
The Role of Fatty Acids in Non-Alcoholic Fatty Liver Disease Progression: An Update

 , ,
, ,  and
and
Abstract
:1. Introduction
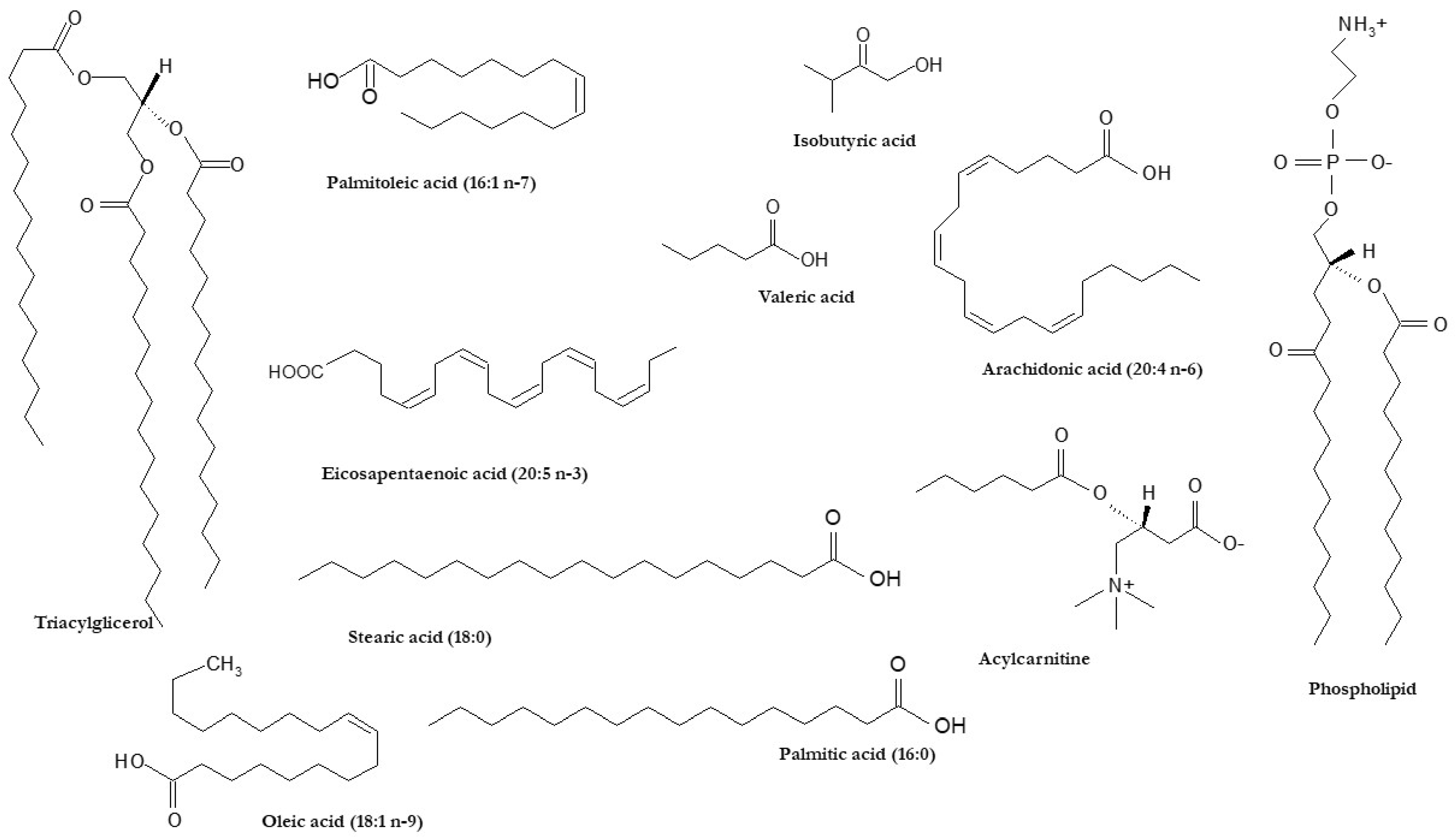
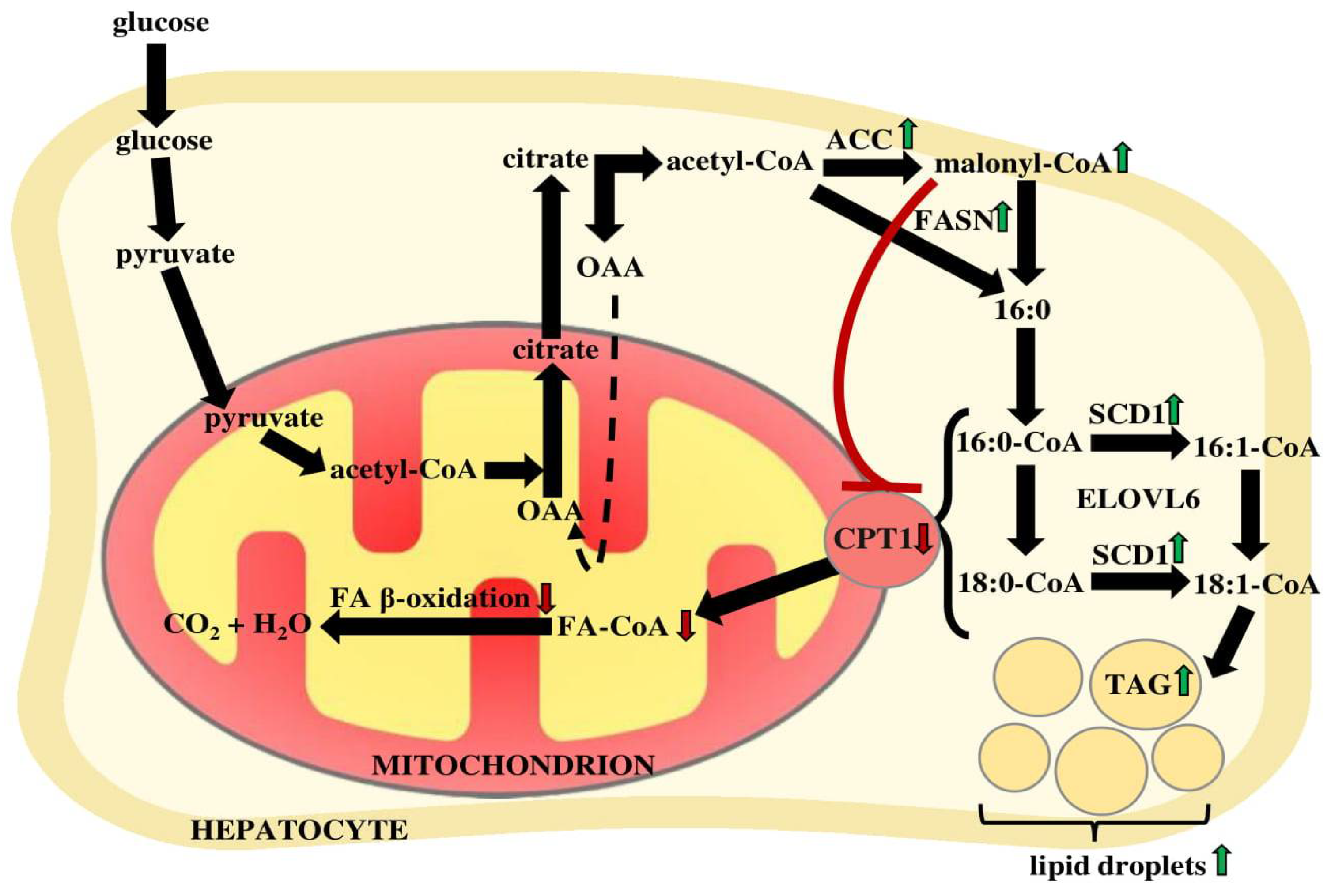
2. Alterations of Hepatic FA Profile in NAFLD Pathogenesis
3. Circulating FAs as Potential Biomarkers of NAFLD
4. Alterations of the Expression of Genes Related to NAFLD
5. Gut Microbiota-Derived Short-Chain Fatty Acids and NAFLD
6. Hepatocellular Carcinoma and Lipid Alterations
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis. 2008, 28, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar] [CrossRef] [Green Version]
- Clouston, A.D.; Powell, E.E. Nonalcoholic fatty liver disease: Is all the fat bad? Intern. Med. J. 2004, 34, 187–191. [Google Scholar] [CrossRef]
- Sanyal, A.J. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology 2002, 123, 1705–1725. [Google Scholar] [CrossRef]
- Peters, R.L.; Gay, T.; Reynolds, T.B. Post jejunoileal bypass hepatic disease. Its similarity to alcoholic hepatic disease. Am. J. Clin. Pathol. 1975, 63, 318–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drenick, E.J.; Simmons, F.; Murphy, J.F. Effect on Hepatic Morphology of Treatment of Obesity by Fasting, Reducing Diets and Small-Bowel Bypass. N. Engl. J. Med. 1970, 282, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.M.; Brunt, E.M. Pathology of nonalcoholic fatty liver disease. Am. J. Clin. Pathol. 2007, 128, 837–847. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Das, U. Essential Fatty Acids—A Review. Curr. Pharm. Biotechnol. 2006, 7, 467–482. [Google Scholar] [CrossRef]
- Calder, P.C. Functional Roles of Fatty Acids and Their Effects on Human Health. J. Parenter. Enter. Nutr. 2015, 39, 18S–32S. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Czumaj, A.; Śledziński, T. Biological role of unsaturated fatty acid desaturases in health and disease. Nutrients 2020, 12, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, U.N. Essential fatty acids: Biochemistry, physiology and pathology. Biotechnol. J. 2006, 1, 420–439. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Bailey, N.J.C.; Johnson, H.E. Measuring the metabolome: Current analytical technologies. Analyst 2005, 130, 606–625. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, Y.; Fu, Y.; Guo, G.; Zhang, X. Liver fatty acid composition in mice with or without nonalcoholic fatty liver disease. Lipids Health Dis. 2011, 10, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuipers, E.J.; Yang, V.W.; Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. REVIEWS IN BASIC AND CLINICAL GASTROENTEROLOGY AND HEPATOLOGY Bioactive Lipid Species and Metabolic Pathways in Progression and Resolution of Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 282–302.e8. [Google Scholar] [CrossRef]
- Peng, C.; Stewart, A.G.; Woodman, O.L.; Ritchie, R.H.; Qin, C.X. Non-Alcoholic Steatohepatitis: A Review of Its Mechanism, Models and Medical Treatments. Front. Pharmacol. 2020, 11, 1864. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.-X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of Hepatic Insulin Resistance in Non-alcoholic Fatty Liver Disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446. [Google Scholar] [CrossRef] [Green Version]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á.M. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Yamada, K.; Mizukoshi, E.; Sunagozaka, H.; Arai, K.; Yamashita, T.; Takeshita, Y.; Misu, H.; Takamura, T.; Kitamura, S.; Zen, Y.; et al. Characteristics of hepatic fatty acid compositions in patients with nonalcoholic steatohepatitis. Liver Int. 2015, 35, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Hardy, S.; El-Assaad, W.; Przybytkowski, E.; Joly, E.; Prentki, M.; Langelier, Y. Saturated Fatty Acid-induced Apoptosis in MDA-MB-231 Breast Cancer Cells a role for cardiolipin. J. Biol. Chem. 2003, 278, 31861–31870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, I.J.; Ginsberg, H.N. Ins and Outs Modulating Hepatic Triglyceride and Development of Nonalcoholic Fatty Liver Disease. Gastroenterology 2006, 130, 1343–1346. [Google Scholar] [CrossRef]
- Pfaffenbach, K.T.; Gentile, C.L.; Nivala, A.M.; Wang, D.; Wei, Y.; Pagliassotti, M.J. Linking endoplasmic reticulum stress to cell death in hepatocytes: Roles of C/EBP homologous protein and chemical chaperones in palmitate-mediated cell death. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1027–E1035. [Google Scholar] [CrossRef] [Green Version]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallée, J.-C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juárez-Hernández, E.; Chávez-Tapia, N.C.; Uribe, M.; Barbero-Becerra, V.J. Role of bioactive fatty acids in nonalcoholic fatty liver disease. Nutr. J. 2016, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bonsembiante, L.; Targher, G.; Maffeis, C. Non-alcoholic fatty liver disease in obese children and adolescents: A role for nutrition? Eur. J. Clin. Nutr. 2021, 1–12. [Google Scholar] [CrossRef]
- Kew, M.C. Serum aminotransferase concentration as evidence of hepatocellular damage. Lancet 2000, 355, 591–592. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef] [Green Version]
- Long, M.T.; Gandhi, S.; Loomba, R. Advances in non-invasive biomarkers for the diagnosis and monitoring of non-alcoholic fatty liver disease. Metabolism 2020, 111, S:154259. [Google Scholar] [CrossRef]
- Wong, V.W.S.; Adams, L.A.; de Lédinghen, V.; Wong, G.L.H.; Sookoian, S. Noninvasive biomarkers in NAFLD and NASH—current progress and future promise. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 461–478. [Google Scholar] [CrossRef]
- Ferraioli, G.; Monteiro, L.B.S. Ultrasound-based techniques for the diagnosis of liver steatosis. World J. Gastroenterol. 2019, 25, 6053–6062. [Google Scholar] [CrossRef]
- Masarone, M.; Troisi, J.; Aglitti, A.; Torre, P.; Colucci, A.; Dallio, M.; Federico, A.; Balsano, C.; Persico, M. Untargeted metabolomics as a diagnostic tool in NAFLD: Discrimination of steatosis, steatohepatitis and cirrhosis. Metabolomics 2021, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Tavares De Almeida, I.; Cortez-Pinto, H.; Fidalgo, G.; Rodrigues, D.; Camilo, M.E. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clin. Nutr. 2002, 21, 219–223. [Google Scholar] [CrossRef]
- Zhou, Y.; Orešič, M.; Leivonen, M.; Gopalacharyulu, P.; Hyysalo, J.; Arola, J.; Verrijken, A.; Francque, S.; Van Gaal, L.; Hyötyläinen, T.; et al. Noninvasive Detection of Nonalcoholic Steatohepatitis Using Clinical Markers and Circulating Levels of Lipids and Metabolites. Clin. Gastroenterol. Hepatol. 2016, 14, 1463–1472.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, R.; Luo, C.; Li, C.; Du, S.; Okekunle, A.P.; Li, Y.; Chen, Y.; Zi, T.; Niu, Y. Free fatty acids profile among lean, overweight and obese non-alcoholic fatty liver disease patients: A case-Control study. Lipids Health Dis. 2017, 16. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H.; et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, K.A.; Walker, C.L.; Pavlina, T.M.; Xu, Z.; Zaloga, G.P.; Siddiqui, R.A. Long-chain saturated fatty acids induce pro-inflammatory responses and impact endothelial cell growth. Clin. Nutr. 2010, 29, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.T.; Elfert, A.; Helal, M.; Salama, I.; El-Said, H.; Fiehn, O. Remodeling lipids in the transition from chronic liver disease to hepatocellular carcinoma. Cancers 2021, 13, 88. [Google Scholar] [CrossRef]
- Diraison, F.; Moulin, P.H.; Beylot, M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003, 29, 478–485. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Coste, A.; Poordad, F.; Alkhouri, N.; Loo, N.; McColgan, B.J.; Tarrant, J.M.; Nguyen, T.; Han, L.; Chung, C.; et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2018, 16, 1983–1991.e3. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwood, H.J. Treating the meytabolic syndrome: Acetyl-CoA carboxylase inhibition. Expert Opin. Ther. Targets 2005, 9, 267–281. [Google Scholar] [CrossRef]
- Loomba, R.; Kayali, Z.; Noureddin, M.; Ruane, P.; Lawitz, E.J.; Bennett, M.; Wang, L.; Harting, E.; Tarrant, J.M.; McColgan, B.J.; et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1463–1473.e6. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Chakravarthy, M.; Previs, S.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 394–406.e6. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, M.; Flowers, M.T.; Sampath, H.; Chu, K.; Otzelberger, C.; Liu, X.; Ntambi, J.M. Hepatic Stearoyl-CoA Desaturase-1 Deficiency Protects Mice from Carbohydrate-Induced Adiposity and Hepatic Steatosis. Cell Metab. 2007, 6, 484–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao-Borengasser, A.; Rassouli, N.; Varma, V.; Bodles, A.M.; Rasouli, N.; Unal, R.; Phanavanh, B.; Ranganathan, G.; McGehee, R.E.; Kern, P.A. Stearoyl-coenzyme A desaturase 1 gene expression increases after pioglitazone treatment and is associated with peroxisomal proliferator-activated receptor-γ responsiveness. J. Clin. Endocrinol. Metab. 2008, 93, 4431–4439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flowers, J.B.; Rabaglia, M.E.; Schueler, K.L.; Flowers, M.T.; Lan, H.; Keller, M.P.; Ntambi, J.M.; Attie, A.D. Loss of Stearoyl-CoA Desaturase-1 Improves Insulin Sensitivity in Lean Mice but Worsens Diabetes in Leptin-Deficient Obese Mice. Diabetes 2007, 56, 1228–1239. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Li, Z.; Liu, F.; Ellsworth, K.; Dallas-Yang, Q.; Wu, M.; Ronan, J.; Esau, C.; Murphy, C.; Szalkowski, D.; et al. Prevention of obesity in mice by antisense oligonucleotide inhibitors of stearoyl-CoA desaturase-1. J. Clin. Investig. 2005, 115, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Flowers, M.T.; Miyazaki, M.; Liu, X.; Ntambi, J.M. Probing the role of stearoyl-CoA desaturase-1 in hepatic insulin resistance. J. Clin. Investig. 2006, 116, 1478–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Moraes Domingos, M.; Fernanda, M.; Rodrigues, C.; Stotzer, U.S.; Rodrigues Bertucci, D.; Vinicius, M.; Souza, C.; Adorna, D.; Camila, M. Resistance training restores the gene expression of molecules related to fat oxidation and lipogenesis in the liver of ovariectomized rats. Eur. J. Appl. Physiol. 2012, 112, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Czumaj, A.; Śledziński, T.; Carrero, J.J.; Stepnowski, P.; Sikorska-Wisniewska, M.; Chmielewski, M.; Mika, A. Alterations of Fatty Acid Profile May Contribute to Dyslipidemia in Chronic Kidney Disease by Influencing Hepatocyte Metabolism. Int. J. Mol. Sci. 2019, 20, 2470. [Google Scholar] [CrossRef] [Green Version]
- Czumaj, A.; Mika, A.; Chmielewski, M.; Sledzinski, T. Cyclopropaneoctanoic Acid 2-Hexyl Upregulates the Expression of Genes Responsible for Lipid Synthesis and Release in Human Hepatic HepG2 Cells. Lipids 2018, 53, 345–351. [Google Scholar] [CrossRef]
- Catanzaro, G.; Filardi, T.; Sabato, C.; Vacca, A.; Migliaccio, S.; Morano, S.; Ferretti, E. Tissue and circulating microRNAs as biomarkers of response to obesity treatment strategies. J. Endocrinol. Investig. 2021, 44, 1159–1174. [Google Scholar] [CrossRef]
- Cannataro, R.; Perri, M.; Gallelli, L.; Caroleo, M.C.; De Sarro, G.; Cione, E. Ketogenic Diet Acts on Body Remodeling and MicroRNAs Expression Profile. MicroRNA 2018, 8, 116–126. [Google Scholar] [CrossRef]
- Kumashiro, N.; Yoshimura, T.; Cantley, J.L.; Majumdar, S.K.; Guebre-Egziabher, F.; Kursawe, R.; Vatner, D.F.; Fat, I.; Kahn, M.; Erion, D.M.; et al. Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology 2013, 57, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Verrijken, A.; Beckers, S.; Francque, S.; Hilden, H.; Caron, S.; Zegers, D.; Ruppert, M.; Hubens, G.; Van Marck, E.; Michielsen, P.; et al. A gene variant of PNPLA3, but not of APOC3, is associated with histological parameters of NAFLD in an obese population. Obesity 2013, 21, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Yamaguchi, K.; Itoh, Y. The genetic backgrounds in nonalcoholic fatty liver disease. Clin. J. Gastroenterol. 2018, 11, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernaez, R. Genetic factors associated with the presence and progression of nonalcoholic fatty liver disease: A narrative review. Gastroenterol. Hepatol. 2012, 35, 32–41. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; Mcgilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Masujima, Y.; Ushiroda, C.; Mizushima, R.; Taira, S.; Ohue-Kitano, R.; Kimura, I. Dietary short-chain fatty acid intake improves the hepatic metabolic condition via FFAR3. Sci. Rep. 2019, 9, 16574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollica, M.P.; Raso, G.M.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Guida, F.D.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; Bleeker, A.; Gerding, A.; Van Eunen, K.; Havinga, R.; Van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-chain fatty acids protect against high-fat diet-induced obesity via a pparg-dependent switch from lipogenesis to fat oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Pan, Q.; Xin, F.Z.; Zhang, R.N.; He, C.X.; Chen, G.Y.; Liu, C.; Chen, Y.W.; Fan, J.G. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J. Gastroenterol. 2017, 23, 60–75. [Google Scholar] [CrossRef]
- Weitkunat, K.; Schumann, S.; Nickel, D.; Kappo, K.A.; Petzke, K.J.; Kipp, A.P.; Blaut, M.; Klaus, S. Importance of propionate for the repression of hepatic lipogenesis and improvement of insulin sensitivity in high-fat diet-induced obesity. Mol. Nutr. Food Res. 2016, 60, 2611–2621. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.-H.; Wang, Z.-X.; Zhou, D.; Han, Y.; Ma, F.; Hu, Z.; Xin, F.-Z.; Liu, X.-L.; Ren, T.-Y.; Zhang, F.; et al. Sodium butyrate supplementation inhibits hepatic steatosis by stimulating liver kinase B1 and insulin-induced gene. Cell. Mol. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Müller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M.; et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Haga, S.; Aoyama, Y.; Kiriyama, S. Biochemical and Molecular Action of Nutrients Short-Chain Fatty Acids Suppress Cholesterol Synthesis in Rat Liver and Intestine. J. Nutr. 1999, 129, 942–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, S.; Qin, S.; Li, L.; Zhu, L.; Zou, Z.; Wang, L. Dietary butyrate suppresses inflammation through modulating gut microbiota in high-fat diet-fed mice. FEMS Microbiol. Lett. 2019, 366, 153. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tenen, D.G.; Chai, L.; Tan, J.L. Metabolic alterations and vulnerabilities in hepatocellular carcinoma. Gastroenterol. Rep. 2021, 9, 1–13. [Google Scholar] [CrossRef]
- Yaligar, J.; Teoh, W.W.; Othman, R.; Verma, S.K.; Phang, B.H.; Lee, S.S.; Wang, W.W.; Toh, H.C.; Gopalan, V.; Sabapathy, K.; et al. Longitudinal metabolic imaging of hepatocellular carcinoma in transgenic mouse models identifies acylcarnitine as a potential biomarker for early detection. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Hayata, Y.; Kawamura, S.; Yamada, T.; Fujiwara, N.; Koike, K. Lipid metabolic reprogramming in hepatocellular carcinoma. Cancers 2018, 10, 447. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Henry, L. Epidemiology of Non-alcoholic Fatty Liver Disease and Hepatocellular Carcinoma. JHEP Rep. 2021, 100305. [Google Scholar] [CrossRef]
- Zoller, H.; Tilg, H. Nonalcoholic fatty liver disease and hepatocellular carcinoma. Metabolism. 2016, 65, 1151–1160. [Google Scholar] [CrossRef]
- Baffy, G. Hepatocellular carcinoma in non-alcoholic fatty liver disease: Epidemiology, pathogenesis, and prevention. J. Clin. Transl. Hepatol. 2013, 1, 131–137. [Google Scholar] [PubMed] [Green Version]
- Wang, M.; Han, J.; Xing, H.; Zhang, H.; Li, Z.; Liang, L.; Li, C.; Dai, S.; Wu, M.; Shen, F.; et al. Dysregulated fatty acid metabolism in hepatocellular carcinoma. Hepatic Oncol. 2016, 3, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Q.; Yin, P.; Xing, W.; Wu, Z.; Chen, S.; Lu, X.; Zhang, Y.; Lin, X.; Xu, G. Serum metabolomics reveals the deregulation of fatty acids metabolism in hepatocellular carcinoma and chronic liver diseases. Anal. Bioanal. Chem. 2012, 403, 203–213. [Google Scholar] [CrossRef]
- Qiu, J.F.; Zhang, K.L.; Zhang, X.J.; Hu, Y.J.; Li, P.; Shang, C.Z.; Wan, J.B. Abnormalities in Plasma Phospholipid Fatty Acid Profiles of Patients with Hepatocellular Carcinoma. Lipids 2015, 50, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Vlock, E.M.; Karanjit, S.; Talmon, G.; Farazi, P.A. Reduction of polyunsaturated fatty acids with tumor progression in a lean non-alcoholic steatohepatitis- associated hepatocellular carcinoma mouse model. J. Cancer 2020, 11, 5536–5546. [Google Scholar] [CrossRef] [PubMed]
- Passos-Castilho, A.M.; Carvalho, V.M.; Cardozo, K.H.M.; Kikuchi, L.; Chagas, A.L.; Gomes-Gouvêa, M.S.; Malta, F.; de Seixas-Santos Nastri, A.C.; Pinho, J.R.R.; Carrilho, F.J.; et al. Serum lipidomic profiling as a useful tool for screening potential biomarkers of hepatitis B-related hepatocellular carcinoma by ultraperformance liquid chromatography-mass spectrometry. BMC Cancer 2015, 15, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, M.P.; Liesa, M. The Role of Mitochondrial Fat Oxidation in Cancer Cell Proliferation and Survival. Cells 2020, 9, 2600. [Google Scholar] [CrossRef] [PubMed]
- Mika, A.M.; Duzowska, K.; Halinski, L.P.; Pakiet, A.; Czumaj, A.; Rostkowska, O.; Dobrzycka, M.; Kobiela, J.; Sledzinski, T. Rearrangements of blood and tissue fatty acid profile in colorectal cancer—Molecular mechanism and diagnostic potential. Front. Oncol. 2021, 11, 1898. [Google Scholar] [CrossRef]
- Lin, L.; Ding, Y.; Wang, Y.; Wang, Z.; Yin, X.; Yan, G.; Zhang, L.; Yang, P.; Shen, H. Functional lipidomics: Palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology 2017, 66, 432–448. [Google Scholar] [CrossRef]
- Lin, J. Commentary: Anti-tumor Effect of Oleic Acid in Hepatocellular Carcinoma Cell Lines via Autophagy Reduction. Front. Cell Dev. Biol. 2021, 9, 901. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Lipid Molecule | Used Matrix | Patients/ Experimental Model | Direction of Change | Reference |
|---|---|---|---|---|
| 16:0 MUFA 16:1 | FAs in plasma phospholipids | HCC patients | ↓ | [91] |
| tumor tissue | HCC patients | ↑ | [91] | |
| FFAs in plasma | HCC patients | ↑ | [90] | |
| 18:1 n-9 PUFA | FAs in plasma phospholipids | HCC patients | ↓ | [91] |
| tumor tissue | HCC patients | ↓ | [91] | |
| 18:2 n-6 18:2 n-6 18:3 n-3 20:4 n-6 | FAs in plasma phospholipids | HCC patients | ↓ | [91] |
| FFAs in plasma | mouse model | ↓ | [92] | |
| FFAs in plasma | mouse model | ↓ | [92] | |
| FAs in plasma phospholipids | HCC patients | ↓ | [91] | |
| 20:5 n-3 22:6 n-3 | FFAs in plasma | mouse model | ↓ | [92] |
| FFAs in plasma | mouse model | ↓ | [92] | |
| acylcarnitines | serum samples | HCC patients | ↑ | [83,90] |
| phosphatidylcholines (PCs) | serum samples | HCC patients | ↓ | [93] |
| phosphatidylserines (PSs) | serum samples | HCC patients | ↓ | [93] |
| phosphatidylinositols (PIs) | serum samples | HCC patients | ↓ | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hliwa, A.; Ramos-Molina, B.; Laski, D.; Mika, A.; Sledzinski, T. The Role of Fatty Acids in Non-Alcoholic Fatty Liver Disease Progression: An Update. Int. J. Mol. Sci. 2021, 22, 6900. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136900
Hliwa A, Ramos-Molina B, Laski D, Mika A, Sledzinski T. The Role of Fatty Acids in Non-Alcoholic Fatty Liver Disease Progression: An Update. International Journal of Molecular Sciences. 2021; 22(13):6900. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136900
Chicago/Turabian StyleHliwa, Aleksandra, Bruno Ramos-Molina, Dariusz Laski, Adriana Mika, and Tomasz Sledzinski. 2021. "The Role of Fatty Acids in Non-Alcoholic Fatty Liver Disease Progression: An Update" International Journal of Molecular Sciences 22, no. 13: 6900. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136900