
Differential Expression of Inflammasome-Related Genes in Induced Pluripotent Stem-Cell-Derived Retinal Pigment Epithelial Cells with or without History of Age-Related Macular Degeneration

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
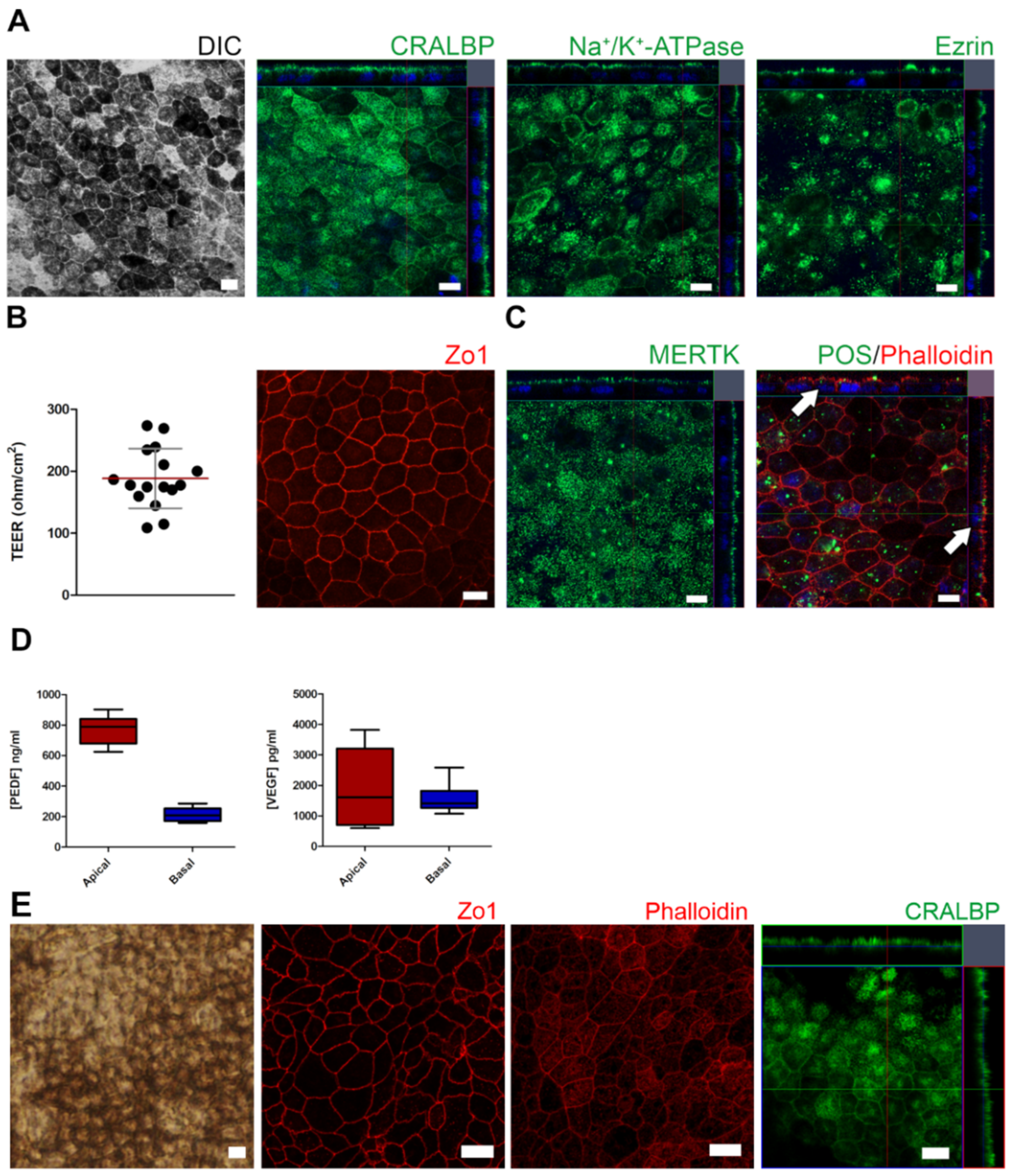
2.1. Control and AMD-Patient-Derived iPSC-RPE Monolayers Showed Mature RPE Characteristics
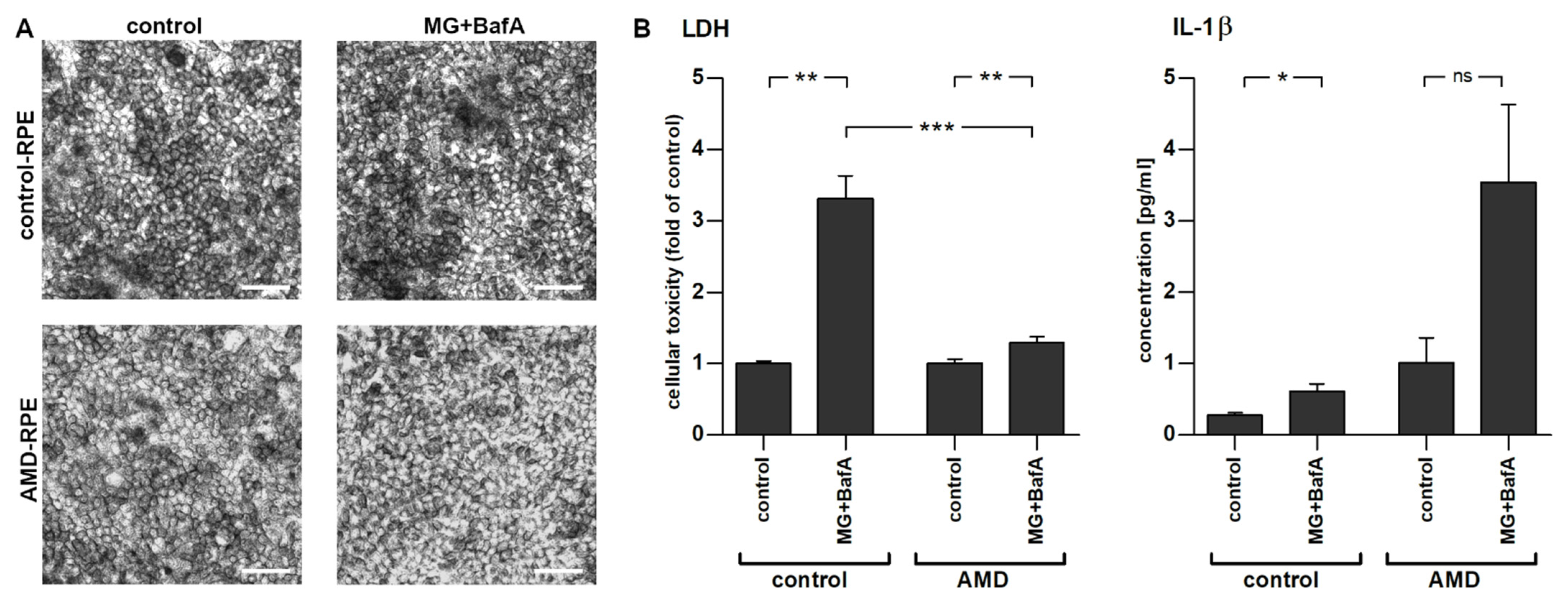
2.2. Inflammasome Activation in AMD- and Control-Patient-Derived iPSC-RPE Cell Lines
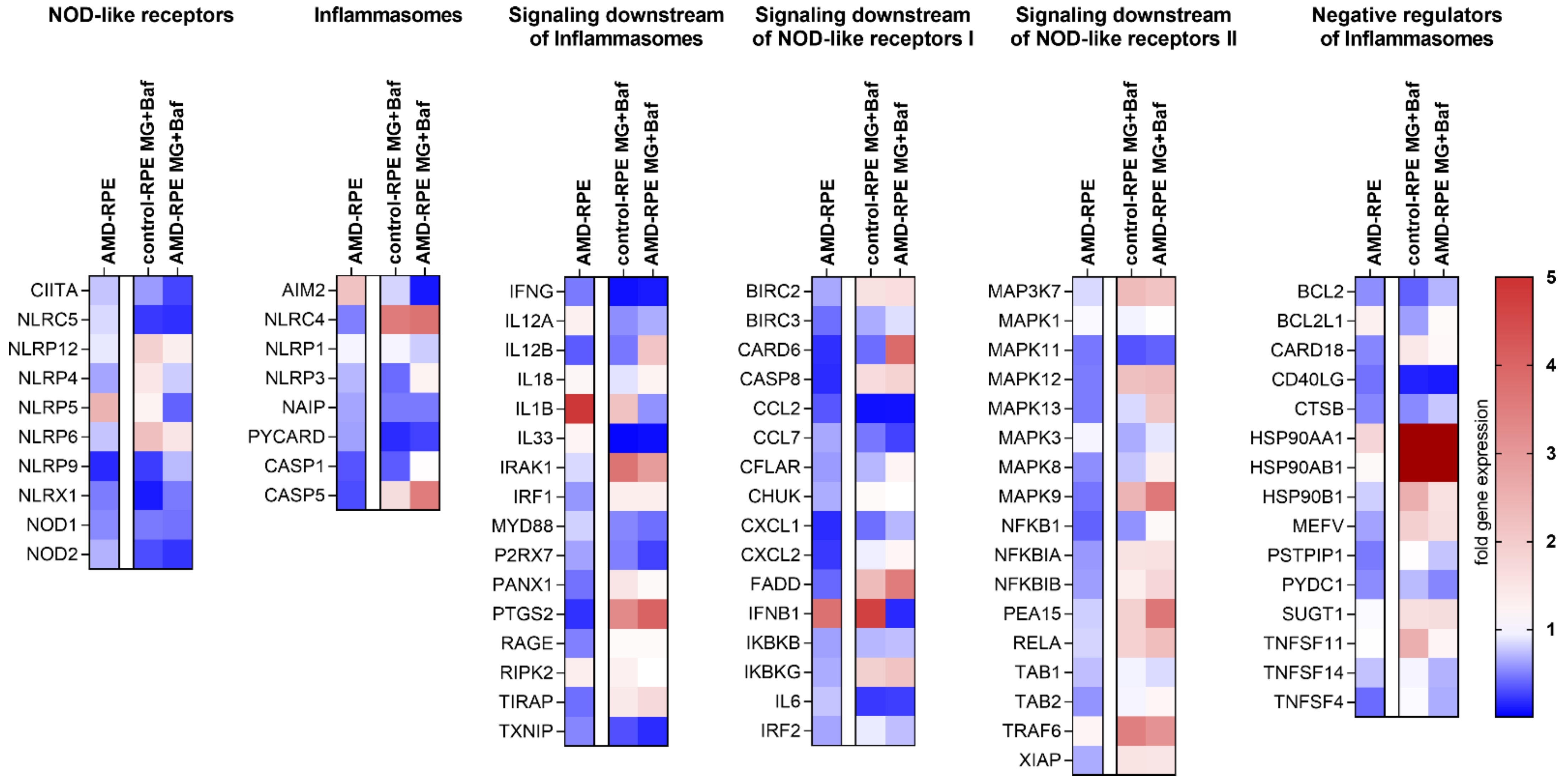
2.3. Inflammasome-Related Gene Expression in iPSC-Derived RPE Cells
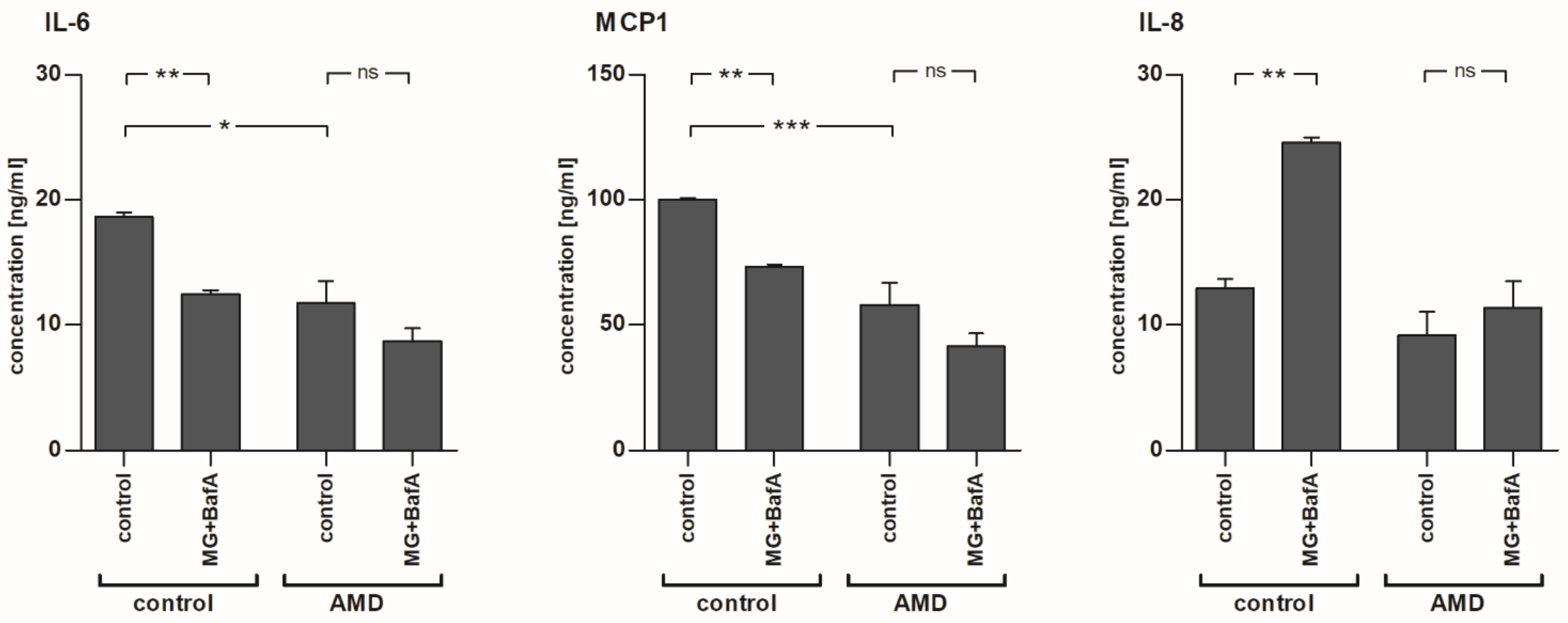
2.4. The Levels of IL-6 and MCP-1, but Not IL-8, Were Reduced after MG-132 and Bafilomycin A1 Exposure in Control-RPE Cells
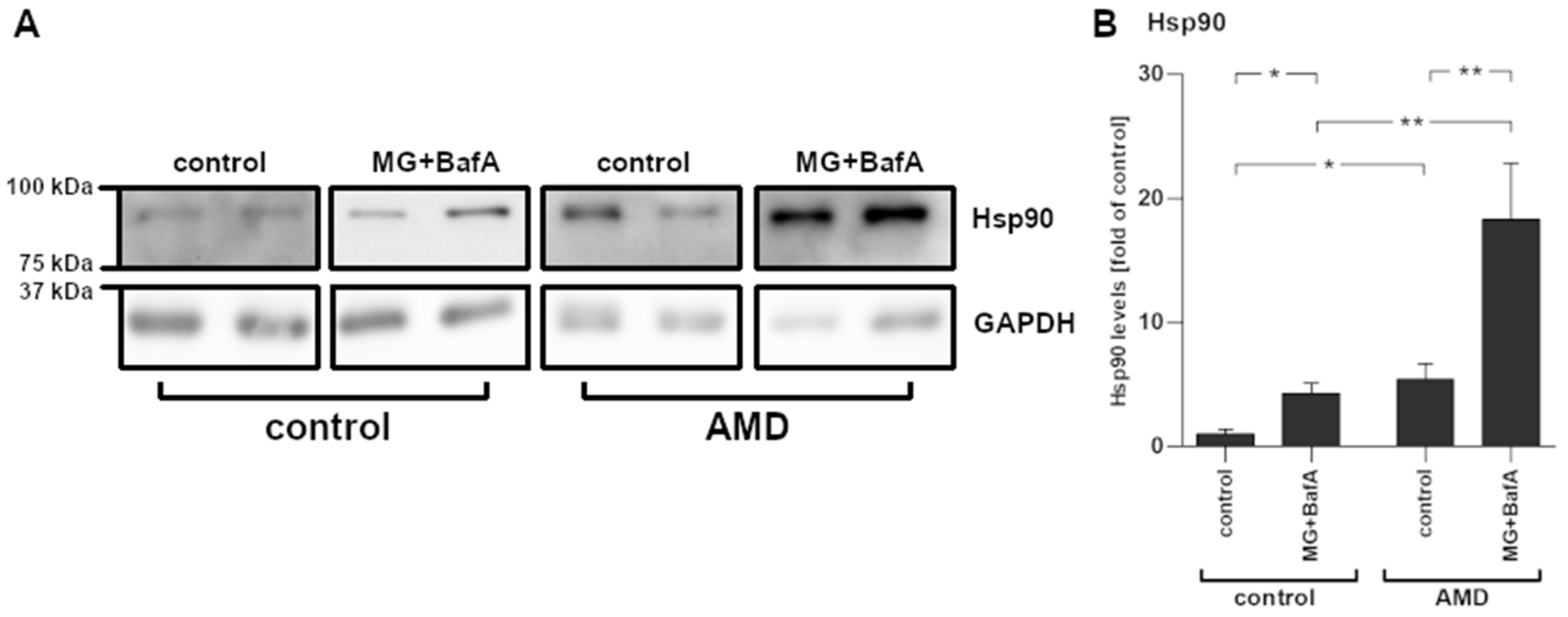
2.5. Increased Hsp90 mRNA Expression Levels Corresponded with Protein Levels
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Lactate Dehydrogenase (LDH) Assay
4.3. Enzyme-Linked Immunosorbent Assay (ELISA)
4.4. Polymerase Chain Reaction (PCR) Array
4.5. Western Blotting
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colijn, J.M.; Buitendijk, G.H.S.; Prokofyeva, E.; Alves, D.; Cachulo, M.L.; Khawaja, A.P.; Cougnard-Gregoire, A.; Merle, B.M.J.; Korb, C.; Erke, M.G.; et al. Prevalence of Age-Related Macular Degeneration in Europe: The Past and the Future. Ophthalmology 2017, 124, 1753–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, J.B.; Cheung, C.M.G.; Panda-Jonas, S. Updates on the Epidemiology of Age-Related Macular Degeneration. Asia Pac. J. Ophthalmol. 2017, 6, 493–497. [Google Scholar] [CrossRef]
- Kauppinen, A. Introduction to the multi-author review on macular degeneration. Cell. Mol. Life Sci. 2020, 77, 779–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [Green Version]
- Ferrington, D.A.; Fisher, C.R.; Kowluru, R.A. Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol. Med. 2020, 26, 105–118. [Google Scholar] [CrossRef]
- Blasiak, J.; Pawlowska, E.; Szczepanska, J.; Kaarniranta, K. Interplay between Autophagy and the Ubiquitin-Proteasome System and Its Role in the Pathogenesis of Age-Related Macular Degeneration. Int. J. Mol. Sci. 2019, 20, 210. [Google Scholar] [CrossRef] [Green Version]
- Abokyi, S.; To, C.H.; Lam, T.T.; Tse, D.Y. Central Role of Oxidative Stress in Age-Related Macular Degeneration: Evidence from a Review of the Molecular Mechanisms and Animal Models. Oxid. Med. Cell Longev. 2020, 2020, 7901270. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Li, Y.; Chan, L.; Tsai, Y.T.; Wu, W.H.; Nguyen, H.V.; Hsu, C.W.; Li, X.; Brown, L.M.; Egli, D.; et al. Validation of genome-wide association study (GWAS)-identified disease risk alleles with patient-specific stem cell lines. Hum. Mol. Genet. 2014, 23, 3445–3455. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, W.H.; Hsu, C.W.; Nguyen, H.V.; Tsai, Y.T.; Chan, L.; Nagasaki, T.; Maumenee, I.H.; Yannuzzi, L.A.; Hoang, Q.V.; et al. Gene therapy in patient-specific stem cell lines and a preclinical model of retinitis pigmentosa with membrane frizzled-related protein defects. Mol. Ther. 2014, 22, 1688–1697. [Google Scholar] [CrossRef] [Green Version]
- Lukovic, D.; Artero Castro, A.; Delgado, A.B.; Bernal Mde, L.; Luna Pelaez, N.; Díez Lloret, A.; Perez Espejo, R.; Kamenarova, K.; Fernandez Sanchez, L.; Cuenca, N.; et al. Human iPSC derived disease model of MERTK-associated retinitis pigmentosa. Sci. Rep. 2015, 5, 12910. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Shen, W.; Kuai, D.; Martin, J.M.; Guo, X.; Smith, M.A.; Perez, E.T.; Phillips, M.J.; Simonett, J.M.; Wallace, K.A.; et al. iPS cell modeling of Best disease: Insights into the pathophysiology of an inherited macular degeneration. Hum. Mol. Genet. 2013, 22, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kuai, D.; Guziewicz, K.E.; Meyer, J.; Wilson, M.; Lu, J.; Smith, M.; Clark, E.; Verhoeven, A.; Aguirre, G.D.; et al. Pharmacological Modulation of Photoreceptor Outer Segment Degradation in a Human iPS Cell Model of Inherited Macular Degeneration. Mol. Ther. 2015, 23, 1700–1711. [Google Scholar] [CrossRef] [Green Version]
- May-Simera, H.L.; Wan, Q.; Jha, B.S.; Hartford, J.; Khristov, V.; Dejene, R.; Chang, J.; Patnaik, S.; Lu, Q.; Banerjee, P.; et al. Primary Cilium-Mediated Retinal Pigment Epithelium Maturation Is Disrupted in Ciliopathy Patient Cells. Cell Rep. 2018, 22, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Polinati, P.P.; Ilmarinen, T.; Trokovic, R.; Hyotylainen, T.; Otonkoski, T.; Suomalainen, A.; Skottman, H.; Tyni, T. Patient-Specific Induced Pluripotent Stem Cell-Derived RPE Cells: Understanding the Pathogenesis of Retinopathy in Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3371–3382. [Google Scholar] [CrossRef] [Green Version]
- Kiamehr, M.; Klettner, A.; Richert, E.; Koskela, A.; Koistinen, A.; Skottman, H.; Kaarniranta, K.; Aalto-Setälä, K.; Juuti-Uusitalo, K. Compromised Barrier Function in Human Induced Pluripotent Stem-Cell-Derived Retinal Pigment Epithelial Cells from Type 2 Diabetic Patients. Int. J. Mol. Sci. 2019, 20, 3773. [Google Scholar] [CrossRef] [Green Version]
- Hallam, D.; Collin, J.; Bojic, S.; Chichagova, V.; Buskin, A.; Xu, Y.; Lafage, L.; Otten, E.G.; Anyfantis, G.; Mellough, C.; et al. An Induced Pluripotent Stem Cell Patient Specific Model of Complement Factor H (Y402H) Polymorphism Displays Characteristic Features of Age-Related Macular Degeneration and Indicates a Beneficial Role for UV Light Exposure. Stem Cells 2017, 35, 2305–2320. [Google Scholar] [CrossRef] [Green Version]
- Saini, J.S.; Corneo, B.; Miller, J.D.; Kiehl, T.R.; Wang, Q.; Boles, N.C.; Blenkinsop, T.A.; Stern, J.H.; Temple, S. Nicotinamide Ameliorates Disease Phenotypes in a Human iPSC Model of Age-Related Macular Degeneration. Cell Stem Cell 2017, 20, 635–647.e7. [Google Scholar] [CrossRef] [Green Version]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med. 2016, 14, 344. [Google Scholar] [CrossRef] [Green Version]
- Piippo, N.; Korkmaz, A.; Hytti, M.; Kinnunen, K.; Salminen, A.; Atalay, M.; Kaarniranta, K.; Kauppinen, A. Decline in cellular clearance systems induces inflammasome signaling in human ARPE-19 cells. Biochim. Biophys. Acta 2014, 1843, 3038–3046. [Google Scholar] [CrossRef] [Green Version]
- Piippo, N.; Korhonen, E.; Hytti, M.; Kinnunen, K.; Kaarniranta, K.; Kauppinen, A. Oxidative Stress is the Principal Contributor to Inflammasome Activation in Retinal Pigment Epithelium Cells with Defunct Proteasomes and Autophagy. Cell Physiol. Biochem. 2018, 49, 359–367. [Google Scholar] [CrossRef]
- Piippo, N.; Korhonen, E.; Hytti, M.; Skottman, H.; Kinnunen, K.; Josifovska, N.; Petrovski, G.; Kaarniranta, K.; Kauppinen, A. Hsp90 inhibition as a means to inhibit activation of the NLRP3 inflammasome. Sci. Rep. 2018, 8, 6720. [Google Scholar] [CrossRef] [Green Version]
- Litwinska, Z.; Sobus, A.; Luczkowska, K.; Grabowicz, A.; Mozolewska-Piotrowska, K.; Safranow, K.; Kawa, M.P.; Machalinski, B.; Machalinska, A. The Interplay Between Systemic Inflammatory Factors and MicroRNAs in Age-Related Macular Degeneration. Front. Aging Neurosci. 2019, 11, 286. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; George, S.; Rosner, B.; Rifai, N. Progression of age-related macular degeneration: Prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch. Ophthalmol. 2005, 123, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Xu, H. Parainflammation, chronic inflammation, and age-related macular degeneration. J. Leukoc. Biol. 2015, 98, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Ko, A.; Partanen, M.; Pakzad-Vaezi, K.; Merkur, A.B.; Albiani, D.A.; Kirker, A.W.; Wang, A.; Cui, J.Z.; Forooghian, F.; et al. Relationship between systemic cytokines and complement factor H Y402H polymorphism in patients with dry age-related macular degeneration. Am. J. Ophthalmol. 2013, 156, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassar, K.; Grisanti, S.; Elfar, E.; Lüke, J.; Lüke, M.; Grisanti, S. Serum cytokines as biomarkers for age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Celkova, L.; Doyle, S.L.; Campbell, M. NLRP3 Inflammasome and Pathobiology in AMD. J. Clin. Med. 2015, 4, 172–192. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Liu, R.T.; Cao, S.; Cui, J.Z.; Wang, A.; To, E.; Matsubara, J.A. NLRP3 inflammasome: Activation and regulation in age-related macular degeneration. Mediat. Inflamm. 2015, 2015, 690243. [Google Scholar] [CrossRef] [Green Version]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef]
- Viiri, J.; Hyttinen, J.M.T.; Ryhänen, T.; Rilla, K.; Paimela, T.; Kuusisto, E.; Siitonen, A.; Urtti, A.; Salminen, A.; Kaarniranta, K. p62/sequestosome 1 as a regulator of proteasome inhibitor-induced autophagy in human retinal pigment epithelial cells. Mol. Vis. 2010, 16, 1399–1414. [Google Scholar] [CrossRef]
- Juuti-Uusitalo, K.; Koskela, A.; Kivinen, N.; Viiri, J.; Hyttinen, J.M.T.; Reinisalo, M.; Koistinen, A.; Uusitalo, H.; Sinha, D.; Skottman, H.; et al. Autophagy Regulates Proteasome Inhibitor-Induced Pigmentation in Human Embryonic Stem Cell-Derived Retinal Pigment Epithelial Cells. Int. J. Mol. Sci. 2017, 18, 1089. [Google Scholar] [CrossRef] [Green Version]
- Hongisto, H.; Ilmarinen, T.; Vattulainen, M.; Mikhailova, A.; Skottman, H. Xeno- and feeder-free differentiation of human pluripotent stem cells to two distinct ocular epithelial cell types using simple modifications of one method. Stem Cell Res. Ther. 2017, 8, 291. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chan, L.; Nguyen, H.V.; Tsang, S.H. Personalized Medicine: Cell and Gene Therapy Based on Patient-Specific iPSC-Derived Retinal Pigment Epithelium Cells. Adv. Exp. Med. Biol. 2016, 854, 549–555. [Google Scholar] [CrossRef]
- Greene, W.A.; Kaini, R.R.; Wang, H.C. Utility of Induced Pluripotent Stem Cell-Derived Retinal Pigment Epithelium for an In Vitro Model of Proliferative Vitreoretinopathy. Adv. Exp. Med. Biol. 2019, 1186, 33–53. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef]
- Zuo, Y.; Wang, J.; Liao, F.; Yan, X.; Li, J.; Huang, L.; Liu, F. Inhibition of Heat Shock Protein 90 by 17-AAG Reduces Inflammation via P2X7 Receptor/NLRP3 Inflammasome Pathway and Increases Neurogenesis After Subarachnoid Hemorrhage in Mice. Front. Mol. Neurosci. 2018, 11, 401. [Google Scholar] [CrossRef] [Green Version]
- Decanini, A.; Nordgaard, C.L.; Feng, X.; Ferrington, D.A.; Olsen, T.W. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am. J. Ophthalmol. 2007, 143, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Kosmidou, C.; Efstathiou, N.E.; Hoang, M.V.; Notomi, S.; Konstantinou, E.K.; Hirano, M.; Takahashi, K.; Maidana, D.E.; Tsoka, P.; Young, L.; et al. Issues with the Specificity of Immunological Reagents for NLRP3: Implications for Age-related Macular Degeneration. Sci. Rep. 2018, 8, 461. [Google Scholar] [CrossRef]
- Kerur, N.; Fukuda, S.; Banerjee, D.; Kim, Y.; Fu, D.; Apicella, I.; Varshney, A.; Yasuma, R.; Fowler, B.J.; Baghdasaryan, E.; et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 2018, 24, 50–61. [Google Scholar] [CrossRef]
- Dang, E.V.; McDonald, J.G.; Russell, D.W.; Cyster, J.G. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 2017, 171, 1057–1071.e11. [Google Scholar] [CrossRef] [Green Version]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Balacco Gabrieli, C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [Green Version]
- Kenney, M.C.; Atilano, S.R.; Boyer, D.; Chwa, M.; Chak, G.; Chinichian, S.; Coskun, P.; Wallace, D.C.; Nesburn, A.B.; Udar, N.S. Characterization of retinal and blood mitochondrial DNA from age-related macular degeneration patients. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4289–4297. [Google Scholar] [CrossRef]
- Hytti, M.; Korhonen, E.; Hyttinen, J.M.T.; Roehrich, H.; Kaarniranta, K.; Ferrington, D.A.; Kauppinen, A. Antimycin A-Induced Mitochondrial Damage Causes Human RPE Cell Death despite Activation of Autophagy. Oxid. Med. Cell Longev. 2019, 2019, 1583656. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 100858. [Google Scholar] [CrossRef]
- Luhmann, U.F.; Robbie, S.; Munro, P.M.; Barker, S.E.; Duran, Y.; Luong, V.; Fitzke, F.W.; Bainbridge, J.W.; Ali, R.R.; MacLaren, R.E. The drusenlike phenotype in aging Ccl2-knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5934–5943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Forrester, J.V.; Xu, H. Dysregulation in retinal para-inflammation and age-related retinal degeneration in CCL2 or CCR2 deficient mice. PLoS ONE 2011, 6, e22818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sennlaub, F.; Auvynet, C.; Calippe, B.; Lavalette, S.; Poupel, L.; Hu, S.J.; Dominguez, E.; Camelo, S.; Levy, O.; Guyon, E.; et al. CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol. Med. 2013, 5, 1775–1793. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Staurenghi, G.; Lepre, T.; Missiroli, F.; Zampatti, S.; Cascella, R.; Borgiani, P.; Marsella, L.T.; Eandi, C.M.; Cusumano, A.; et al. Haplotypes in IL-8 Gene Are Associated to Age-Related Macular Degeneration: A Case-Control Study. PLoS ONE 2013, 8, e66978. [Google Scholar] [CrossRef]
- Fodor, M.; Facska, A.; Rajnavölgyi, E.; Harsfalvi, J.; Bessenyei, E.; Kardos, L.; Berta, A. Enhanced release of IL-6 and IL-8 into tears in various anterior segment eye diseases. Ophthalmic Res. 2006, 38, 182–188. [Google Scholar] [CrossRef]
- Leung, K.W.; Barnstable, C.J.; Tombran-Tink, J. Bacterial endotoxin activates retinal pigment epithelial cells and induces their degeneration through IL-6 and IL-8 autocrine signaling. Mol. Immunol. 2009, 46, 1374–1386. [Google Scholar] [CrossRef]
- Afarid, M.; Azimi, A.; Malekzadeh, M. Evaluation of serum interferons in patients with age-related macular degeneration. J. Res. Med. Sci. 2019, 24, 24. [Google Scholar] [CrossRef]
- Cerniauskas, E.; Kurzawa-Akanbi, M.; Xie, L.; Hallam, D.; Moya-Molina, M.; White, K.; Steel, D.; Doherty, M.; Whitfield, P.; Al-Aama, J.; et al. Complement modulation reverses pathology in Y402H-retinal pigment epithelium cell model of age-related macular degeneration by restoring lysosomal function. Stem Cells Transl. Med. 2020, 12, 1585–1603. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| iPS-RPE Line | Clinical Diagnosis | Donor Age (Gender) | History of Smoking | Additional Medication (Duration/Number) |
|---|---|---|---|---|
| AMD-RPE | AMD wet one eye, dry one eye | 71 (M) | Yes, not current | Blood pressure medication (5 years) Anti-VEGF injections: 2 |
| control-RPE | Control | 73 (M) | No | Blood pressure medication anticoagulants (both 12 years) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hytti, M.; Korhonen, E.; Hongisto, H.; Kaarniranta, K.; Skottman, H.; Kauppinen, A. Differential Expression of Inflammasome-Related Genes in Induced Pluripotent Stem-Cell-Derived Retinal Pigment Epithelial Cells with or without History of Age-Related Macular Degeneration. Int. J. Mol. Sci. 2021, 22, 6800. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136800
Hytti M, Korhonen E, Hongisto H, Kaarniranta K, Skottman H, Kauppinen A. Differential Expression of Inflammasome-Related Genes in Induced Pluripotent Stem-Cell-Derived Retinal Pigment Epithelial Cells with or without History of Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2021; 22(13):6800. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136800
Chicago/Turabian StyleHytti, Maria, Eveliina Korhonen, Heidi Hongisto, Kai Kaarniranta, Heli Skottman, and Anu Kauppinen. 2021. "Differential Expression of Inflammasome-Related Genes in Induced Pluripotent Stem-Cell-Derived Retinal Pigment Epithelial Cells with or without History of Age-Related Macular Degeneration" International Journal of Molecular Sciences 22, no. 13: 6800. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136800