
MUC16 Is Overexpressed in Idiopathic Pulmonary Fibrosis and Induces Fibrotic Responses Mediated by Transforming Growth Factor-β1 Canonical Pathway


Abstract
:1. Introduction
2. Results
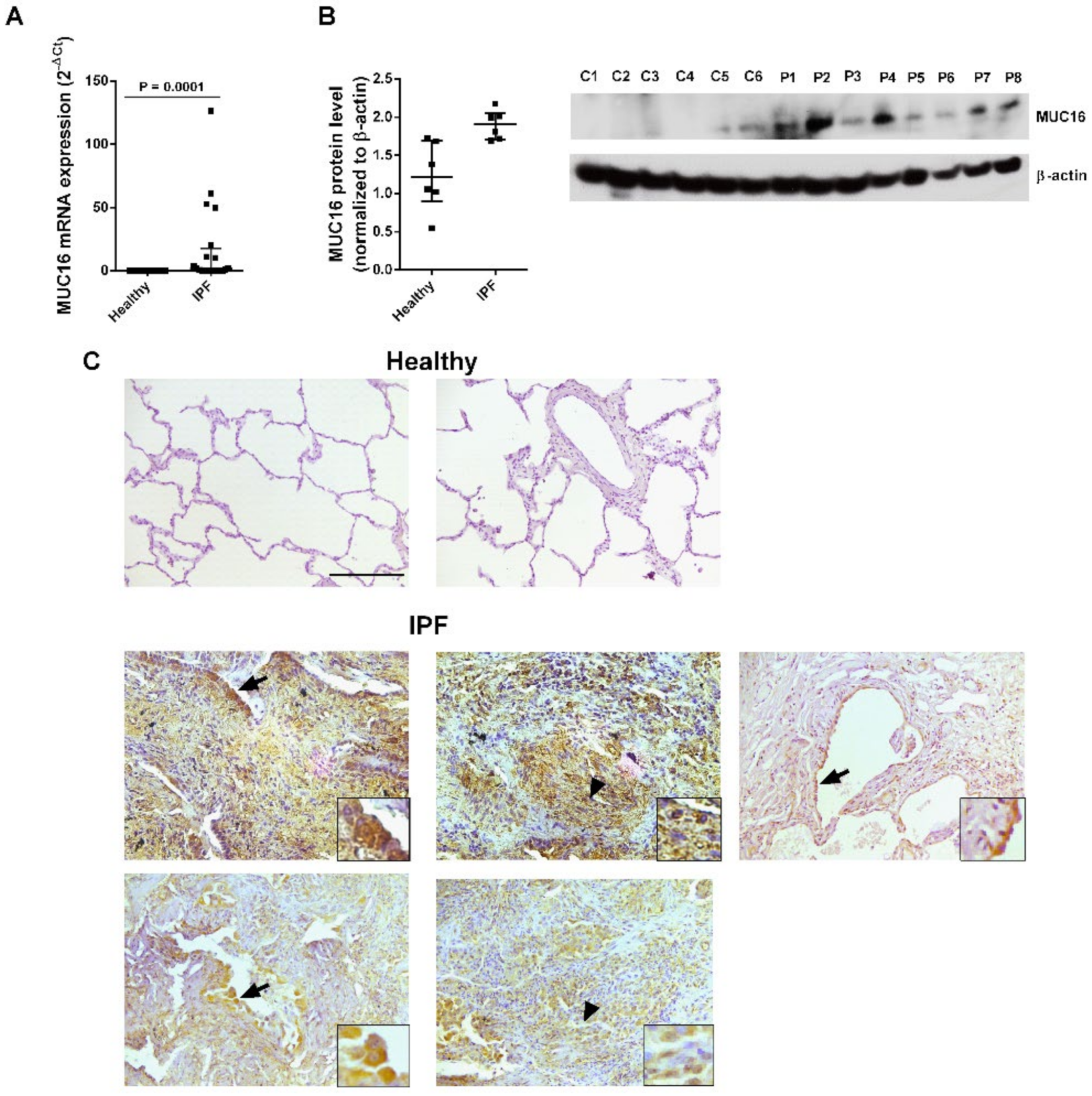
2.1. MUC16 Is Overexpressed in IPF Lung Tissue
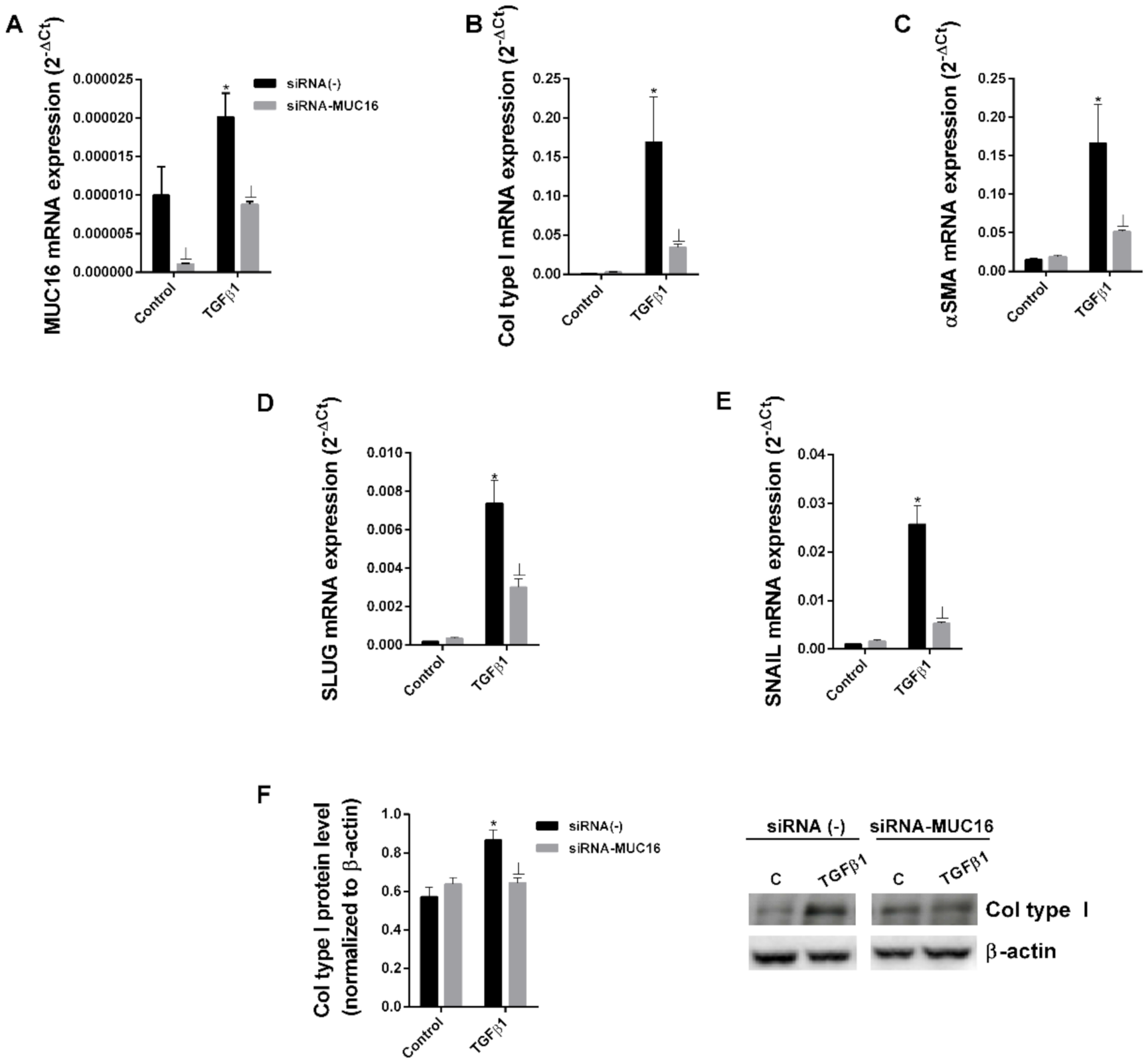
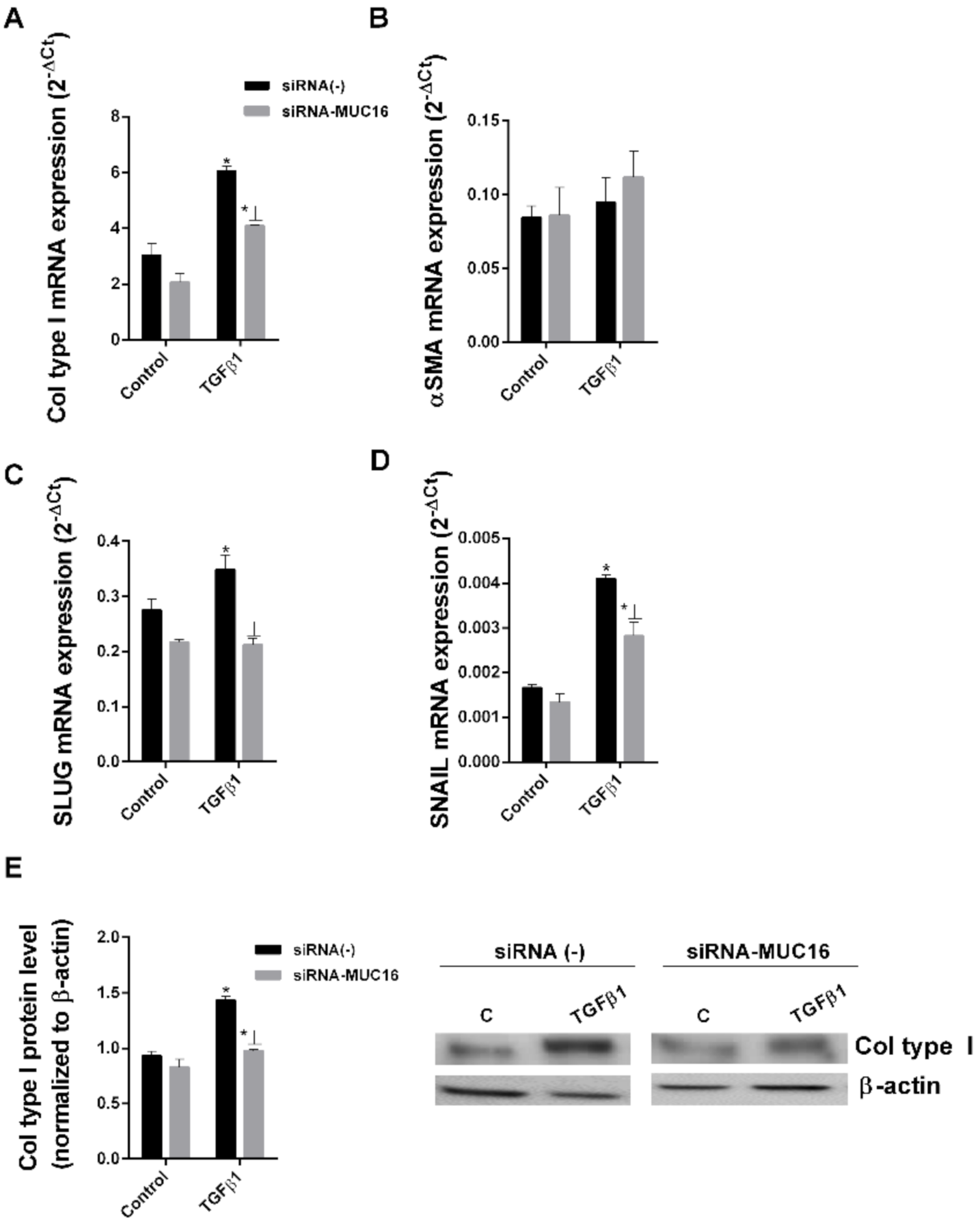
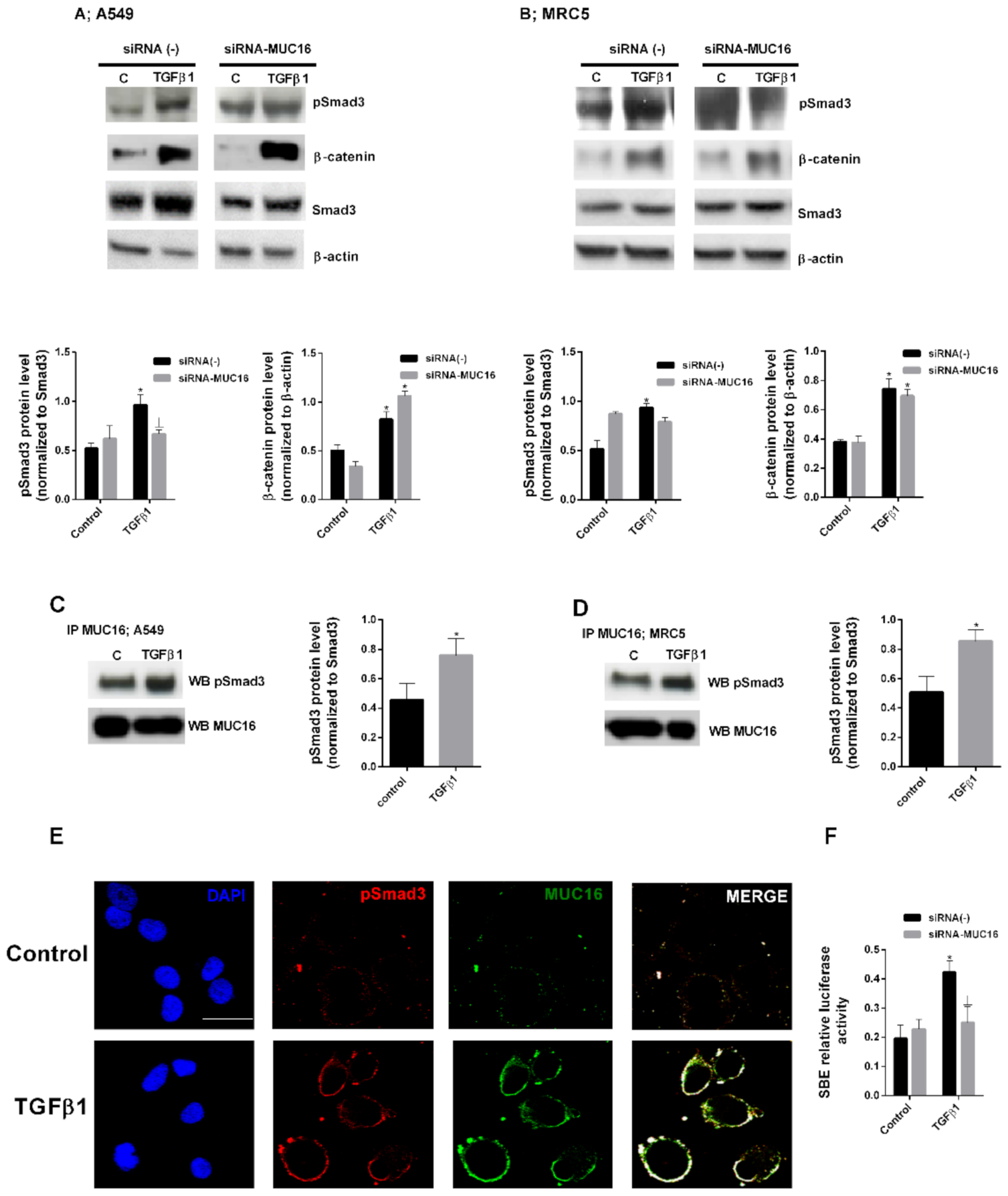
2.2. MUC16 Collaborates with TGF-β1 to Promote the Alveolar Type II to Mesenchymal and Fibroblast to Myofibroblast Transitions
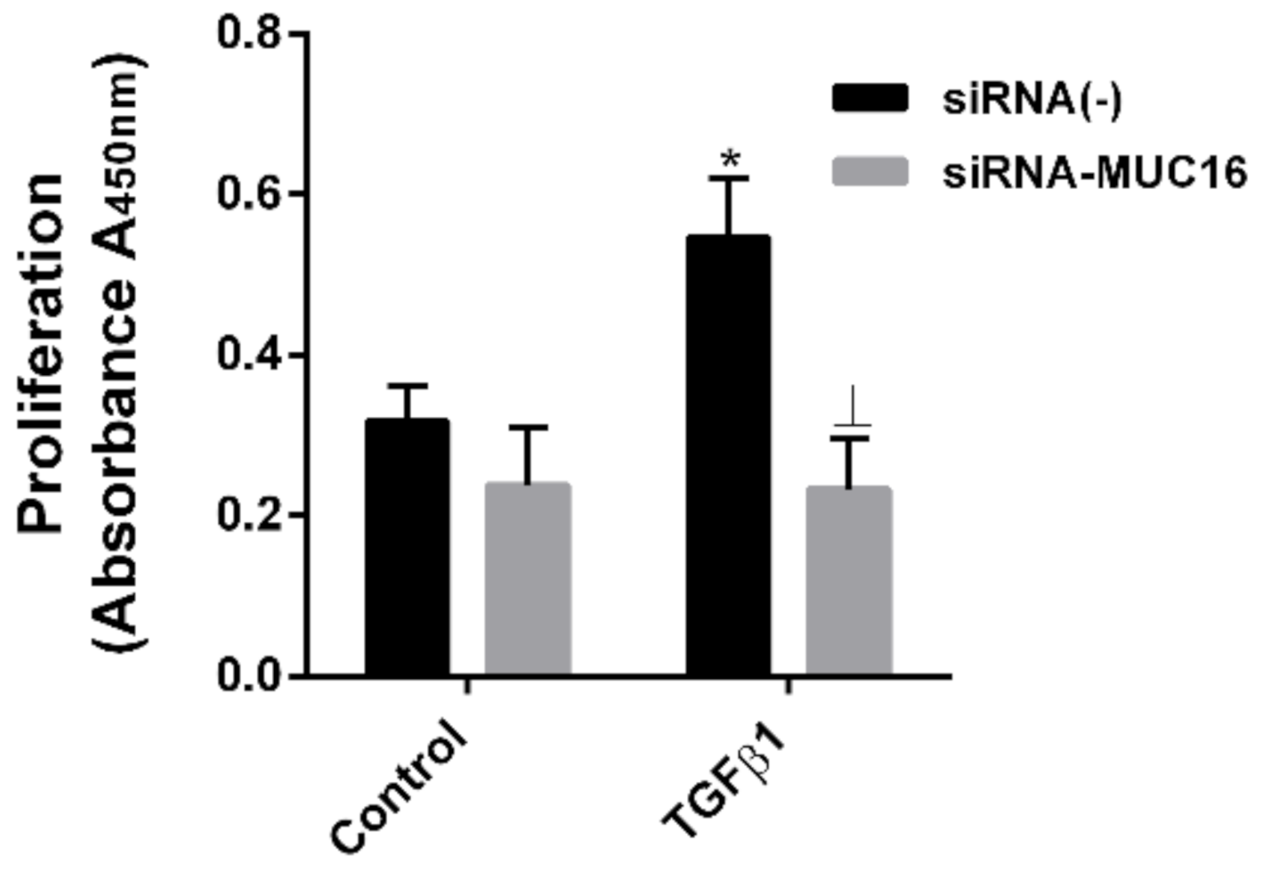
2.3. MUC16 Mediates TGF-β1-Induced Lung Fibroblast Proliferation
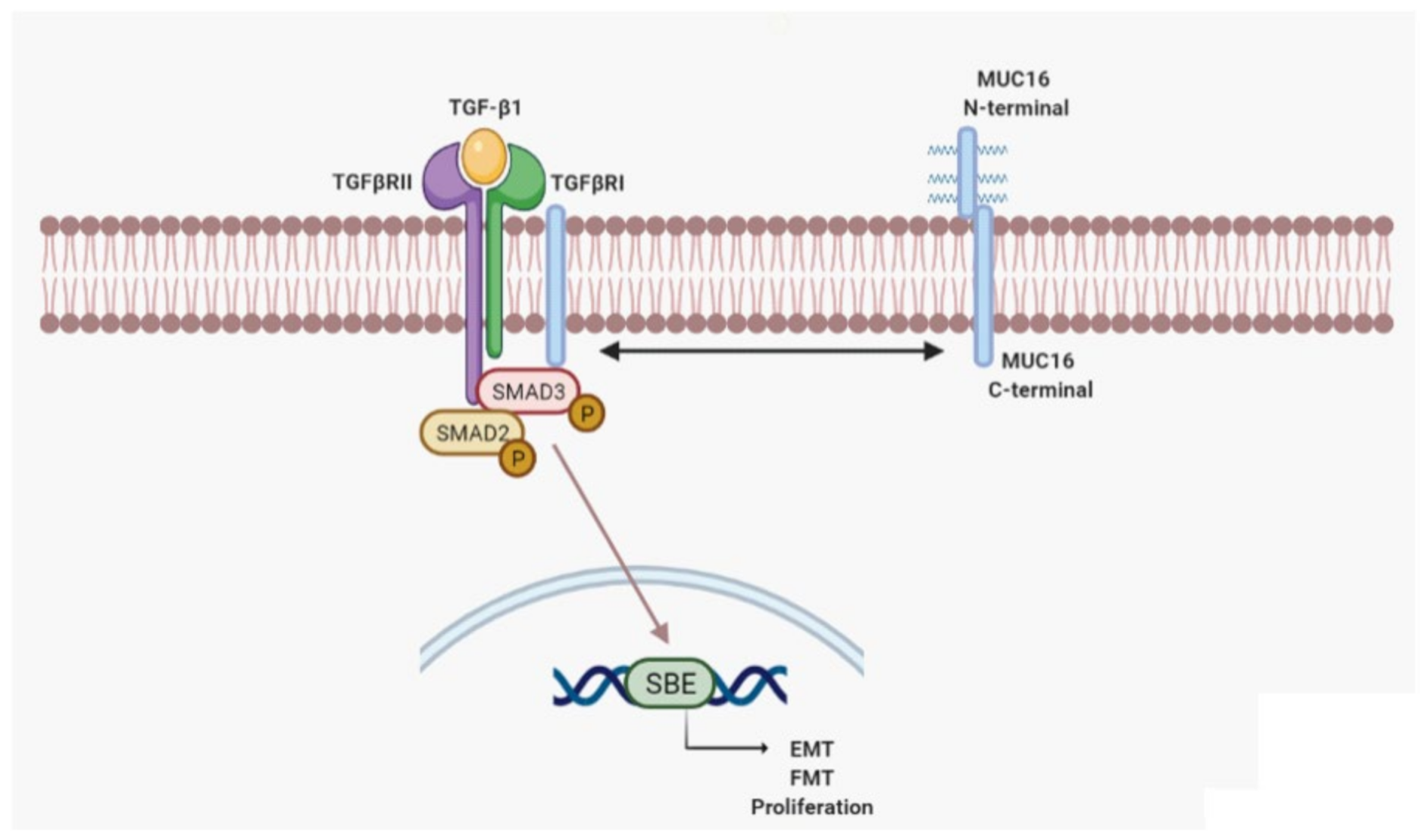
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Culture of A549 and MRC5 Cells
4.3. SiRNA Transfection of A549 and MRC5 Cells
4.4. Western Blotting Analysis
4.5. Real-Time RT-PCR
4.6. Proliferation
4.7. Immunoprecipitation
4.8. Histological, Immunohistochemical and Immunofluorescence Studies
4.9. SBE Assay in siRNA Transfected Cells
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Strieter, R.M.; Mehrad, B. New mechanisms of pulmonary fibrosis. Chest 2009, 136, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiners, S.; Eickelberg, O.; Konigshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Fabrellas, E.; Peris Sanchez, R.; Sabater Abad, C.; Juan Samper, G. Prognosis and Follow-Up of Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.R.; Hall, I.P. Airway myofibroblasts and their relationship with airway myocytes and fibroblasts. Proc. Am. Thorac Soc. 2008, 5, 127–132. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [Green Version]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: The myofibroblast in focus. Chest 2007, 132, 1311–1321. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Toews, G.B.; White, E.S.; Lynch, J.P., 3rd; Martinez, F.J. Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 2004, 55, 395–417. [Google Scholar] [CrossRef]
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2015, 24, 102–114. [Google Scholar] [CrossRef]
- Richeldi, L.; Kolb, M.; Jouneau, S.; Wuyts, W.A.; Schinzel, B.; Stowasser, S.; Quaresma, M.; Raghu, G. Efficacy and safety of nintedanib in patients with advanced idiopathic pulmonary fibrosis. BMC Pulm. Med. 2020, 20, 3. [Google Scholar] [CrossRef] [Green Version]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. New Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G. Pharmacotherapy for idiopathic pulmonary fibrosis: Current landscape and future potential. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2017, 26. [Google Scholar] [CrossRef]
- Ballester, B.; Milara, J.; Cortijo, J. Mucins as a New Frontier in Pulmonary Fibrosis. J. Clin. Med. 2019, 8, 1447. [Google Scholar] [CrossRef] [Green Version]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. New Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo-Salazar, J.M.; Ma, S.F.; Jou, J.; Hou, P.C.; Guillen-Guio, B.; Allen, R.J.; Jenkins, R.G.; Wain, L.V.; Oldham, J.M.; Noth, I.; et al. Novel idiopathic pulmonary fibrosis susceptibility variants revealed by deep sequencing. ERJ Open Res. 2019, 5. [Google Scholar] [CrossRef]
- Milara, J.; Ballester, B.; Montero, P.; Escriva, J.; Artigues, E.; Alos, M.; Pastor-Clerigues, A.; Morcillo, E.; Cortijo, J. MUC1 intracellular bioactivation mediates lung fibrosis. Thorax 2020, 75, 132–142. [Google Scholar] [CrossRef]
- Milara, J.; Ballester, B.; Safont, M.J.; Artigues, E.; Escriva, J.; Morcillo, E.; Cortijo, J. MUC4 is overexpressed in idiopathic pulmonary fibrosis and collaborates with transforming growth factor beta inducing fibrotic responses. Mucosal Immunol. 2021, 14, 377–388. [Google Scholar] [CrossRef]
- Ishikawa, N.; Hattori, N.; Yokoyama, A.; Kohno, N. Utility of KL-6/MUC1 in the clinical management of interstitial lung diseases. Respir. Investig. 2012, 50, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, K.; Nagata, N.; Kumazoe, H.; Oda, K.; Ishimoto, H.; Yoshimi, M.; Takata, S.; Hamada, M.; Koreeda, Y.; Takakura, K.; et al. Prognostic value of serial serum KL-6 measurements in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2017, 55, 16–23. [Google Scholar] [CrossRef]
- Maher, T.M.; Oballa, E.; Simpson, J.K.; Porte, J.; Habgood, A.; Fahy, W.A.; Flynn, A.; Molyneaux, P.L.; Braybrooke, R.; Divyateja, H.; et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: An analysis from the multicentre PROFILE cohort study. Lancet Respir. Med. 2017, 5, 946–955. [Google Scholar] [CrossRef] [Green Version]
- Rusanov, V.; Kramer, M.R.; Raviv, Y.; Medalion, B.; Guber, A.; Shitrit, D. The significance of elevated tumor markers among patients with idiopathic pulmonary fibrosis before and after lung transplantation. Chest 2012, 141, 1047–1054. [Google Scholar] [CrossRef]
- Zheng, M.; Lou, A.; Zhang, H.; Zhu, S.; Yang, M.; Lai, W. Serum KL-6, CA19-9, CA125 and CEA are Diagnostic Biomarkers for Rheumatoid Arthritis-Associated Interstitial Lung Disease in the Chinese Population. Rheumatol. Ther. 2021, 8, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Liu, J.; Liang, L.; Ban, C.; Jiang, J.; Liu, Y.; Ye, Q.; Wang, C. Increased lung cancer risk in patients with interstitial lung disease and elevated CEA and CA125 serum tumour markers. Respirology 2014, 19, 707–713. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.J.; Beard, J.B.; Underwood, L.J.; Dennis, R.A.; Santin, A.D.; York, L. The CA 125 gene: An extracellular superstructure dominated by repeat sequences. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2001, 22, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Giamougiannis, P.; Martin-Hirsch, P.L.; Martin, F.L. The evolving role of MUC16 (CA125) in the transformation of ovarian cells and the progression of neoplasia. Carcinogenesis 2021, 42, 327–343. [Google Scholar] [CrossRef]
- Maeda, T.; Inoue, M.; Koshiba, S.; Yabuki, T.; Aoki, M.; Nunokawa, E.; Seki, E.; Matsuda, T.; Motoda, Y.; Kobayashi, A.; et al. Solution structure of the SEA domain from the murine homologue of ovarian cancer antigen CA125 (MUC16). J. Biol. Chem. 2004, 279, 13174–13182. [Google Scholar] [CrossRef] [Green Version]
- Aithal, A.; Junker, W.M.; Kshirsagar, P.; Das, S.; Kaur, S.; Orzechowski, C.; Gautam, S.K.; Jahan, R.; Sheinin, Y.M.; Lakshmanan, I.; et al. Development and characterization of carboxy-terminus specific monoclonal antibodies for understanding MUC16 cleavage in human ovarian cancer. PLoS ONE 2018, 13, e0193907. [Google Scholar] [CrossRef]
- Das, S.; Majhi, P.D.; Al-Mugotir, M.H.; Rachagani, S.; Sorgen, P.; Batra, S.K. Membrane proximal ectodomain cleavage of MUC16 occurs in the acidifying Golgi/post-Golgi compartments. Sci. Rep. 2015, 5, 9759. [Google Scholar] [CrossRef] [Green Version]
- Mai, P.L.; Wentzensen, N.; Greene, M.H. Challenges related to developing serum-based biomarkers for early ovarian cancer detection. Cancer Prev. Res. 2011, 4, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Einama, T.; Kamachi, H.; Nishihara, H.; Homma, S.; Kanno, H.; Takahashi, K.; Sasaki, K.; Tahara, M.; Okada, K.; Muraoka, S.; et al. Co-expression of mesothelin and CA125 correlates with unfavorable patient outcome in pancreatic ductal adenocarcinoma. Pancreas 2011, 40, 1276–1282. [Google Scholar] [CrossRef]
- Streppel, M.M.; Vincent, A.; Mukherjee, R.; Campbell, N.R.; Chen, S.H.; Konstantopoulos, K.; Goggins, M.G.; van Seuningen, I.; Maitra, A.; Montgomery, E.A. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum. Pathol. 2012, 43, 1755–1763. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, A.; Hirono, S.; Tani, M.; Kawai, M.; Okada, K.; Miyazawa, M.; Kitahata, Y.; Nakamura, Y.; Noda, T.; Yokoyama, S.; et al. Coexpression of MUC16 and mesothelin is related to the invasion process in pancreatic ductal adenocarcinoma. Cancer Sci. 2012, 103, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.R.; Kirkham, S.; Svitacheva, N.; Thornton, D.J.; Carlstedt, I. MUC16 is produced in tracheal surface epithelium and submucosal glands and is present in secretions from normal human airway and cultured bronchial epithelial cells. Int. J. Biochem. Cell Biol. 2007, 39, 1943–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bafna, S.; Kaur, S.; Batra, S.K. Membrane-bound mucins: The mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene 2010, 29, 2893–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E.; Jr Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Agostini, C.; Antoniou, K.M.; Bouros, D.; Chambers, R.C.; Cottin, V.; Egan, J.J.; Lambrecht, B.N.; Lories, R.; Parfrey, H.; et al. The pathogenesis of pulmonary fibrosis: A moving target. Eur. Respir. J. 2013, 41, 1207–1218. [Google Scholar] [CrossRef]
- Bonniaud, P.; Margetts, P.J.; Ask, K.; Flanders, K.; Gauldie, J.; Kolb, M. TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis. J. Immunol. 2005, 175, 5390–5395. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.G.; Huang, X.; Kaminski, N.; Wang, Y.; Shapiro, S.D.; Dolganov, G.; Glick, A.; Sheppard, D. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 2003, 422, 169–173. [Google Scholar] [CrossRef]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.C.; Yi, M.J.; Ran, N.; Wang, C.; Fu, P.; Feng, X.Y.; Xu, L.; Qu, Z.-H. Transforming growth factor-beta1 induces bronchial epithelial cells to mesenchymal transition by activating the Snail pathway and promotes airway remodeling in asthma. Mol. Med. Rep. 2013, 8, 1663–1668. [Google Scholar] [CrossRef]
- Michalik, M.; Wojcik-Pszczola, K.; Paw, M.; Wnuk, D.; Koczurkiewicz, P.; Sanak, M.; Pękala, E.; Madeja, Z. Fibroblast-to-myofibroblast transition in bronchial asthma. Cell. Mol. Life Sci. CMLS 2018, 75, 3943–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escrivá, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Jelovac, D.; Armstrong, D.K. Recent progress in the diagnosis and treatment of ovarian cancer. CA A Cancer J. Clin. 2011, 61, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor biomarker to cancer therapy, a work in progress. Mol. Cancer 2014, 13, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berek, J.S.; Taylor, P.T.; Gordon, A.; Cunningham, M.J.; Finkler, N.; Orr, J., Jr.; Rivkin, S.; Schultes, B.C.; Whiteside, T.L.; Nicodemus, C.F. Randomized, placebo-controlled study of oregovomab for consolidation of clinical remission in patients with advanced ovarian cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 3507–3516. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.; Harter, P.; Scambia, G.; Sehouli, J.; Meier, W.; Wimberger, P.; Baumann, K.H.; Kurzeder, C.; Schmalfeldt, B.; Cibula, D.; et al. Abagovomab as maintenance therapy in patients with epithelial ovarian cancer: A phase III trial of the AGO OVAR, COGI, GINECO, and GEICO--the MIMOSA study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 1554–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blalock, T.D.; Spurr-Michaud, S.J.; Tisdale, A.S.; Gipson, I.K. Release of membrane-associated mucins from ocular surface epithelia. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1864–1871. [Google Scholar] [CrossRef]
- Govindarajan, B.; Menon, B.B.; Spurr-Michaud, S.; Rastogi, K.; Gilmore, M.S.; Argueso, P.; Gipson, I.K. A metalloproteinase secreted by Streptococcus pneumoniae removes membrane mucin MUC16 from the epithelial glycocalyx barrier. PLoS ONE 2012, 7, e32418. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Rachagani, S.; Torres-Gonzalez, M.P.; Lakshmanan, I.; Majhi, P.D.; Smith, L.M.; Wagner, K.-U.; Batra, S.K. Carboxyl-terminal domain of MUC16 imparts tumorigenic and metastatic functions through nuclear translocation of JAK2 to pancreatic cancer cells. Oncotarget 2015, 6, 5772–5787. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Cheng, Z.; Luo, L.; Yang, Y.; Zhang, Z.; Ma, H.; Chen, T.; Huang, X.; Lin, S.; Jin, M.; et al. C-terminus of MUC16 activates Wnt signaling pathway through its interaction with beta-catenin to promote tumorigenesis and metastasis. Oncotarget 2016, 7, 36800–36813. [Google Scholar] [CrossRef] [Green Version]
- Argueso, P.; Guzman-Aranguez, A.; Mantelli, F.; Cao, Z.; Ricciuto, J.; Panjwani, N. Association of cell surface mucins with galectin-3 contributes to the ocular surface epithelial barrier. J. Biol. Chem. 2009, 284, 23037–23045. [Google Scholar] [CrossRef] [Green Version]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Van Putten, J.P.M.; Strijbis, K. Transmembrane Mucins: Signaling Receptors at the Intersection of Inflammation and Cancer. J. Innate Immun. 2017, 9, 281–299. [Google Scholar] [CrossRef]
- Kesimer, M.; Ehre, C.; Burns, K.A.; Davis, C.W.; Sheehan, J.K.; Pickles, R.J. Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal Immunol. 2013, 6, 379–392. [Google Scholar] [CrossRef]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef] [Green Version]
- Kramann, R.; DiRocco, D.P.; Humphreys, B.D. Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J. Pathol. 2013, 231, 273–289. [Google Scholar] [CrossRef]
- Evans, J.N.; Kelley, J.; Krill, J.; Low, R.B.; Adler, K.B. The myofibroblast in pulmonary fibrosis. Chest 1983, 83, 97S–98S. [Google Scholar] [CrossRef] [Green Version]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.W.; Xie, Q.M.; Chen, J.Q.; Deng, Y.M.; Tang, H.F. TGF-beta1 induces alveolar epithelial to mesenchymal transition in vitro. Life Sci. 2004, 76, 29–37. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Hung, C.; Linn, G.; Chow, Y.H.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 820–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolak, J.S.; Jagirdar, R.; Surolia, R.; Karki, S.; Oliva, O.; Hock, T.; Guroji, P.; Ding, Q.; Liu, R.; Bolisetty, S.; et al. Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am. J. Pathol. 2013, 182, 1239–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasreen, N.; Mohammed, K.A.; Mubarak, K.K.; Baz, M.A.; Akindipe, O.A.; Fernandez-Bussy, S.; Antony, V.B. Pleural mesothelial cell transformation into myofibroblasts and haptotactic migration in response to TGF-beta1 in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L115–L124. [Google Scholar] [CrossRef] [Green Version]
- Mubarak, K.K.; Montes-Worboys, A.; Regev, D.; Nasreen, N.; Mohammed, K.A.; Faruqi, I.; Hensel, E.; Baz, M.A.; Akindipe, O.A.; Fernandez-Bussy, S.; et al. Parenchymal trafficking of pleural mesothelial cells in idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 39, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Hattrup, C.L.; Gendler, S.J. Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol. 2008, 70, 431–457. [Google Scholar] [CrossRef]
- Waters, D.W.; Blokland, K.E.C.; Pathinayake, P.S.; Burgess, J.K.; Mutsaers, S.E.; Prele, C.M.; Schuliga, M.; Grainge, C.L.; Knight, D.A. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L162–L172. [Google Scholar] [CrossRef] [Green Version]
- DePianto, D.J.; Vander Heiden, J.A.; Morshead, K.B.; Sun, K.H.; Modrusan, Z.; Teng, G.; Wolters, P.; Arron, J.R. Molecular mapping of interstitial lung disease reveals a phenotypically distinct senescent basal epithelial cell population. JCI Insight 2021, 6, e143626. [Google Scholar] [CrossRef]
- Placido, L.; Romero, Y.; Maldonado, M.; Toscano-Marquez, F.; Ramirez, R.; Calyeca, J.; Mora, A.L.; Selman, M.; Pardo, A. Loss of MT1-MMP in Alveolar Epithelial Cells Exacerbates Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 2923. [Google Scholar] [CrossRef]
- Jager, B.; Seeliger, B.; Terwolbeck, O.; Warnecke, G.; Welte, T.; Muller, M.; Bode, C.; Prasse, A. The NLRP3-Inflammasome-Caspase-1 Pathway Is Upregulated in Idiopathic Pulmonary Fibrosis and Acute Exacerbations and Is Inducible by Apoptotic A549 Cells. Front. Immunol. 2021, 12, 642855. [Google Scholar] [CrossRef]
- Geng, J.; Huang, X.; Li, Y.; Xu, X.; Li, S.; Jiang, D.; Liang, J.; Jiang, D.; Wang, C.; Dai, H. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 124. [Google Scholar] [CrossRef] [Green Version]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef]
- Mata, M.; Sarria, B.; Buenestado, A.; Cortijo, J.; Cerda, M.; Morcillo, E.J. Phosphodiesterase 4 inhibition decreases MUC5AC expression induced by epidermal growth factor in human airway epithelial cells. Thorax 2005, 60, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Milara, J.; Navarro, R.; Juan, G.; Peiro, T.; Serrano, A.; Ramon, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Donor Subjects (n = 17) | IPF Patients (n = 20) | Mean Difference (CI) | p-Value | |
|---|---|---|---|---|
| Age (yr) | 59 (53.5–62) | 61 (55.5–64.5) | 2 (−7 to 2) | 0.2516 |
| Sex (Male/Female) | 13/4 | 15/5 | ||
| Smoking | ||||
| Never smoked/Smokers | 3/14 | 4/16 | ||
| Pack-year | 25 (20–30) | 26 (10.5–32.5) | 1 (−8 to 4) | 0.4782 |
| FEV1, pred | ND | 62 (56.5–70.5) | ||
| FVC, % pred | ND | 55 (51–65.5) | ||
| DLco, % pred | ND | 34 (25–44) | ||
| PaO2, mmHg | 94 (90.5–95) | 51 (45.5–60.5) | 41.8 (35 to 46) | <0.001 * |
| NAC (y/n) | 0 | 16/6 | ||
| Pirfenidone (y/n) | 0 | 6/16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballester, B.; Milara, J.; Montero, P.; Cortijo, J. MUC16 Is Overexpressed in Idiopathic Pulmonary Fibrosis and Induces Fibrotic Responses Mediated by Transforming Growth Factor-β1 Canonical Pathway. Int. J. Mol. Sci. 2021, 22, 6502. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126502
Ballester B, Milara J, Montero P, Cortijo J. MUC16 Is Overexpressed in Idiopathic Pulmonary Fibrosis and Induces Fibrotic Responses Mediated by Transforming Growth Factor-β1 Canonical Pathway. International Journal of Molecular Sciences. 2021; 22(12):6502. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126502
Chicago/Turabian StyleBallester, Beatriz, Javier Milara, Paula Montero, and Julio Cortijo. 2021. "MUC16 Is Overexpressed in Idiopathic Pulmonary Fibrosis and Induces Fibrotic Responses Mediated by Transforming Growth Factor-β1 Canonical Pathway" International Journal of Molecular Sciences 22, no. 12: 6502. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126502