidence for decreased neuronal somal size and increased neuronal density. Biol. Psychiatry 2003, 53, 1086–1098. [Google Scholar] [CrossRef]
- Maes, M.; Yirmyia, R.; Noraberg, J.; Brene, S.; Hibbeln, J.; Perini, G.; Kubera, M.; Bob, P.; Lerer, B.; Maj, M. The inflammatory & neurodegenerative (i&nd) hypothesis of depression: Leads for future research and new drug developments in depression. Metab. Brain Dis. 2009, 24, 27–53. [Google Scholar]
- Rajkowska, G.; Selemon, L.D.; Goldman-Rakic, P.S. Neuronal and glial somal size in the prefrontal cortex: A postmortem morphometric study of schizophrenia and huntington disease. Arch. Gen. Psychiatry 1998, 55, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Selemon, L.D.; Rajkowska, G.; Goldman-Rakic, P.S. Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: Application of a three-dimensional, stereologic counting method. J. Comparat. Neurol. 1998, 392, 402–412. [Google Scholar] [CrossRef]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of hdac inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi, T.; Ikuta, T.; Matsuda, Y.; Sakuma, K.; Okuya, M.; Mishima, K.; Iwata, N. Mood stabilizers and/or antipsychotics for bipolar disorder in the maintenance phase: A systematic review and network meta-analysis of randomized controlled trials. Mol. Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Sakuma, K.; Okuya, M.; Matsuda, Y.; Esumi, S.; Hashimoto, Y.; Hatano, M.; Miyake, N.; Miura, I.; Mishima, K.; et al. Effects of a conventional mood stabilizer alone or in combination with second-generation antipsychotics on recurrence rate and discontinuation rate in bipolar i disorder in the maintenance phase: A systematic review and meta-analysis of randomized, placebo-controlled trials. Bipolar Disord. 2021. [Google Scholar] [CrossRef]
- Azorin, J.M.; Simon, N. Dopamine receptor partial agonists for the treatment of bipolar disorder. Drugs 2019, 79, 1657–1677. [Google Scholar] [CrossRef] [PubMed]
- Vieta, E.; Sachs, G.; Chang, D.; Hellsten, J.; Brewer, C.; Peters-Strickland, T.; Hefting, N. Two randomized, double-blind, placebo-controlled trials and one open-label, long-term trial of brexpiprazole for the acute treatment of bipolar mania. J. Psychopharmacol. 2021, 269881120985102. [Google Scholar] [CrossRef]
- Yatham, L.N.; Kennedy, S.H.; Parikh, S.V.; Schaffer, A.; Bond, D.J.; Frey, B.N.; Sharma, V.; Goldstein, B.I.; Rej, S.; Beaulieu, S.; et al. Canadian network for mood and anxiety treatments (canmat) and international society for bipolar disorders (isbd) 2018 guidelines for the management of patients with bipolar disorder. Bipolar Disord. 2018, 20, 97–170. [Google Scholar] [CrossRef]
- Goodwin, G.M.; Haddad, P.M.; Ferrier, I.N.; Aronson, J.K.; Barnes, T.; Cipriani, A.; Coghill, D.R.; Fazel, S.; Geddes, J.R.; Grunze, H.; et al. Evidence-based guidelines for treating bipolar disorder: Revised third edition recommendations from the british association for psychopharmacology. J. Psychopharmacol. 2016, 30, 495–553. [Google Scholar] [CrossRef] [PubMed]
- Verdolini, N.; Hidalgo-Mazzei, D.; Murru, A.; Pacchiarotti, I.; Samalin, L.; Young, A.H.; Vieta, E.; Carvalho, A.F. Mixed states in bipolar and major depressive disorders: Systematic review and quality appraisal of guidelines. Acta Psychiatr. Scand. 2018, 138, 196–222. [Google Scholar] [CrossRef]
- Vasudev, A.; Chaudhari, S.; Sethi, R.; Fu, R.; Sandieson, R.M.; Forester, B.P. A review of the pharmacological and clinical profile of newer atypical antipsychotics as treatments for bipolar disorder: Considerations for use in older patients. Drugs Aging 2018, 35, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Ronaldson, K.J.; Fitzgerald, P.B.; Taylor, A.J.; Topliss, D.J.; Wolfe, R.; McNeil, J.J. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: A case-control study. Schizophr. Res. 2012, 141, 173–178. [Google Scholar] [CrossRef]
- Schoretsanitis, G.; Paulzen, M.; Unterecker, S.; Schwarz, M.; Conca, A.; Zernig, G.; Grunder, G.; Haen, E.; Baumann, P.; Bergemann, N.; et al. Tdm in psychiatry and neurology: A comprehensive summary of the consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology, update 2017; a tool for clinicians. World J. Biol. Psychiatry 2018, 19, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Hiemke, C.; Bergemann, N.; Clement, H.W.; Conca, A.; Deckert, J.; Domschke, K.; Eckermann, G.; Egberts, K.; Gerlach, M.; Greiner, C.; et al. Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: Update 2017. Pharmacopsychiatry 2018, 51, 9–62. [Google Scholar] [PubMed] [Green Version]
- Fasciani, I.; Temperan, A.; Perez-Atencio, L.F.; Escudero, A.; Martinez-Montero, P.; Molano, J.; Gomez-Hernandez, J.M.; Paino, C.L.; Gonzalez-Nieto, D.; Barrio, L.C. Regulation of connexin hemichannel activity by membrane potential and the extracellular calcium in health and disease. Neuropharmacology 2013, 75, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Brivaracetam prevents astroglial l-glutamate release associated with hemichannel through modulation of synaptic vesicle protein. Biomed. Pharmacother. 2021, 138, 111462. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Yamamura, S.; Ohoyama, K.; Nakagawa, M.; Motomura, E.; Kaneko, S.; Okada, M. Effects of valproate on neurotransmission associated with ryanodine receptors. Neurosci. Res. 2010, 68, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Battino, D.; Andermann, E.; Wada, K.; Kan, R.; Takeda, A.; Nakane, Y.; Ogawa, Y.; Avanzini, G.; Fumarola, C.; et al. Congenital malformations due to antiepileptic drugs. Epilepsy Res. 1999, 33, 145–158. [Google Scholar] [CrossRef]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girao, H. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitterauer, B. Loss of function of glial gap junctions may cause severe cognitive impairments in schizophrenia. Med. Hypotheses 2009, 73, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Gawlik, M.; Wagner, M.; Pfuhlmann, B.; Stober, G. The role of pannexin gene variants in schizophrenia: Systematic analysis of phenotypes. Eur. Arch. Psychiatry Clin. Neurosci. 2016, 266, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Huhn, M.; Nikolakopoulou, A.; Schneider-Thoma, J.; Krause, M.; Samara, M.; Peter, N.; Arndt, T.; Backers, L.; Rothe, P.; Cipriani, A.; et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: A systematic review and network meta-analysis. Lancet 2019, 394, 939–951. [Google Scholar] [CrossRef] [Green Version]
- Tiihonen, J.; Mittendorfer-Rutz, E.; Majak, M.; Mehtala, J.; Hoti, F.; Jedenius, E.; Enkusson, D.; Leval, A.; Sermon, J.; Tanskanen, A.; et al. Real-world effectiveness of antipsychotic treatments in a nationwide cohort of 29823 patients with schizophrenia. JAMA Psychiatry 2017, 74, 686–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostacher, M.; Ng-Mak, D.; Patel, P.; Ntais, D.; Schlueter, M.; Loebel, A. Lurasidone compared to other atypical antipsychotic monotherapies for bipolar depression: A systematic review and network meta-analysis. World J. Biol. Psychiatry 2018, 19, 586–601. [Google Scholar] [CrossRef] [PubMed]
- Fornaro, M.; Carvalho, A.F.; Fusco, A.; Anastasia, A.; Solmi, M.; Berk, M.; Sim, K.; Vieta, E.; de Bartolomeis, A. The concept and management of acute episodes of treatment-resistant bipolar disorder: A systematic review and exploratory meta-analysis of randomized controlled trials. J. Affect. Disord. 2020, 276, 970–983. [Google Scholar] [CrossRef]
- Alper, K.; Schwartz, K.A.; Kolts, R.L.; Khan, A. Seizure incidence in psychopharmacological clinical trials: An analysis of food and drug administration (fda) summary basis of approval reports. Biol. Psychiatry 2007, 62, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Wang, S.C.; Yeh, I.J.; Liu, S.K. Comparative risk of seizure with use of first- and second-generation antipsychotics in patients with schizophrenia and mood disorders. J. Clin. Psychiatry 2016, 77, e573–e579. [Google Scholar] [CrossRef]
- Walrave, L.; Vinken, M.; Leybaert, L.; Smolders, I. Astrocytic connexin43 channels as candidate targets in epilepsy treatment. Biomolecules 2020, 10, 1578. [Google Scholar] [CrossRef]
- Hussein, A.M.; Ghalwash, M.; Magdy, K.; Abulseoud, O.A. Beta lactams antibiotic ceftriaxone modulates seizures, oxidative stress and connexin 43 expression in hippocampus of pentylenetetrazole kindled rats. J. Epilepsy Res. 2016, 6, 8–15. [Google Scholar] [CrossRef]
- Das, A.; Wallace, G.C.; Holmes, C.; McDowell, M.L.; Smith, J.A.; Marshall, J.D.; Bonilha, L.; Edwards, J.C.; Glazier, S.S.; Ray, S.K.; et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 2012, 220, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbelli, R.; Frassoni, C.; Condorelli, D.F.; Trovato Salinaro, A.; Musso, N.; Medici, V.; Tassi, L.; Bentivoglio, M.; Spreafico, R. Expression of connexin 43 in the human epileptic and drug-resistant cerebral cortex. Neurology 2011, 76, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Dopamine d2 and serotonin 5-ht1a receptors mediate the actions of aripiprazole in mesocortical and mesoaccumbens transmission. Neuropharmacology 2012, 62, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Park, S.; Kim, D.; Kwon, J.W. Risk of seizures associated with antipsychotic treatment in pediatrics with psychiatric disorders: A nested case-control study in korea. Eur. Child Adolesc. Psychiatry 2021, 30, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, K.D.; Bruhn, C.H.; Pagsberg, A.K.; Fink-Jensen, A.; Nielsen, J. Neurological, metabolic, and psychiatric adverse events in children and adolescents treated with aripiprazole. J. Clin. Psychopharmacol. 2016, 36, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.C.; Pessin, J.E. Ins (endocytosis) and outs (exocytosis) of glut4 trafficking. Curr. Opin. Cell Biol. 2007, 19, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Emamian, E.S.; Hall, D.; Birnbaum, M.J.; Karayiorgou, M.; Gogos, J.A. Convergent evidence for impaired akt1-gsk3beta signaling in schizophrenia. Nat. Genet. 2004, 36, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Bonnafous, S.; Johnston, A.M.; Chaussade, C.; Portis, F.; Van Obberghen, E. Phosphoinositide 3-kinase-mediated reduction of insulin receptor substrate-1/2 protein expression via different mechanisms contributes to the insulin-induced desensitization of its signaling pathways in l6 muscle cells. J. Biol. Chem. 2003, 278, 15641–15651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panariello, F.; Perruolo, G.; Cassese, A.; Giacco, F.; Botta, G.; Barbagallo, A.P.; Muscettola, G.; Beguinot, F.; Formisano, P.; de Bartolomeis, A. Clozapine impairs insulin action by up-regulating akt phosphorylation and ped/pea-15 protein abundance. J. Cell. Physiol. 2012, 227, 1485–1492. [Google Scholar] [CrossRef] [Green Version]
- Frampton, J.E. Brexpiprazole: A review in schizophrenia. Drugs 2019, 79, 189–200. [Google Scholar] [CrossRef]
- Maeda, K.; Sugino, H.; Akazawa, H.; Amada, N.; Shimada, J.; Futamura, T.; Yamashita, H.; Ito, N.; McQuade, R.D.; Mork, A.; et al. Brexpiprazole i: In vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J. Pharmacol. Exp. Ther. 2014, 350, 589–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.M.; Sotnikova, T.D.; Yao, W.D.; Kockeritz, L.; Woodgett, J.R.; Gainetdinov, R.R.; Caron, M.G. Lithium antagonizes dopamine-dependent behaviors mediated by an akt/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. USA 2004, 101, 5099–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.M.; Sotnikova, T.D.; Marion, S.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. An akt/beta-arrestin 2/pp2a signaling complex mediates dopaminergic neurotransmission and behavior. Cell 2005, 122, 261–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Administration | CLZ | QTP | BPZ | Figure | |
|---|---|---|---|---|---|
| Astroglial glutamate release | |||||
| Basal glutamate release | Acute (120 min) | ↑ | → | → | Figure 1 |
| Subchronic (7 days) | → | → | → | Figure 1 | |
| Evoked glutamate release | Acute (120 min) | ↑↑ | ↑ | ↑ | Figure 2 |
| Subchronic (7 days) | ↑↑ | ↑↑ | ↑↑ | Figure 3 | |
| Evoked release | Acute (120 min) | ↑↑ | ↑↑ | ↑↑ | Figure 2 |
| (+subchronic VPA) | Subchronic (7 days) | ↑↑ | ↑↑ | ↑↑ | Figure 3 |
| Akt inhibitor sensitivity of glutamate release | |||||
| Evoked release | Acute (120 min) | ↓ | → | → | Figure 5 |
| (+ subchronic VPA) | Subchronic (7 days) | ↓ | ↓ | → | Figure 5 |
| Cx43 expression in the astroglial plasma membrane | |||||
| (+ subchronic VPA) | Acute (120 min) | ↑↑ | → | → | Figure 7 |
| (+ subchronic VPA) | Subchronic (7 days) | ↑↑ | ↑↑ | ↑↑ | Figure 7 |
| Akt sensitivity of Cx43 expression in the astroglial plasma membrane | |||||
| (+ subchronic VPA) | Acute (120 min) | ↓↓ | → | → | Figure 7 |
| (+ subchronic VPA) | Subchronic (7 days) | ↓↓ | ↓↓ | → | Figure 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuyama, K.; Okada, M. Effects of Atypical Antipsychotics, Clozapine, Quetiapine and Brexpiprazole on Astroglial Transmission Associated with Connexin43. Int. J. Mol. Sci. 2021, 22, 5623. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115623
Fukuyama K, Okada M. Effects of Atypical Antipsychotics, Clozapine, Quetiapine and Brexpiprazole on Astroglial Transmission Associated with Connexin43. International Journal of Molecular Sciences. 2021; 22(11):5623. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115623
Chicago/Turabian StyleFukuyama, Kouji, and Motohiro Okada. 2021. "Effects of Atypical Antipsychotics, Clozapine, Quetiapine and Brexpiprazole on Astroglial Transmission Associated with Connexin43" International Journal of Molecular Sciences 22, no. 11: 5623. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115623