
Mitochondrial Caseinolytic Protease P: A Possible Novel Prognostic Marker and Therapeutic Target in Cancer

Abstract
:1. Introduction
2. The Mitochondrial Quality Control System
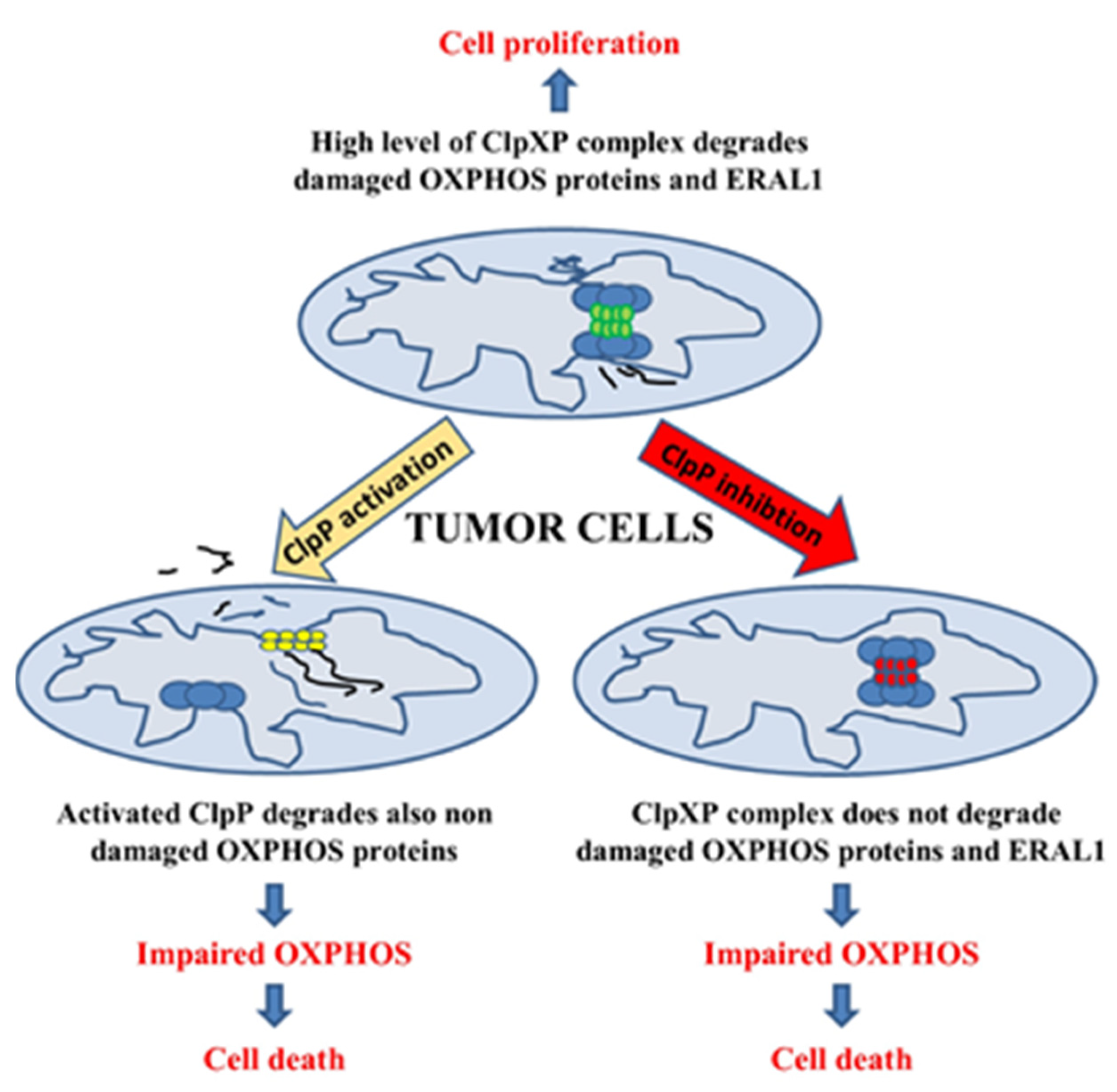
3. The Role of Increased ClpP Expression as a Prognostic Marker in Human Cancer
4. The Influence of Loss of ClpP on Mitochondrial Respiratory Complexes and Cell Viability
5. The Chemical Modulation of ClpP: A Promising Anticancer Therapy
6. Mitochondria and Cisplatin Resistance: The Involvement of ClpP
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Attardi, G. Animal mitochondrial DNA: An extreme example of genetic economy. Int. Rev. Cytol. 1985, 93, 93–145. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K. Mitochondrial free radical generation, oxidative stress and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalska, M.; Piekut, T.; Prendecki, M.; Sodel, A.; Kozubski, W.; Dorszewska, J. Mitochondrial and nuclear DNA oxidative damage in physiological and pathological aging. DNA Cell Biol. 2020, 39, 1410–1420. [Google Scholar] [CrossRef]
- Da Silva, R.R. Controlling proteolysis of Clp peptidase: A possible target for combating mitochondrial diseases. Int. J. Biochem. Cell Biol. 2019, 110, 140–142. [Google Scholar] [CrossRef]
- Gerdes, F.; Tatsuta, T.; Langer, T. Mitochondrial AAA proteases—towards a molecular understanding of membrane-bound proteolytic machines. Biochim. Biophys. Acta 2012, 1823, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Deshwal, S.; Fiedler, K.U.; Langer, T. Mitochondrial proteases: Multifaceted regulators of mitochondrial plasticity. Annu. Rev. Biochem. 2020, 89, 7.1–7.28. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.G.; Dimitrova, M.N.; Ortega, J.; Ginsburg, A.; Maurizi, M.R. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J. Biol. Chem. 2005, 280, 35424–35432. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulos, J.A.; Guarné, A.; Ortega, J. ClpP: A structurally dynamic protease regulated by AAA+ proteins. J. Struct. Biol. 2012, 179, 202–210. [Google Scholar] [CrossRef]
- Wong, K.S.; Houry, W.A. Chemical modulation of human mitochondrial ClpP: Potential application in cancer therapeutics. ACS Chem. Biol. 2019, 14, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Nouri, K.; Feng, Y.; Schimmer, A.D. Mitochondrial ClpP serine protease-biological function and emerging target for cancer therapy. Cell Death Dis. 2020, 11, 841. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y.; Nomura, K.; Katoh, Y.; Yamashita, R.; Kaneko, K.; Furuyama, K. Novel mechanisms for heme-dependent degradation of ALAS1 protein as a component of negative feedback regulation of heme biosynthesis. J. Biol. Chem. 2016, 291, 20516–20529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, A.W.; Grenier, K.; A Aguileta, M.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.; Park, D.S.; A Fon, E. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Esencan, E.; Jiang, Z.; Wang, T.; Zhang, M.; Soylemez-Imamoglu, G.; Seli, E. Impaired mitochondrial stress response due to CLPP deletion is associated with altered mitochondrial dynamics and increased apoptosis in cumulus cells. Reprod. Sci. 2020, 27, 621–630. [Google Scholar] [CrossRef]
- Kasashima, K.; Sumitani, M.; Endo, H. Maintenance of mitochondrial genome distribution by mitochondrial AAA+ protein ClpX. Exp. Cell Res. 2012, 318, 2335–2343. [Google Scholar] [CrossRef]
- Szczepanowska, K.; Maiti, P.; Kukat, A.; Hofsetz, E.; Nolte, H.; Senft, K.; Becker, C.; Ruzzenente, B.; Hornig-Do, H.; Wibom, R.; et al. CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO J. 2016, 35, 2566–2583. [Google Scholar] [CrossRef] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 2016, 39, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, e104705. [Google Scholar] [CrossRef]
- Yoo, S.M.; Jung, Y.K. A molecular approach to mitophagy and mitochondrial dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Jovaisaite, V.; Auwerx, J. The mitochondrial unfolded protein response synchronizing genomes. Curr. Opin. Cell Biol. 2015, 33, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Carvajal, F.; Sanhueza, M. The mitochondrial unfolded protein response: A hinge between healthy and pathological aging. Front. Aging Neurosci. 2020, 12, 581849. [Google Scholar] [CrossRef]
- Qureshi, M.A.; Haynes, C.M.; Pellegrino, M.W. The mitochondrial unfolded protein response: Signaling from the powerhouse. J. Biol. Chem. 2017, 292, 13500–13506. [Google Scholar] [CrossRef] [Green Version]
- Runkel, E.D.; Liu, S.; Baumeister, R.; Schulze, E. Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet. 2013, 9, e1003346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, M.; He, Y.; Zhou, H.; Du, H.; Shao, C.; Yang, J.; Wan, H. Domesticated and optimized mitochondria: Mitochondrial modifications based on energetic status and cellular stress. Life Sci. 2021, 265, 118766. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; MacLean, N.; et al. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.-Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef] [Green Version]
- Cormio, A.; Musicco, C.; Gasparre, G.; Cormio, G.; Pesce, V.; Sardanelli, A.M.; Gadaleta, M.N. Increase in proteins involved in mitochondrial fission, mitophagy, proteolysis and antioxidant response in type I endometrial cancer as an adaptive response to respiratory complex I deficiency. Biochem. Biophys. Res. Commun. 2017, 491, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Kurelac, I.; Cormio, A.; Zuntini, R.; Amato, L.B.; Ceccarelli, C.; Santini, D.; Cormio, G.; Fracasso, F.; Selvaggi, L.; et al. Placing mitochondrial DNA mutations within the progression model of type I endometrial carcinoma. Hum. Mol. Genet. 2011, 20, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Cormio, G.; Musicco, C.; Sardanelli, A.M.; Gasparre, G.; Gadaleta, M.N. Mitochondrial changes in endometrial carcinoma: Possible role in tumor diagnosis and prognosis (review). Oncol. Rep. 2015, 33, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Zeng, B.; Tao, C.; Lu, M.; Ren, G. ClpP regulates breast cancer cell proliferation, invasion and apoptosis by modulating the Src/PI3K/Akt signaling pathway. PeerJ 2020, 8, e8754. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Langer, J.D.; Osiewacz, H.D. Identification of potential mitochondrial CLPXP protease interactors and substrates suggests its central role in energy metabolism. Sci. Rep. 2015, 5, 18375. [Google Scholar] [CrossRef] [Green Version]
- Deepa, S.S.; Bhaskaran, S.; Ranjit, R.; Qaisar, R.; Nair, B.C.; Liu, Y.; Walsh, M.E.; Fok, W.C.; Van Remmen, H. Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic. Biol. Med. 2016, 91, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Sun, X.; Liao, X.; Zhang, X.; Zarabi, S.; Schimmer, A.; Hong, Y.; Ford, C.; Luo, Y.; Qi, X. Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathol. 2019, 137, 939–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanowska, K.; Senft, K.; Heidler, J.; Herholz, M.; Kukat, A.; Höhne, M.N.; Hofsetz, E.; Becker, C.; Kaspar, S.; Giese, H.; et al. A salvage pathway maintains highly functional respiratory complex, I. Nat. Commun. 2020, 11, 1643. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Roessler, M.M. Energy conversion, redox catalysis and generation of reactive oxygen species by respiratory complex I. Biochim. Biophys. Acta (BBA)-Bioenerg 2016, 1857, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Dröse, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef] [Green Version]
- Brodie, E.J.; Zhan, H.; Saiyed, T.; Truscott, K.N.; Dougan, D.A. Perrault syndrome type 3 caused by diverse molecular defects in CLPP. Sci. Rep. 2018, 8, 12862. [Google Scholar] [CrossRef] [Green Version]
- Bottcher, T.; Sieber, S.A. Beta-lactones as specific inhibitors of ClpP attenuate the production of extracellular virulence factors of Staphylococcus aureus. J. Am. Chem. Soc. 2008, 130, 14400–14401. [Google Scholar] [CrossRef] [PubMed]
- Hackl, M.W.; Lakemeyer, M.; Dahmen, M.; Glaser, M.; Pahl, A.; Lorenz-Baath, K.; Menzel, T.; Sievers, S.; Böttcher, T.; Antes, I.; et al. Phenyl esters are potent inhibitors of caseinolytic protease P and reveal a stereogenic switch for deoligomerization. J. Am. Chem. Soc. 2015, 137, 8475–8483. [Google Scholar] [CrossRef]
- Gronauer, T.F.; Mandl, M.M.; Lakemeyer, M.; Hackl, M.W.; Meßner, M.; Korotkov, V.S.; Pachmayr, J.; Sieber, S.A. Design and synthesis of tailored human caseinolytic protease P inhibitors. Chem. Commun. 2018, 54, 9833–9836. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Grouleff, J.J.; Jitkova, Y.; Diaz, D.B.; Griffith, E.C.; Shao, W.; Bogdanchikova, A.F.; Poda, G.; Schimmer, A.D.; Lee, R.E.; et al. De novo design of boron-based peptidomimetics as potent inhibitors of human ClpP in the presence of human, C.l.p.X. J. Med. Chem. 2019, 62, 6377–6390. [Google Scholar] [CrossRef]
- Allen, J.E.; Kline, C.L.B.; Prabhu, V.V.; Wagner, J.; Ishizawa, J.; Madhukar, N.; Lev, A.; Baumeister, M.; Zhou, L.; Lulla, A.; et al. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget 2016, 7, 74380–74392. [Google Scholar] [CrossRef] [Green Version]
- Nii, T.; Prabhu, V.V.; Ruvolo, V.; Madhukar, N.; Zhao, R.; Mu, H.; Heese, L.; Nishida, Y.; Kojima, K.; Garnett, M.J.; et al. Imipridone ONC212 activates orphan G protein-coupled receptor GPR132 and integrated stress response in acute myeloid leukemia. Leukemia 2019, 33, 2805–2816. [Google Scholar] [CrossRef]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer Cell 2019, 35, 721–737.e9. [Google Scholar] [CrossRef] [PubMed]
- Greer, Y.E.; Porat-Shliom, N.; Nagashima, K.; Stuelten, C.; Crooks, D.; Koparde, V.N.; Gilbert, S.F.; Islam, C.; Ubaldini, A.; Ji, Y.; et al. ONC201 kills breast cancer cells in vitro by targeting mitochondria. Oncotarget 2018, 9, 18454–18479. [Google Scholar] [CrossRef] [Green Version]
- Graves, P.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial protease ClpP is a target for the anticancer compounds ONC201 and related analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Pruss, M.; Dwucet, A.; Tanriover, M.; Hlavac, M.; Kast, R.E.; Debatin, K.-M.; Wirtz, C.R.; Halatsch, M.-E.; Siegelin, M.D.; Westhoff, M.-A.; et al. Dual metabolic reprogramming by ONC201/TIC10 and 2-Deoxyglucose induces energy depletion and synergistic anti-cancer activity in glioblastoma. Br. J. Cancer 2020, 122, 1146–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, E.R.; Waszak, S.M.; Grotzer, M.A.; Mueller, S.; Nazarian, J. Mechanisms of imipridones in targeting mitochondrial metabolism in cancer cells. Neuro-Oncol. 2021, 23, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Sirago, G.; Conte, E.; Fracasso, F.; Cormio, A.; Fehrentz, J.-A.; Martinez, J.; Musicco, C.; Camerino, G.M.; Fonzino, A.; Rizzi, L.; et al. Growth hormone secretagogues hexarelin and JMV2894 protect skeletal muscle from mitochondrial damages in a rat model of cisplatin-induced cachexia. Sci. Rep. 2017, 7, 13017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial involvement in cisplatin resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santin, G.; Piccolini, V.M.; Barni, S.; Veneroni, P.; Giansanti, V.; Bo, V.D.; Bernocchi, G.; Bottone, M.G. Mitochondrial fusion: A mechanism of cisplatin-induced resistance in neuroblastoma cells? Neurotoxicology 2013, 34, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Farrand, L.; Byun, S.; Kim, J.Y.; Im-Aram, A.; Lee, J.; Lim, S.; Lee, K.W.; Suh, J.-Y.; Lee, H.J.; Tsang, B.K. Piceatannol enhances cisplatin sensitivity in ovarian cancer via modulation of p53, X-linked inhibitor of apoptosis protein (XIAP), and mitochondrial fission. J. Biol. Chem. 2013, 288, 23740–23750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampieri, L.X.; Grasso, D.; Bouzin, C.; Brusa, D.; Rossignol, R.; Sonveaux, P. Mitochondria participate in chemoresistance to cisplatin in human ovarian cancer cells. Mol. Cancer Res. 2020, 18, 1379–1391. [Google Scholar] [CrossRef]
- Zhang, Y.; Maurizi, M.R. Mitochondrial ClpP activity is required for cisplatin resistance in human cells. Biochim. Biophys. Acta 2016, 1862, 252–264. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate ‘fuel’ tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Tumor Type | Sample | Ref. |
|---|---|---|
| Acute Myeloid Leukemia | non-solid tumor | [32] |
| Uterus | solid tumor | [34,35] |
| Bladder | solid tumor | [34], Cormio et al., unpublished |
| Prostate | solid tumor | [34] |
| Lung | solid tumor | [34] |
| Liver | solid tumor | [34] |
| Colon | solid tumor | [34] |
| Thyroid | solid tumor | [34] |
| Ovary | solid tumor | [34] |
| Testis | solid tumor | [34] |
| Stomach | solid tumor | [34] |
| Lymph nodes | solid tumor | [34] |
| Central nervous system | solid tumor | [34] |
| Uveal melanoma | solid tumor | [34] |
| Breast | solid tumor, ll lines (MCF-7, ZR-75-1, MB-231) | [34,38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cormio, A.; Sanguedolce, F.; Pesce, V.; Musicco, C. Mitochondrial Caseinolytic Protease P: A Possible Novel Prognostic Marker and Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 6228. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126228
Cormio A, Sanguedolce F, Pesce V, Musicco C. Mitochondrial Caseinolytic Protease P: A Possible Novel Prognostic Marker and Therapeutic Target in Cancer. International Journal of Molecular Sciences. 2021; 22(12):6228. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126228
Chicago/Turabian StyleCormio, Antonella, Francesca Sanguedolce, Vito Pesce, and Clara Musicco. 2021. "Mitochondrial Caseinolytic Protease P: A Possible Novel Prognostic Marker and Therapeutic Target in Cancer" International Journal of Molecular Sciences 22, no. 12: 6228. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126228