
Chrysomycin A Attenuates Neuroinflammation by Down-Regulating NLRP3/Cleaved Caspase-1 Signaling Pathway in LPS-Stimulated Mice and BV2 Cells

 and
and 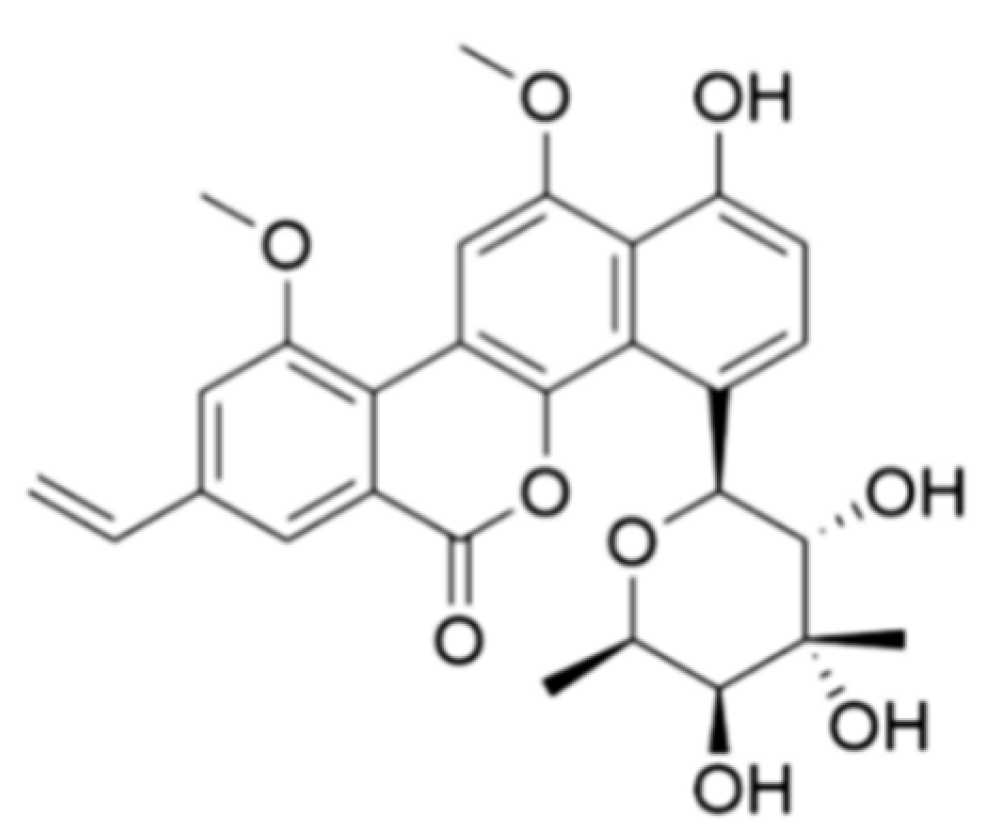{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
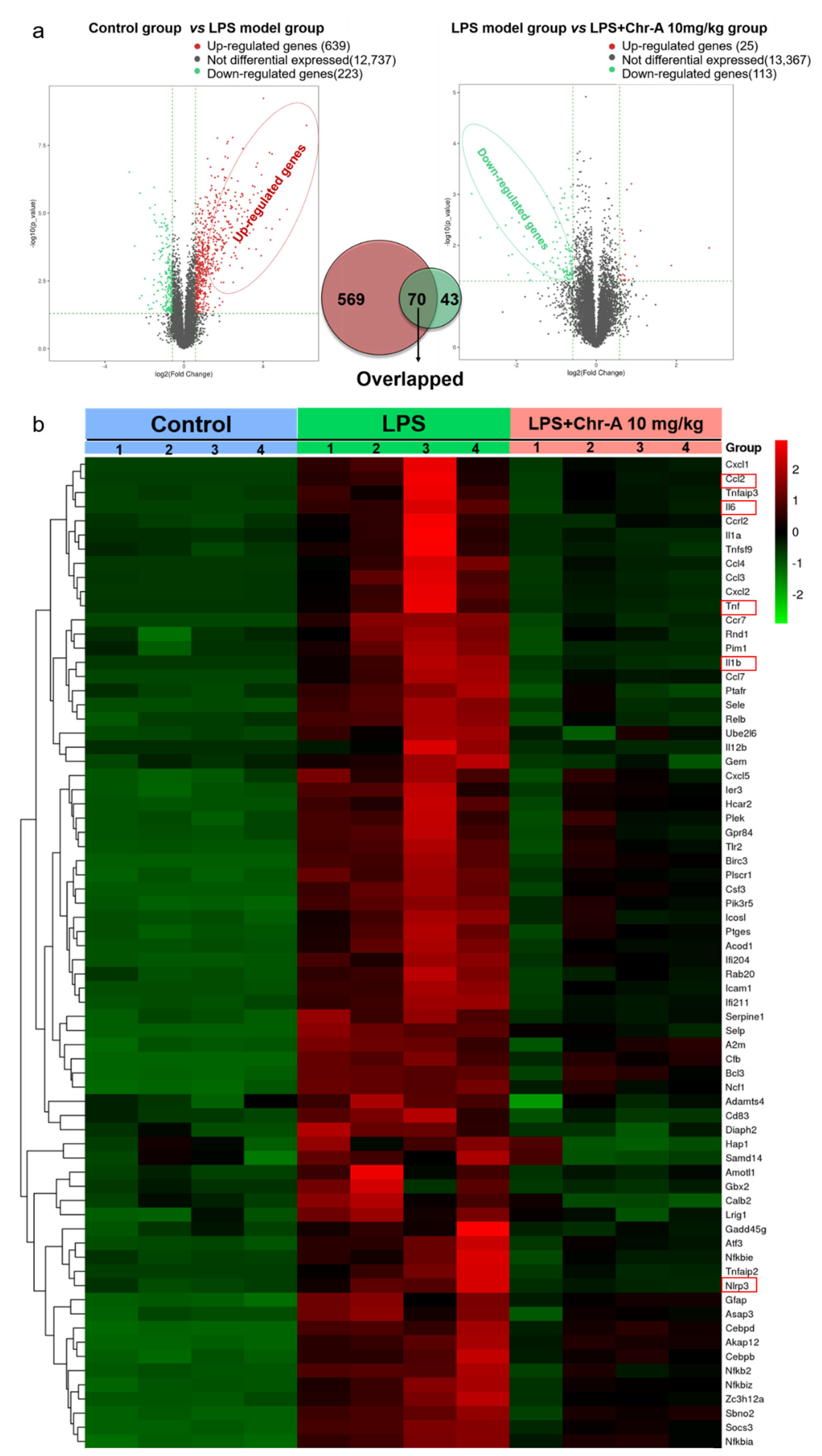
2.1. Identifying Differential Expression Genes after Chr-A Treatment in LPS-Stimulated Mice by RNA-Seq Transcriptome Analysis
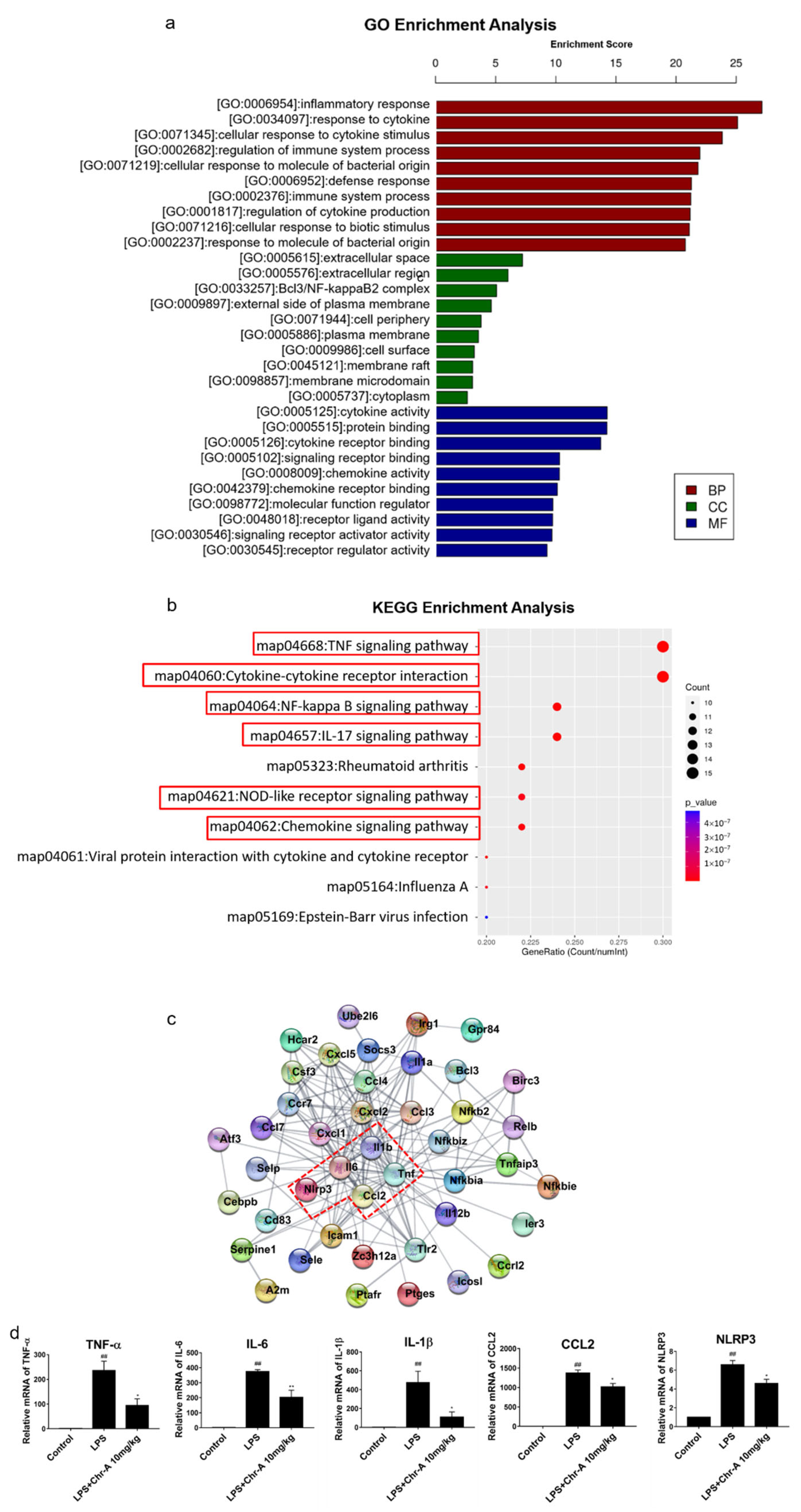
2.2. Enrichment Analysis of Core Genes for Chr-A against Neuroinflammatory
2.3. Chr-A Inhibits Neuroinflammation in the Cortex Tissues of LPS-Induced Mice
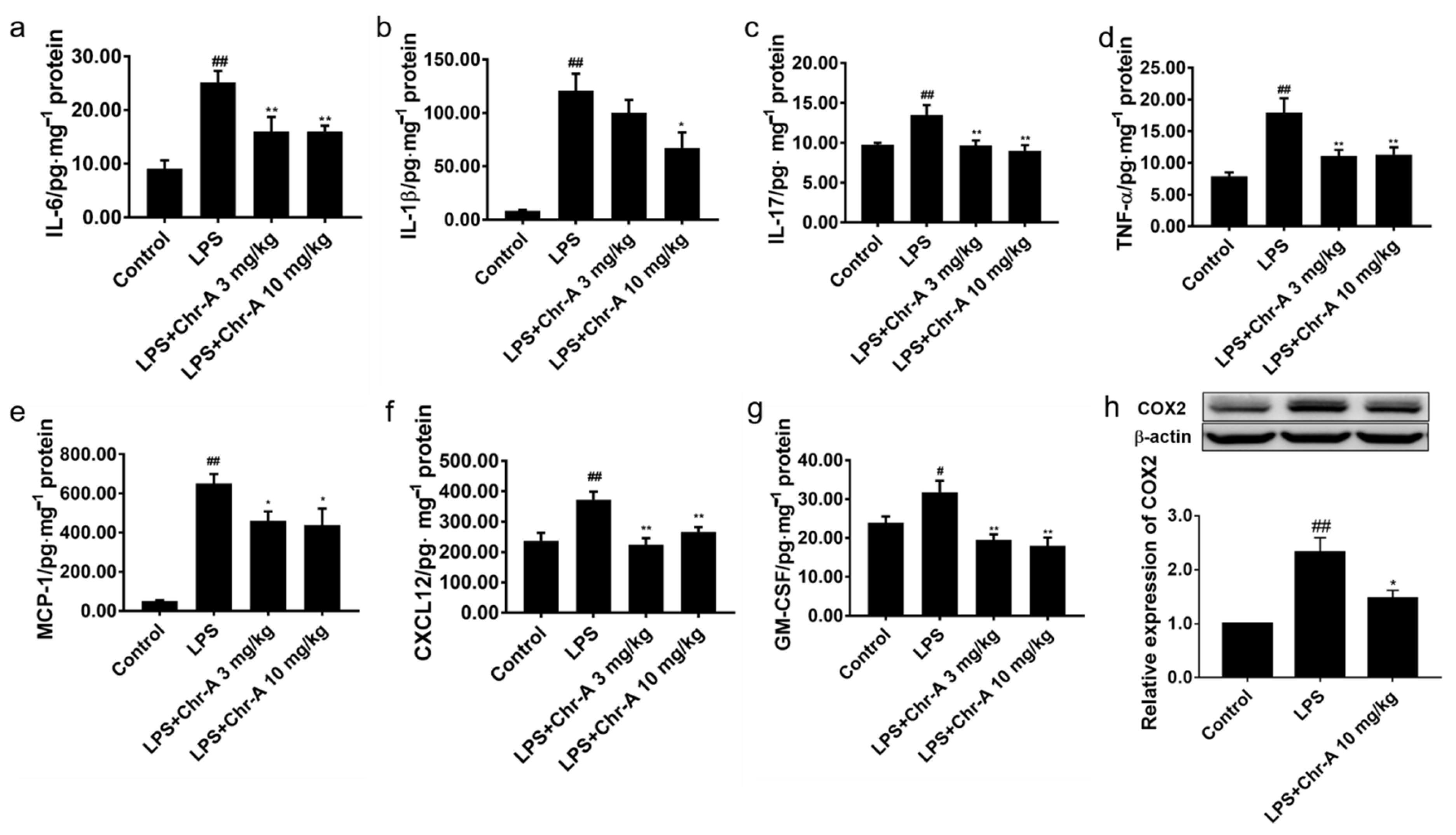
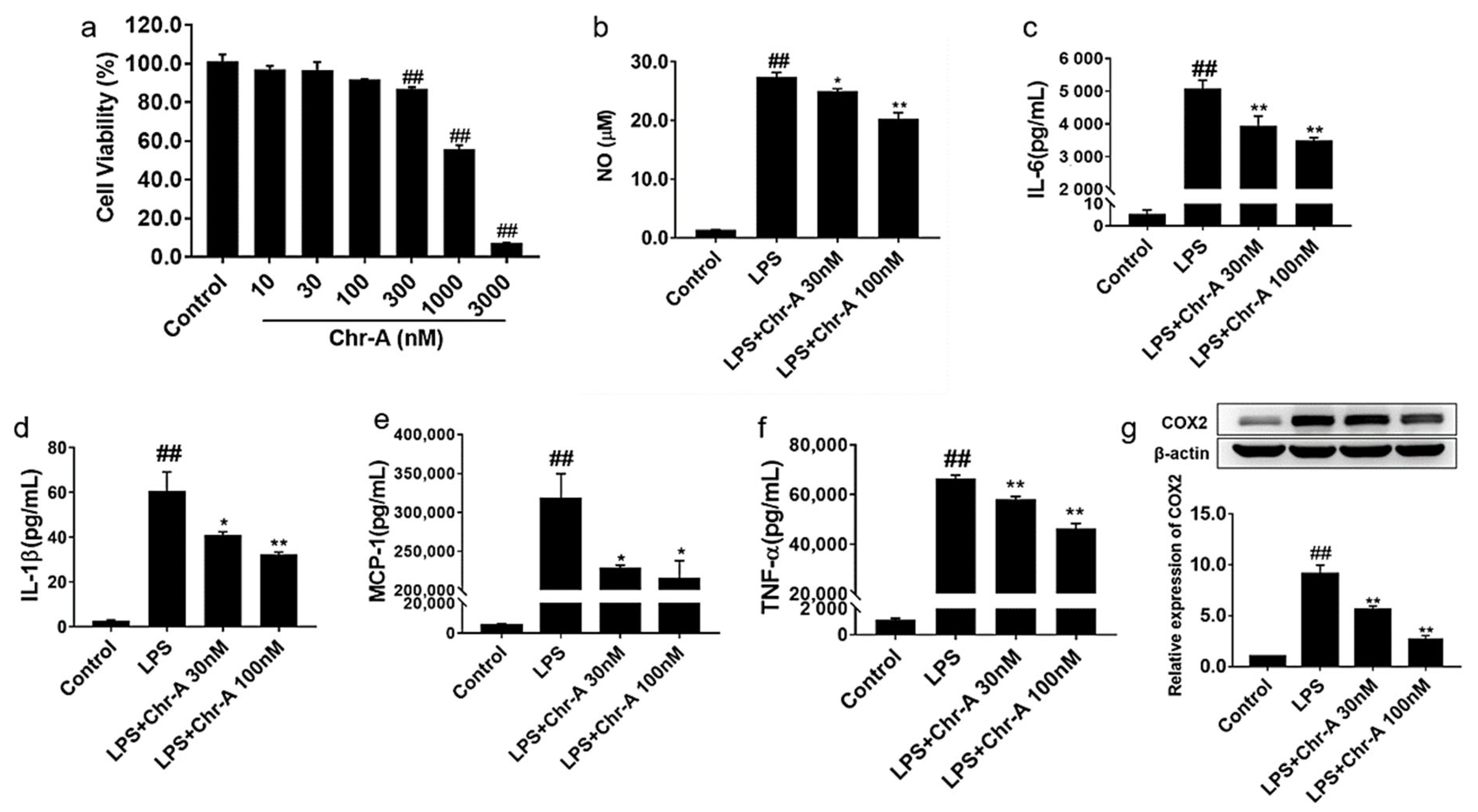
2.4. Chr-A Reduced Inflammatory Factors Production in the Supernatants of LPS-Stimulated BV2 Microglia Cells
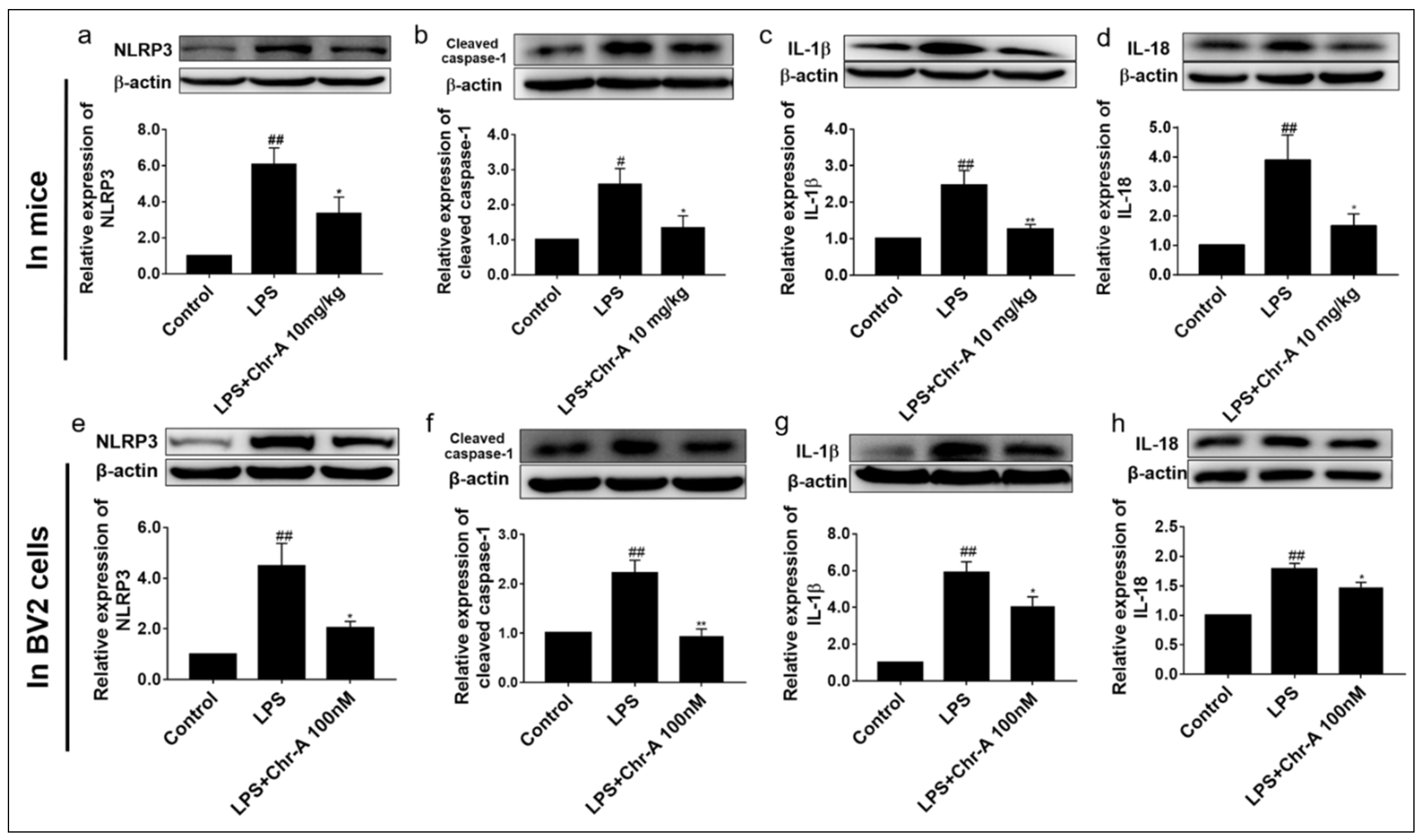
2.5. Chr-A Down-Regulated NLRP3/Cleaved Caspase-1 Signaling Pathway in LPS-Induced Injury Both In Vivo and In Vitro
3. Discussion
4. Materials and Methods
4.1. Regents
4.2. Experimental Procedure of LPS-Stimulated Mice
4.3. RNA-Seq and Enrichment Analysis
4.4. Protein–Protein Interaction (PPI) Network Construction of Core Targets
4.5. BV2 Microglia Cells Culture and Treatment
4.6. NO Determination in the Supernatant of BV2 Cells
4.7. Inflammatory Factors Determined by ELISA Assay
4.8. Real-Time PCR Analysis
4.9. Western Blot Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheng, X.; Yang, Y.-L.; Li, W.-H.; Liu, M.; Wang, Y.-H.; Du, G.-H. Cerebral ischemia-reperfusion aggravated cerebral infarction injury and possible differential genes identified by RNA-Seq in rats. Brain Res. Bull. 2020, 156, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Kustrimovic, N.; Marino, F.; Cosentino, M. Peripheral Immunity, Immunoaging and Neuroinflammation in Parkinson’s Disease. Curr. Med. Chem. 2019, 26, 3719–3753. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflamm. 2012, 9, 155. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Hua, X.; Kong, D.; Stein, D.; Hua, F. Role of Toll-like receptor mediated signaling in traumatic brain injury. Neuropharmacology 2019, 145, 259–267. [Google Scholar] [CrossRef]
- Qin, L.; Li, G.; Qian, X.; Liu, Y.; Wu, X.; Liu, B.; Hong, J.-S.; Block, M.L. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia 2005, 52, 78–84. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Licastro, F.; Pedrini, S.; Caputo, L.; Annoni, G.; Davis, L.J.; Ferri, C.; Casadei, V.; Grimaldi, L.M. Increased plasma levels of interleukin-1, interleukin-6 and alpha-1-antichymotrypsin in patients with Alzheimer’s disease: Peripheral inflammation or signals from the brain? J. Neuroimmunol. 2000, 103, 97–102. [Google Scholar] [CrossRef]
- Huang, W.-X.; Huang, P.; Hillert, J. Increased expression of caspase-1 and interleukin-18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult. Scler. J. 2004, 10, 482–487. [Google Scholar] [CrossRef]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [Green Version]
- Strelitz, F.; Flon, H.; Asheshov, I.N. Chrysomycin: A New Antibiotic Substance for Bacterial Viruses. J. Bacteriol. 1955, 69, 280–283. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.T.; Byrne, K.M.; Warnick-Pickle, D.; Greenstein, M. Studies on the mechanism of action of gilvocarcin V and chrysomycin A. J. Antibiot. 1982, 35, 545–548. [Google Scholar] [CrossRef] [Green Version]
- Weiss, U.; Yoshihira, K.; Highet, R.J.; White, R.J.; Wei, T.T. The chemistry of the antibiotics chrysomycin A and B antitumor activity of chrysomycin A. J. Antibiot. 1982, 35, 1194–1201. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhang, J.; Song, F.; Wang, S.; Guo, H.; Wei, Q.; Dai, H.; Chen, X.; Xia, X.; Liu, X.; et al. Chrysomycin A Derivatives for the Treatment of Multi-Drug-Resistant Tuberculosis. ACS Cent. Sci. 2020, 6, 928–938. [Google Scholar] [CrossRef]
- Pan, D.S.; Liu, W.G.; Yang, X.F.; Cao, F. Inhibitory effect of progesterone on inflammatory factors after experimental traumatic brain injury. Biomed. Environ. Sci. 2007, 20, 432–438. [Google Scholar]
- Fuster-Matanzo, A.; Llorens-Martín, M.; Hernández, F.; Avila, J. Role of neuroinflammation in adult neurogenesis and Alzheimer disease: Therapeutic approaches. Mediat. Inflamm. 2013, 2013, 260925. [Google Scholar] [CrossRef] [Green Version]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell Neurosci. 2000, 16, 724–739. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.L.; Liu, M.; Cheng, X.; Li, W.H.; Zhang, S.S.; Wang, Y.H.; Du, G.H. Myricitrin blocks activation of NF-κB and MAPK signaling pathways to protect nigrostriatum neuron in LPS-stimulated mice. J. Neuroimmunol. 2019, 337, 577049. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Yang, Y.-L.; Yang, H.; Wang, Y.-H.; Du, G.-H. Kaempferol alleviates LPS-induced neuroinflammation and BBB dysfunction in mice via inhibiting HMGB1 release and down-regulating TLR4/MyD88 pathway. Int. Immunopharmacol. 2018, 56, 29–35. [Google Scholar] [CrossRef]
- Yang, H.; Cheng, X.; Yang, Y.L.; Wang, Y.H.; Du, G.H. Ramulus Cinnamomi extract attenuates neuroinflammatory responses via downregulating TLR4/MyD88 signaling pathway in BV2 cells. Neural Regen. Res. 2017, 12, 1860–1864. [Google Scholar]
- Yang, Y.-L.; Cheng, X.; Li, W.-H.; Liu, M.; Wang, Y.-H.; Du, G.-H. Kaempferol Attenuates LPS-Induced Striatum Injury in Mice Involving Anti-Neuroinflammation, Maintaining BBB Integrity, and Down-Regulating the HMGB1/TLR4 Pathway. Int. J. Mol. Sci. 2019, 20, 491. [Google Scholar] [CrossRef] [Green Version]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Asadi, S.; Patel, A.B. Focal brain inflammation and autism. J. Neuroinflamm. 2013, 10, 815–846. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Jin, Y.; Li, J. Protective role of fentanyl in lipopolysaccharide-induced neuroinflammation in BV-2 cells. Exp. Ther. Med. 2018, 16, 3740–3744. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflamm. 2015, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Rawat, C.; Kukal, S.; Dahiya, U.R.; Kukreti, R. Cyclooxygenase-2 (COX-2) inhibitors: Future therapeutic strategies for epilepsy management. J. Neuroinflamm. 2019, 16, 197. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, S.; Shah, F.A.; Naeem, K.; Nadeem, H.; Sarwar, S.; Ashraf, Z.; Imran, M.; Khan, T.; Tayyaba Anwar, T.; Li, S. Succinamide Derivatives Ameliorate Neuroinflammation and Oxidative Stress in Scopolamine-Induced Neurodegeneration. Biomolecules 2020, 10, 443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.M.; Fish, E.N. Chemokines: Attractive mediators of the immune response. Semin. Immunol. 2003, 15, 5–14. [Google Scholar] [CrossRef]
- Olson, T.S.; Ley, K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Integr. Comp. Physiol. 2002, 283, R7–R28. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomerantz, J.L.; Baltimore, D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999, 18, 6694–6704. [Google Scholar] [CrossRef]
- Cortez, D.M.; Feldman, M.D.; Mummidi, S.; Valente, A.J.; Steffensen, B.; Vincenti, M.; Barnes, J.L.; Chandrasekar, B. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta, NF-κappaB, and AP-1 activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3356–H3365. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Verma, I.M. NF-κappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, J.; Jiang, W.; Cao, Z.; Zhao, F.; Cai, T.; Aschner, M.; Luo, W. The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy 2017, 13, 914–927. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Mei, X. Role of NLRP3 Inflammasomes in Neuroinflammation Diseases. Eur. Neurol. 2020, 83, 576–580. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, A.; Lv, J.; Zhang, Q.; Ran, Y.; Wei, C.; Wu, J. Development of small molecule inhibitors targeting NLRP3 inflammasome pathway for inflammatory diseases. Eur. J. Med. Chem. 2020, 185, 111822. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nat. Cell Biol. 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, Y.; Zhou, R.; Li, Y.; Gao, Y.; Tu, D.; Wilson, B.; Song, S.; Feng, J.; Hong, J.S.; et al. A novel role of NLRP3-generated IL-1β in the acute-chronic transition of peripheral lipopolysaccharide-elicited neuroinflammation: Implications for sepsis-associated neurodegeneration. J. Neuroinflamm. 2020, 17, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Zhang, S.-S.; Liu, D.-N.; Yang, Y.-L.; Wang, Y.-H.; Du, G.-H. Chrysomycin A Attenuates Neuroinflammation by Down-Regulating NLRP3/Cleaved Caspase-1 Signaling Pathway in LPS-Stimulated Mice and BV2 Cells. Int. J. Mol. Sci. 2021, 22, 6799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136799
Liu M, Zhang S-S, Liu D-N, Yang Y-L, Wang Y-H, Du G-H. Chrysomycin A Attenuates Neuroinflammation by Down-Regulating NLRP3/Cleaved Caspase-1 Signaling Pathway in LPS-Stimulated Mice and BV2 Cells. International Journal of Molecular Sciences. 2021; 22(13):6799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136799
Chicago/Turabian StyleLiu, Man, Shan-Shan Zhang, Dong-Ni Liu, Ying-Lin Yang, Yue-Hua Wang, and Guan-Hua Du. 2021. "Chrysomycin A Attenuates Neuroinflammation by Down-Regulating NLRP3/Cleaved Caspase-1 Signaling Pathway in LPS-Stimulated Mice and BV2 Cells" International Journal of Molecular Sciences 22, no. 13: 6799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136799