
Diminazene Aceturate Stabilizes Atherosclerotic Plaque and Attenuates Hepatic Steatosis in apoE-Knockout Mice by Influencing Macrophages Polarization and Taurine Biosynthesis

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results
2.1. Influence of DIZE on Atherosclerosis Progression
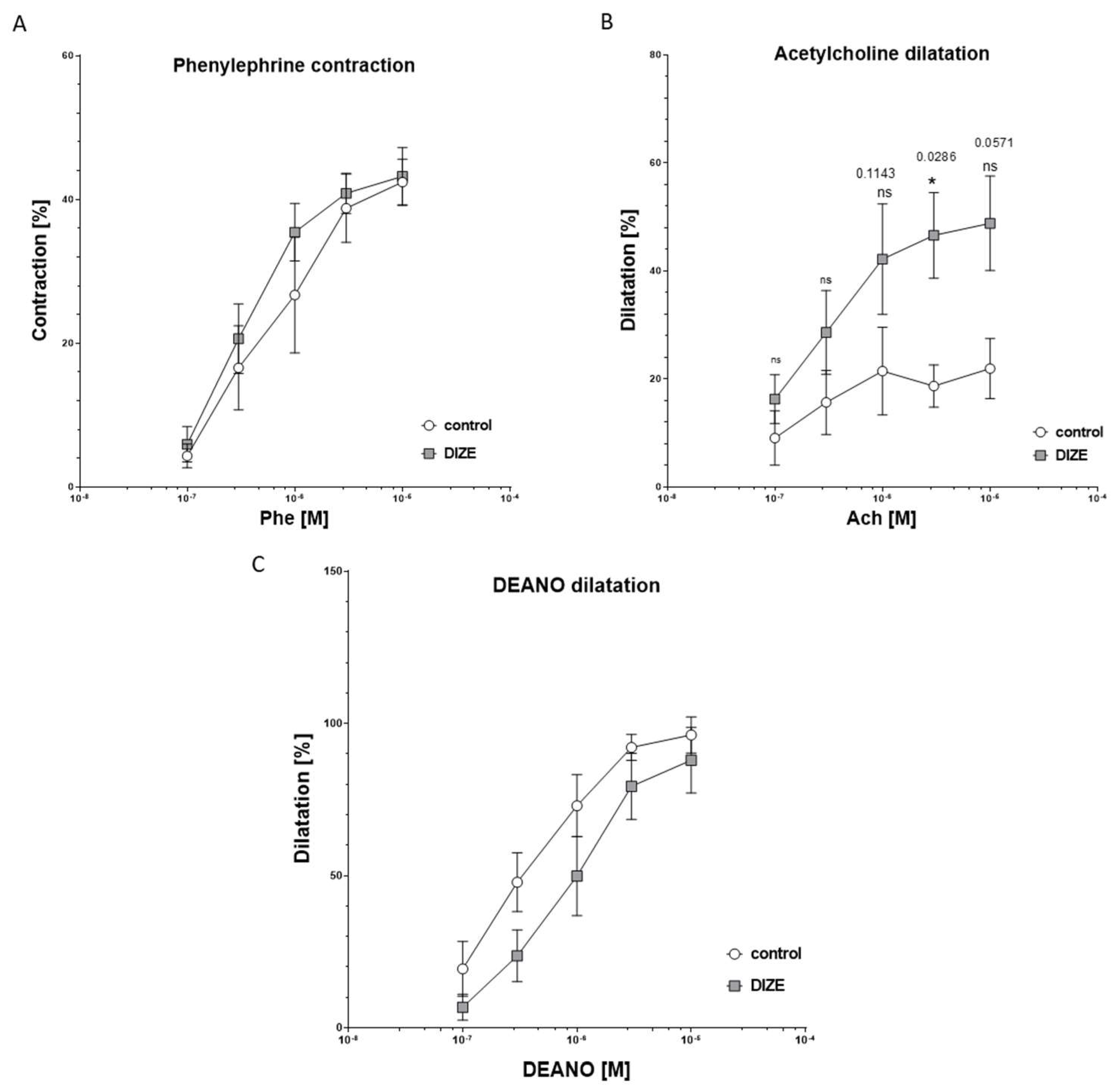
2.2. Influence of DIZE on Mesenteric Arteries Responses Ex Vivo
2.3. Influence of DIZE on Hepatic Steatosis
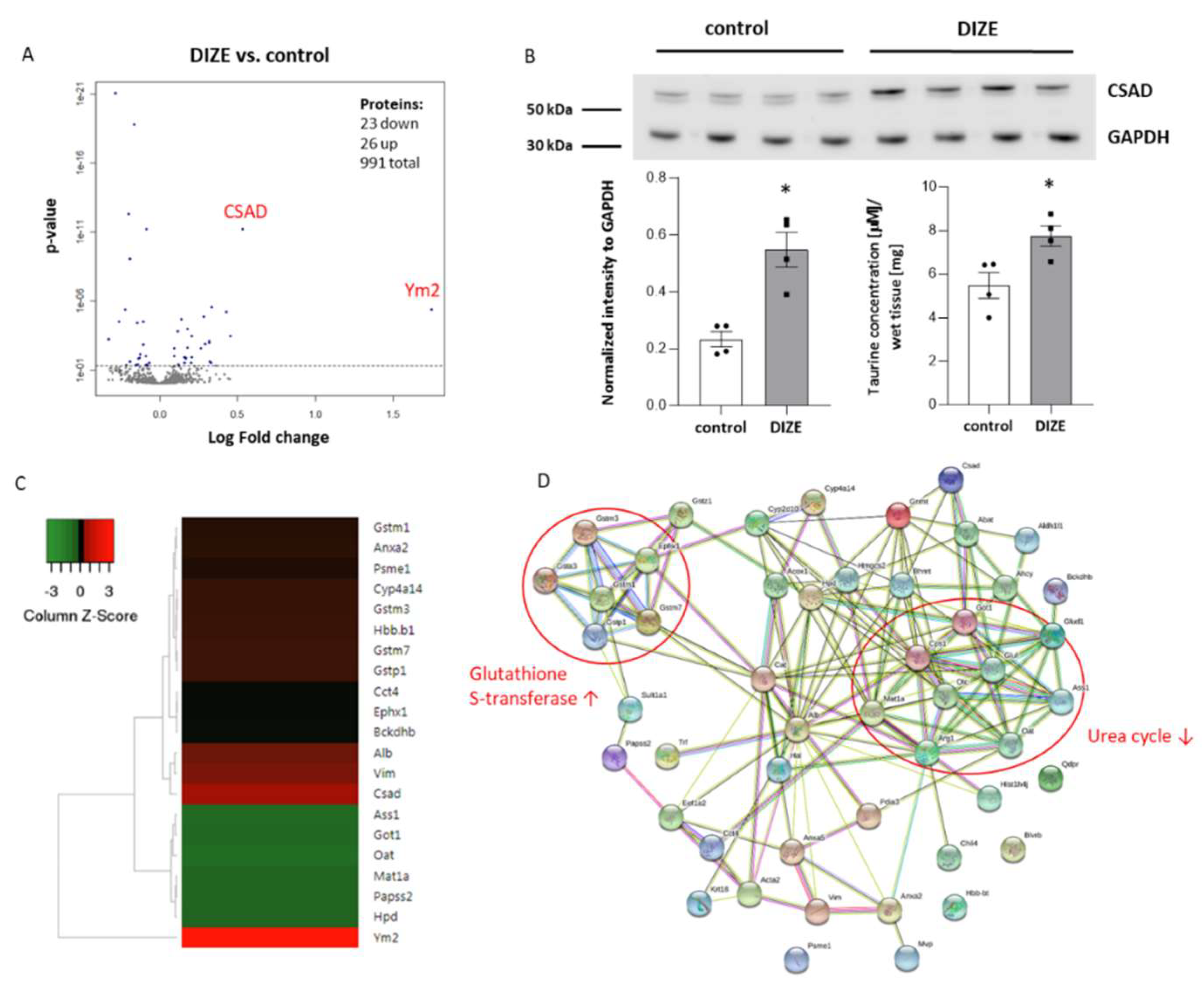
2.4. Influence of DIZE on Proteomic Changes in the Liver
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Atherosclerotic Lesion Assessment
4.3. Immunohistochemical Staining of Aortic Roots
4.4. Histology of the Liver
4.5. Biochemical Measurement
4.6. Western Blot Analysis
4.7. Real-time PCR
4.8. Mesenteric Arteries Preparation
4.9. Proteomics Studies in the Liver
4.10. THP-1 Cell Culture
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE | angiotensin-converting enzyme |
| ACE2 | angiotensin-converting enzyme 2 |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| CSAD | cysteine sulfinic acid decarboxylase |
| DIZE | diminazene aceturate |
| HDL | High-density lipoprotein |
| HE | hematoxylin/eosin |
| HFD | High-fat diet |
| LDL | low-density lipoprotein |
| NAFLD | nonalcoholic fatty liver disease |
| NEP | neprilysin |
| RAS | renin-angiotensin system |
| TG | triglyceride |
References
- Lloyd-Jones, D.M. Cardiovascular Risk Prediction: Basic Concepts, Current Status, and Future Directions. Circulation 2010, 121, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons from Sudden Coronary Death: A Comprehensive Morphological Classification Scheme for Atherosclerotic Lesions. Arter. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Okamoto, Y.; Rocha, V.Z.; Folco, E. Inflammation in Atherosclerosis: Transition from Theory to Practice. Circ. J. Off. J. Jpn. Circ. Soc. 2010, 74, 213–220. [Google Scholar]
- Targher, G. Non-Alcoholic Fatty Liver Disease, the Metabolic Syndrome and the Risk of Cardiovascular Disease: The Plot Thickens. Diabet. Med. J. Br. Diabet. Assoc. 2007, 24, 1–6. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human Fatty Liver Disease: Old Questions and New Insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [Green Version]
- Day, C.P.; James, O.F. Steatohepatitis: A Tale of Two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Da Silva, A.R.; Fraga-Silva, R.A.; Stergiopulos, N.; Montecucco, F.; Mach, F. Update on the Role of Angiotensin in the Pathophysiology of Coronary Atherothrombosis. Eur. J. Clin. Investig. 2015, 45, 274–287. [Google Scholar] [CrossRef]
- Santos, S.H.S.; Andrade, J.M.O. Angiotensin 1–7: A Peptide for Preventing and Treating Metabolic Syndrome. Peptides 2014, 59, 34–41. [Google Scholar] [CrossRef]
- Keidar, S.; Attias, J.; Heinrich, R.; Coleman, R.; Aviram, M. Angiotensin II Atherogenicity in Apolipoprotein E Deficient Mice Is Associated with Increased Cellular Cholesterol Biosynthesis. Atherosclerosis 1999, 146, 249–257. [Google Scholar] [CrossRef]
- Wei, Y.; Clark, S.E.; Morris, E.M.; Thyfault, J.P.; Uptergrove, G.M.E.; Whaley-Connell, A.T.; Ferrario, C.M.; Sowers, J.R.; Ibdah, J.A. Angiotensin II-Induced Non-Alcoholic Fatty Liver Disease Is Mediated by Oxidative Stress in Transgenic TG(mRen2)27(Ren2) Rats. J. Hepatol. 2008, 49, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Keidar, S.; Heinrich, R.; Kaplan, M.; Hayek, T.; Aviram, M. Angiotensin II Administration to Atherosclerotic Mice Increases Macrophage Uptake of Oxidized Ldl: A Possible Role for Interleukin-6. Arter. Thromb. Vasc. Biol. 2001, 21, 1464–1469. [Google Scholar] [CrossRef] [Green Version]
- Nickenig, G.; Harrison, D.G. The AT(1)-Type Angiotensin Receptor in Oxidative Stress and Atherogenesis: Part I: Oxidative Stress and Atherogenesis. Circulation 2002, 105, 393–396. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tikellis, C.; Thomas, M.C.; Golledge, J. Angiotensin Converting Enzyme 2 and Atherosclerosis. Atherosclerosis 2013, 226, 3–8. [Google Scholar] [CrossRef]
- Cao, X.; Yang, F.-Y.; Xin, Z.; Xie, R.-R.; Yang, J.-K. The ACE2/Ang-(1-7)/Mas Axis Can Inhibit Hepatic Insulin Resistance. Mol. Cell. Endocrinol. 2014, 393, 30–38. [Google Scholar] [CrossRef]
- Dong, B.; Zhang, C.; Feng, J.B.; Zhao, Y.X.; Li, S.Y.; Yang, Y.P.; Dong, Q.L.; Deng, B.P.; Zhu, L.; Yu, Q.T.; et al. Overexpression of ACE2 Enhances Plaque Stability in a Rabbit Model of Atherosclerosis. Arter. Thromb. Vasc. Biol. 2008, 28, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Pickering, R.J.; Tsorotes, D.; Koitka, A.; Sheehy, K.; Bernardi, S.; Toffoli, B.; Nguyen-Huu, T.P.; Head, G.A.; Fu, Y.; et al. Genetic Ace2 Deficiency Accentuates Vascular Inflammation and Atherosclerosis in the ApoE Knockout Mouse. Circ. Res. 2010, 107, 888–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Yang, F.; Shi, T.; Yuan, M.; Xin, Z.; Xie, R.; Li, S.; Li, H.; Yang, J.-K. Angiotensin-Converting Enzyme 2/Angiotensin-(1–7)/Mas Axis Activates Akt Signaling to Ameliorate Hepatic Steatosis. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peregrine, A.S.; Mamman, M. Pharmacology of Diminazene: A Review. Acta Trop. 1993, 54, 185–203. [Google Scholar] [CrossRef]
- Shenoy, V.; Gjymishka, A.; Jarajapu, Y.P.; Qi, Y.; Afzal, A.; Rigatto, K.; Ferreira, A.J.; Fraga-Silva, R.A.; Kearns, P.; Douglas, J.Y.; et al. Diminazene Attenuates Pulmonary Hypertension and Improves Angiogenic Progenitor Cell Functions in Experimental Models. Am. J. Respir. Crit. Care Med. 2013, 187, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Qaradakhi, T.; Gadanec, L.K.; McSweeney, K.R.; Tacey, A.; Apostolopoulos, V.; Levinger, I.; Rimarova, K.; Egom, E.E.; Rodrigo, L.; Kruzliak, P.; et al. The Potential Actions of Angiotensin-Converting Enzyme II (ACE2) Activator Diminazene Aceturate (DIZE) in Various Diseases. Clin. Exp. Pharm. Physiol. 2020, 47, 751–758. [Google Scholar] [CrossRef]
- Bruce, E.B.; Sakarya, Y.; Kirichenko, N.; Toklu, H.Z.; Sumners, C.; Morgan, D.; Tümer, N.; Scarpace, P.J.; Carter, C.S. ACE2 Activator Diminazene Aceturate Reduces Adiposity but Preserves Lean Mass in Young and Old Rats. Exp. Gerontol. 2018, 111, 133–140. [Google Scholar] [CrossRef]
- Joviano-Santos, J.V.; Santos-Miranda, A.; Joca, H.C.; Cruz, J.S.; Ferreira, A.J. Diminazene Aceturate (DIZE) Has Cellular and in Vivo Antiarrhythmic Effects. Clin. Exp. Pharm. Physiol. 2020, 47, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, S.E.; Zhang, X.; Howatt, D.A.; Lu, H.; Gurley, S.B.; Daugherty, A.; Cassis, L.A. ACE2 Deficiency in Whole Body or Bone Marrow-Derived Cells Increases Atherosclerosis in LDL Receptor −/− Mice. Arter. Thromb. Vasc. Biol. 2011, 31, 758–765. [Google Scholar] [CrossRef] [Green Version]
- Fraga-Silva, R.A.; Montecucco, F.; Costa-Fraga, F.P.; Nencioni, A.; Caffa, I.; Bragina, M.E.; Mach, F.; Raizada, M.K.; Santos, R.A.S.; da Silva, R.F.; et al. Diminazene Enhances Stability of Atherosclerotic Plaques in ApoE-Deficient Mice. Vasc. Pharm. 2015, 74, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, A. Control of Macrophage Activation and Function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.Y.; Miyoshi, H.; Kuroda, S.; Yasuda, H.; Kamiyama, K.; Nakagawara, J.; Takigami, M.; Kondo, T.; Atsumi, T. The Phenotype of Infiltrating Macrophages Influences Arteriosclerotic Plaque Vulnerability in the Carotid Artery. J. Stroke Cereb. Dis. Off. J. Natl. Stroke Assoc. 2013, 22, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.N.; Shaked, I.; Hubbeling, H.G.; Punt, J.A.; Wu, R.; Herrley, E.; Zaugg, C.; Pei, H.; Geissmann, F.; Ley, K.; et al. NR4A1 (Nur77) Deletion Polarizes Macrophages toward an Inflammatory Phenotype and Increases Atherosclerosis. Circ. Res. 2012, 110, 416–427. [Google Scholar] [CrossRef]
- Sharma, M.; Gupta, S.; Singh, K.; Mehndiratta, M.; Gautam, A.; Kalra, O.P.; Shukla, R.; Gambhir, J.K. Association of Glutathione-S-Transferase with Patients of Type 2 Diabetes Mellitus with and without Nephropathy. Diabetes Metab. Syndr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Cardilo-Reis, L.; Gruber, S.; Schreier, S.M.; Drechsler, M.; Papac-Milicevic, N.; Weber, C.; Wagner, O.; Stangl, H.; Soehnlein, O.; Binder, C.J. Interleukin-13 Protects from Atherosclerosis and Modulates Plaque Composition by Skewing the Macrophage Phenotype. EMBO Mol. Med. 2012, 4, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, U.O.; Kintscher, U. ACE2 and SARS-CoV-2—Tissue or Plasma, Good or Bad? Am. J. Hypertens. 2021. [Google Scholar] [CrossRef]
- Kuriakose, S.; Muleme, H.; Onyilagha, C.; Okeke, E.; Uzonna, J.E. Diminazene Aceturate (Berenil) Modulates LPS Induced pro-Inflammatory Cytokine Production by Inhibiting Phosphorylation of MAPKs and STAT Proteins. Innate Immun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Qaradakhi, T.; Gadanec, L.; Matsoukas, J.; Apostolopoulos, V.; Zulli, A. Could DIZE Be the Answer to COVID-19? Maturitas 2020, 140, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Feltenberger, J.D.; Andrade, J.M.O.; Paraíso, A.; Barros, L.O.; Filho, A.B.M.; Sinisterra, R.D.; Sousa, F.B.; Guimaraes, A.; De Paula, A.; Campagnole-Santos, M.; et al. Oral Formulation of Angiotensin-(1–7) Improves Lipid Metabolism and Prevents High-Fat Diet–Induced Hepatic Steatosis and Inflammation in Mice. Hypertension 2013, 62, 324–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, C.C.L.; Lourenço, F.C.; Mario, É.G.; Santos, R.A.S.; Botion, L.M.; Chaves, V.E. Long-Term Effects of Angiotensin-(1-7) on Lipid Metabolism in the Adipose Tissue and Liver. Peptides 2017, 92, 16–22. [Google Scholar] [CrossRef]
- Sutherland, T.E. Chitinase-like Proteins as Regulators of Innate Immunity and Tissue Repair: Helpful Lessons for Asthma? Biochem. Soc. Trans. 2018, 46, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Available online: https://www.hindawi.com/journals/mi/2015/816460/ (accessed on 16 November 2020).
- Murakami, S. Taurine and Atherosclerosis. Amino Acids 2014, 46, 73–80. [Google Scholar] [CrossRef]
- Murakami, S.; Ono, A.; Kawasaki, A.; Takenaga, T.; Ito, T. Taurine Attenuates the Development of Hepatic Steatosis through the Inhibition of Oxidative Stress in a Model of Nonalcoholic Fatty Liver Disease in Vivo and in Vitro. Amino Acids 2018, 50, 1279–1288. [Google Scholar] [CrossRef]
- Lin, S.; Hirai, S.; Yamaguchi, Y.; Goto, T.; Takahashi, N.; Tani, F.; Mutoh, C.; Sakurai, T.; Murakami, S.; Yu, R.; et al. Taurine Improves Obesity-Induced Inflammatory Responses and Modulates the Unbalanced Phenotype of Adipose Tissue Macrophages. Mol. Nutr. Food Res. 2013, 57, 2155–2165. [Google Scholar] [CrossRef]
- Sartório, C.L.; Pimentel, E.B.; dos Santos, R.L.; Rouver, W.N.; Mill, J.G. Acute Hypotensive Effect of Diminazene Aceturate in Spontaneously Hypertensive Rats: Role of NO and Mas Receptor. Clin. Exp. Pharm. Physiol. 2020, 47, 1723–1730. [Google Scholar] [CrossRef]
- Guizoni, D.M.; Vettorazzi, J.F.; Carneiro, E.M.; Davel, A.P. Modulation of Endothelium-Derived Nitric Oxide Production and Activity by Taurine and Taurine-Conjugated Bile Acids. Nitric Oxide Biol. Chem. 2020, 94, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qiu, L.; Li, M.; Dürrnagel, S.; Orser, B.A.; Xiong, Z.-G.; MacDonald, J.F. Diarylamidines: High Potency Inhibitors of Acid-Sensing Ion Channels. Neuropharmacology 2010, 58. [Google Scholar] [CrossRef] [Green Version]
- Velkoska, E.; Patel, S.K.; Griggs, K.; Pickering, R.J.; Tikellis, C.; Burrell, L.M. Short-Term Treatment with Diminazene Aceturate Ameliorates the Reduction in Kidney ACE2 Activity in Rats with Subtotal Nephrectomy. PLoS ONE 2015, 10, e0118758. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.K.; Ye, M.; Wysocki, J.; Maier, C.; Haque, S.K.; Batlle, D. ACE2-Independent Action Of Presumed ACE2 Activators: Studies In Vivo, Ex Vivo and In Vitro. Hypertension 2014, 63, 774–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamman, M.; Gettinby, G.; Murphy, N.B.; Kemei, S.; Peregrine, A.S. Frequency of Diminazene-Resistant Trypanosomes in Populations of Trypanosoma Congolense Arising in Infected Animals Following Treatment with Diminazene Aceturate. Antimicrob. Agents Chemother. 1995, 39, 1107–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachowicz, A.; Wiśniewska, A.; Kuś, K.; Kiepura, A.; Gębska, A.; Gajda, M.; Białas, M.; Totoń-Żurańska, J.; Stachyra, K.; Suski, M.; et al. The Influence of Trehalose on Atherosclerosis and Hepatic Steatosis in Apolipoprotein E Knockout Mice. Int. J. Mol. Sci. 2019, 20, 1552. [Google Scholar] [CrossRef] [Green Version]
- Suski, M.; Kiepura, A.; Wiśniewska, A.; Kuś, K.; Skałkowska, A.; Stachyra, K.; Stachowicz, A.; Gajda, M.; Korbut, R.; Olszanecki, R. Anti-Atherosclerotic Action of GW9508—Free Fatty Acid Receptors Activator—In apoE-Knockout Mice. Pharm. Rep. Pr. 2019, 71, 551–555. [Google Scholar] [CrossRef]
- Stachowicz, A.; Olszanecki, R.; Suski, M.; Wiśniewska, A.; Totoń-Żurańska, J.; Madej, J.; Jawień, J.; Białas, M.; Okoń, K.; Gajda, M.; et al. Mitochondrial Aldehyde Dehydrogenase Activation by Alda-1 Inhibits Atherosclerosis and Attenuates Hepatic Steatosis in Apolipoprotein E-Knockout Mice. J. Am. Heart Assoc. 2014, 3, e001329. [Google Scholar] [CrossRef] [Green Version]
- Wiśniewski, J.R.; Zougman, A.; Mann, M. Combination of FASP and StageTip-Based Fractionation Allows in-Depth Analysis of the Hippocampal Membrane Proteome. J. Proteome Res. 2009, 8, 5674–5678. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Rakus, D. Multi-Enzyme Digestion FASP and the “Total Protein Approach”-based Absolute Quantification of the Escherichia Coli Proteome. J. Proteom. 2014, 109, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taverner, T.; Karpievitch, Y.V.; Polpitiya, A.D.; Brown, J.N.; Dabney, A.R.; Anderson, G.A.; Smith, R.D. DanteR: An Extensible R-Based Tool for Quantitative Analysis of -Omics Data. Bioinforma. Oxf. Engl. 2012, 28, 2404–2406. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProtKB ID. | Gene Name | Protein Name | Fold Change |
|---|---|---|---|
| Q91Z98 | Chil4/Ym2 | Chitinase-like protein 4 | 3.36 |
| Q9DBE0 | Csad | Cysteine sulfinic acid decarboxylase | 1.45 |
| P20152 | Vim | Vimentin | 1.37 |
| P07724 | Alb | Serum albumin | 1.35 |
| P19157 | Gstp1 | Glutathione S-transferase P 1 | 1.26 |
| Q80W21 | Gstm7 | Glutathione S-transferase Mu 7 | 1.26 |
| P02088 | Hbb-b1 | Hemoglobin subunit beta-1 | 1.25 |
| P19639 | Gstm3 | Glutathione S-transferase Mu 3 | 1.25 |
| O35728 | Cyp4a14 | Cytochrome P450 4A14 | 1.25 |
| P07356 | Anxa2 | Annexin A2 | 1.22 |
| P10649 | Gstm1 | Glutathione S-transferase Mu 1 | 1.22 |
| P97371 | Psme1 | Proteasome activator complex subunit 1 | 1.20 |
| Q9D379 | Ephx1 | Epoxide hydrolase 1 | 1.15 |
| P80315 | Cct4 | T-complex protein 1 subunit delta | 1.15 |
| Q6P3A8 | Bckdhb | 2-oxoisovalerate dehydrogenase subunit beta, mitochondrial | 1.15 |
| P48036 | Anxa5 | Annexin A5 | 1.13 |
| Q921I1 | Tf | Serotransferrin | 1.13 |
| Q923D2 | Blvrb | Flavin reductase (NADPH) | 1.12 |
| P24456 | Cyp2d10 | Cytochrome P450 2D10 | 1.12 |
| Q9EQK5 | Mvp | Major vault protein | 1.12 |
| P62806 | Hist1h4a | Histone H4 | 1.12 |
| P62737 | Acta2 | Actin, aortic smooth muscle | 1.10 |
| P15105 | Glul | Glutamine synthetase | 1.09 |
| P54869 | Hmgcs2 | Hydroxymethylglutaryl-CoA synthase, mitochondrial | 1.09 |
| P27773 | Pdia3 | Protein disulfide-isomerase A3 | 1.07 |
| P26443 | Glud1 | Glutamate dehydrogenase 1, mitochondrial | 1.07 |
| P11725 | Otc | Ornithine carbamoyltransferase, mitochondrial | −1.04 |
| Q9R0H0 | Acox1 | Peroxisomal acyl-coenzyme A oxidase 1 | −1.06 |
| P05784 | Krt18 | Keratin, type I cytoskeletal 18 | −1.06 |
| Q8C196 | Cps1 | Carbamoyl-phosphate synthase [ammonia], mitochondrial | −1.06 |
| P24270 | Cat | Catalase | −1.06 |
| Q9WVL0 | Gstz1 | Maleylacetoacetate isomerase | −1.06 |
| Q61176 | Arg1 | Arginase-1 | −1.07 |
| P50247 | Ahcy | Adenosylhomocysteinase | −1.08 |
| P30115 | Gsta3 | Glutathione S-transferase A3 | −1.09 |
| Q8BVI4 | Qdpr | Dihydropteridine reductase | −1.09 |
| P61922 | Abat | 4-aminobutyrate aminotransferase, mitochondrial | −1.10 |
| P35492 | Hal | Histidine ammonia-lyase | −1.10 |
| Q9QXF8 | Gnmt | Glycine N-methyltransferase | −1.10 |
| P52840 | Sult1a1 | Sulfotransferase 1A1 | −1.11 |
| Q8R0Y6 | Aldh1l1 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | −1.12 |
| P62631 | Eef1a2 | Elongation factor 1-alpha 2 | −1.14 |
| O35490 | Bhmt | Betaine-homocysteine S-methyltransferase 1 | −1.14 |
| P49429 | Hpd | 4-hydroxyphenylpyruvate dioxygenase | −1.15 |
| O88428 | Papss2 | Bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthase 2 | −1.16 |
| Q91X83 | Mat1a | S-adenosylmethionine synthase isoform type-1 | −1.17 |
| P05201 | Got1 | Aspartate aminotransferase, cytoplasmic | −1.20 |
| P16460 | Ass1 | Argininosuccinate synthase | −1.22 |
| P29758 | Oat | Ornithine aminotransferase, mitochondrial | −1.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stachowicz, A.; Wiśniewska, A.; Kuś, K.; Białas, M.; Łomnicka, M.; Totoń-Żurańska, J.; Kiepura, A.; Stachyra, K.; Suski, M.; Bujak-Giżycka, B.; et al. Diminazene Aceturate Stabilizes Atherosclerotic Plaque and Attenuates Hepatic Steatosis in apoE-Knockout Mice by Influencing Macrophages Polarization and Taurine Biosynthesis. Int. J. Mol. Sci. 2021, 22, 5861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115861
Stachowicz A, Wiśniewska A, Kuś K, Białas M, Łomnicka M, Totoń-Żurańska J, Kiepura A, Stachyra K, Suski M, Bujak-Giżycka B, et al. Diminazene Aceturate Stabilizes Atherosclerotic Plaque and Attenuates Hepatic Steatosis in apoE-Knockout Mice by Influencing Macrophages Polarization and Taurine Biosynthesis. International Journal of Molecular Sciences. 2021; 22(11):5861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115861
Chicago/Turabian StyleStachowicz, Aneta, Anna Wiśniewska, Katarzyna Kuś, Magdalena Białas, Magdalena Łomnicka, Justyna Totoń-Żurańska, Anna Kiepura, Kamila Stachyra, Maciej Suski, Beata Bujak-Giżycka, and et al. 2021. "Diminazene Aceturate Stabilizes Atherosclerotic Plaque and Attenuates Hepatic Steatosis in apoE-Knockout Mice by Influencing Macrophages Polarization and Taurine Biosynthesis" International Journal of Molecular Sciences 22, no. 11: 5861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115861